
Abstract
The function of Casein kinase 2 beta (CSNK2B) in human malignancies has drawn increasing attention in recent years. However, its role in colorectal cancer (CRC) remains unclear. In the present study, we aimed to explore the expression and biological functions of CSNK2B in CRC. Public gene expression microarray data from online database and immunohistochemistry analysis demonstrated that CSNK2B was highly expressed in CRC tissues than in normal tissues. In vitro and in vivo cellular functional experiments showed that increased CSNK2B expression promoted CRC cell viability and tumorigenesis of CRC. Further western blots and rescue experiments confirmed that CSNK2B promoted CRC cell proliferation mainly by activating the mTOR signaling pathway. These findings identified CSNK2B as a novel oncogene contributing to the development of CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the third most common malignancy worldwide with increasing incidence annually (Bray et al. 2018; Siegel et al. 2020). Although combined therapies including surgical resection, chemotherapy, targeted therapy and radiotherapy have effectively prolonged the overall survival (OS) of patients with early CRC, the long-term prognosis of patients with advanced CRC remains poor due to postoperative recurrence and high metastatic potential of this disease (Aparicio et al. 2020; Shuford et al. 2020). Therefore, a better understanding of the mechanisms underlying CRC tumorigenesis and progression is critical for developing effective therapeutic strategies.
Casein kinase 2 beta (CSNK2B) is a gene encoding the beta subunit of casein kinase II (CK2) (Heller-Harrison et al. 1989), a ubiquitous protein kinase which regulates a plethora of biological functions including cell proliferation, differentiation and survival by phosphorylating serine and threonine residues of its target proteins (Litchfield 2003; Silva-Pavez and Tapia 2020). CK2 is composed of 2 alpha subunits and a dimer of beta subunits. While the catalytic subunit of CK2 controls the kinase activity of CK2, CSNK2B, is thought to regulate the substrate-specific targeting of catalytic subunits (Guerra and Issinger 1999).
In the area of cancer research, emerging evidence has gradually suggested a critical role of CSNK2B in various malignancies (Filhol et al. 2015; Pallares et al. 2009). For example, overexpression of CSNK2B has been found to activate the NF-κB signaling in hepatocellular carcinoma (HCC), thus promoting cell proliferation and inhibiting cell apoptosis of HCC (Xiao et al. 2020). In another study of breast cancer (BC), higher CSNK2B expression was significantly correlated with worse prognosis of patients suffering from BC (Wang et al. 2019). However, the role of CSNK2B, and its mechanism of functions in CRC are still obscure.
In the present study, we resorted to explore the expression patterns and functions of CSNK2B in CRC. Our results indicated that CSNK2B expression is frequently over-expressed in CRC tissues than in normal tissues, and CSNK2B promotes CRC cell proliferation in vitro and in vivo via activating the mTOR signaling pathway.
Materials and methods
Cell culture and reagents
Human CRC cell lines HT29, LoVo, DLD1, SW620, SW480, Caco-2, HCT116 and RKO were purchased from Shanghai Cell Bank of Chinese Academy of Science (Shanghai, China). Cell line authenticity and mycoplasma contamination were routinely checked by PCR-based assays and STR genotyping, respectively, in 6–10-month intervals. Cells were cultured with Dulbecco's modified Eagle medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% penicillin/streptomycin (#15140122, Thermo Fisher Scientific, USA) in a humidified incubator with 5% CO2 at 37 °C. All cell line experiments were conducted within 10 passages after thawing. Rapamycin was supplied by Selleck Chemicals (#S1039, Shanghai, China), and the working concentration was 20 nM.
Immunohistochemical analysis
To evaluate CSNK2B protein expression in CRC, a tissue microarray (TMA) containing 50 paired CRC tumor tissues and adjacent normal tissues was obtained from Alenabio Biotech Company (#DC-Col02006, Xian, China), and immunohistochemical (IHC) analysis was performed following standard staining procedures with a specific antibody against CSNK2B (1:200, #20234-1-AP, ProteinTech, Wuhan, China). Briefly, the paraffin-embedded sections were deparaffinized by xylene, followed by re-hydration using graded ethanol and then rinsed with deionized water. 3% hydrogen peroxide was used to block endogenous peroxidase activity. For antigen retrieval, the sections were high-pressure-treated and boiled in a 10 mM citrate buffer (pH 6.0) for 4 min. Nonspecific binding was blocked with 5% normal goat serum for 30 min at room temperature. After which, the sections were incubated with primary antibodies overnight at 4℃ and subjected to incubation with horseradish peroxidase-conjugated secondary antibody. Finally, the sections were stained with the DAB substrate at room temperature. The final IHC scores were analyzed by two pathologists blinded to the clinical information independently based on the percentage of stained cells and staining intensity. Briefly, the percentage of CSNK2B-positive cells was classified into 5 groups: < 10% (0), 10–25% (1), 25–50% (2), 50–75 (3), and > 75% (4). The staining intensity was divided into 4 groups: no staining (0), light brown (1), brown (2), and dark brown (3). The overall IHC scores of CSNK2B were calculated using the following formula: overall score = percentage score × intensity score. The samples with an overall score ≤ 6 were defined as weak staining, and > 6 were defined as strong staining. Ethical approval was granted by the ethics committee of Shanghai East Hospital, Tongji University School of Medicine, China.
Online gene expression analysis of gene expression omnibus datasets
To verify the expression of CSNK2B in CRC tissues, previously published microarray data from Gene Expression Omnibus (GEO) datasets (GEO accession number: GSE6988, GSE8671 GSE20842 and GSE20916) were reanalyzed using GEO2R, a R-based publicly online tool for analyzing GEO-deposited gene expression data (http://www.ncbi.nlm.nih.gov/geo/geo2r).
Plasmid and small-inference RNA transfection
CSNK2B-expressing plasmid containing a Flag tag (pCSNK2B, GeneBank Accession Number: NM_001320) was purchased from Vigene Biosciences, Shandong, China (#CH817776), and small inference RNAs (siRNAs) specifically against CSNK2B (siCSNK2B-1 and siCSNK2B-2) were designed and synthesized by GenePharma, an empty plasmid pcDNA3.1 (pVEC) and negative control siRNA (siNC) served as their control groups, respectively. Cell transfection was performed using Lipofectamine 3000 Transfection Reagent (Invitrogen, CA, USA) in accordance with the instructions. To generate stable CSNK2B-overexpression or knockdown cell lines, human embryonic kidney (HEK)-293T cells were used to package the recombinant lentivirus containing CSNK2B gene and shRNAs against CSNK2B (shCSNK2B). The CRC cell lines were infected with lentivirus particles in the presence of Polybrene (4 μg/ml). After 48 h, puromycin (2.5 μg/ml, Sigma, MO, USA) was added to the medium to select stably infected cells. The sequences of siRNAs are as follows (5′–3′): siCSNK2B-1: CUCCGUGGCAAUGAAUUCUdTdT, siCSNK2B-2: GUCAAGACGAUUCGCUGAUdTdT, siNC: UUCUCCGAACGUGUCACGUdTdT, among which siCSNK2B-2 was chosen to construct shCSNK2B.
Western blot analysis
Total cellular proteins were extracted using RIPA lysis buffer (#20-188, Millipore, USA) containing protease inhibitor cocktail (#P8340, Signa-Akdrich, USA) and phosphatase inhibitor cockatil (#P2850, Signa-Akdrich, USA), protein concentration was determined with BCA Protein Assay Kit (#23225, Thermo Fisher Scientific, USA) according to the manufacturer’s protocol. Protein samples were boiled in 1× sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer (#KGP101, KeyGENBio, Nanjing, China) and equal amounts of them were subjected to 10% SDS-PAGE (30 μg of protein were loaded per lane, 5% laminating gel, 10% separation gel, laminated glue voltage 80 V, separation gel voltage 150 V, electrophoresis time 60 min) followed by transfer onto a nitrocellulose membrane (1 h transfer at constant current of 250 mA). The membrane was then soaked in 5% non-fat milk for 1 h to block unspecific binding and subjected to incubation with diluted primary antibodies overnight at 4 °C, after which the membrane was incubated with secondary antibodies for 1 h at room temperature. The bands were visualized using a LI-COR Odyssey imaging system (LI-COR Biosciences, USA). Specific antibodies used in this study are as follows: anti-CSNK2B (1:2000, #20234-1-AP, ProteinTech, Wuhan, China), anti-Flag tag (1:1000, #abs137958 Absin Bioscience, Shanghai, China), anti-β-actin (1:500, #sc-47778, Santa Cruz Biotechnology, CA, USA), anti-mTOR (1:1000, #2983S, Cell Signaling Technology, MA, USA), anti-P-mTOR (1:1000, #5536S, Cell Signaling Technology, MA, USA), anti-STAT3 (1:1000, #9139S, Cell Signaling Technology, MA, USA), anti-P-STAT3 (1:1000, #9145S, Cell Signaling Technology, MA, USA), anti-FAK (1:500, # 12636-1-AP, ProteinTech, Wuhan, China), anti-p53 (1:200, #sc-126, Santa Cruz Biotechnology, CA, USA), anti-cyclin D1 (1:800, #55506S, Cell Signaling Technology, MA, USA), anti-CDK4 (1:500, #11026-1-AP, ProteinTech, Wuhan, China), anti-NRF2 (1:500, # 16396-1-AP, ProteinTech, Wuhan, China), anti-p70S6K (1:1000, #2708S, Cell Signaling Technology, MA, USA), anti-P-p70S6K (1:500, #9234, Cell Signaling Technology, MA, USA), anti-4E-BP1 (1:800, #9644, Cell Signaling Technology, MA, USA) and anti-P-4EBP1 (1:500, #2855, Cell Signaling Technology, MA, USA). Immunoblots shown are representative of at least three independent biological replicates.
Cell proliferation assay
CRC cell proliferation abilities were assessed using Cell Counting Kit-8 (CCK-8) (#CK04, Dojindo Laboratories, Kumamoto, Japan). Briefly, the indicated CRC cell lines were seeded into a 96-well plate with a density of 3 × 103 cells/well. After adherence, 10 μl of CCK-8 reagent was added into each well at the corresponding time points (0, 1, 2, 3, 4 and 5 days) and incubated for 1 h. Values were acquired as absorbance at 450 nm on a SpectraMax M5 microplate reader (Molecular Device, CA, USA). Each experiment was done in triplicate and repeated at least 3 times independently.
Colony formation assay
Colony-forming capacities were evaluated by colony formation assays. Briefly, the indicated CRC cell lines were seeded into a 6-well plate with a density of 1 × 103 cells/well and cultured for 2 weeks under normal conditions. Subsequently, formed colonies were fixed using 4% paraformaldehyde for 15 min and stained using 0.5% crystal violet for 10 min at room temperature. The stained colonies were then photographed and counted using Image J software.
EdU incorporation assay
EdU incorporation assay was conducted using an EdU labeling kit (#C10310-1, RiboBio, Guangzhou, China) according to the manufacturer’s instructions. In brief, cells were seeded into a 24-well plate with cell density at 60% and incubated for 24 h. Next, 50 μM of EdU was added into each well and incubated for 2 h. After fixation with 4% paraformaldehyde, incubation with glycine (2 mg/ml) and subsequent incubation with 1× Apollo reaction cocktail, the plate was protected from light and stained with Hoechst 33342 (5 μg/ml) for 30 min at room temperature. Finally, the stained cells were visualized under a fluorescence microscope (Leica Microsystems, Wetzlar, Germany). 5 fields were randomly selected and the EdU incorporation rates were quantified as the ratio of EdU-positive cells (red) to Hoechst 33342-positive cells (blue).
Animal experiments
4–6 weeks old BALB/c nude mice were purchased from SLAC Laboratory Animal Co., Ltd., Shanghai, China. A subcutaneous xenograft tumor model of nude mice was established for in vivo tumor growth assays. 2 × 106 HCT116 cells stably overexpressing CSNK2B and SW620 cells with stable knockdown of CSNK2B were inoculated subcutaneously into left or right flank of nude mice (n = 6), respectively. After 3 (HCT116) or 5 (SW620) weeks, the nude mice were euthanized, tumors were removed, photographed and weighted. The nude mice were housed in the SPF animal laboratory at Tongji University, which provided a 12-h light/dark cycle and nude mice were allowed ad libitum access to food and water. All animal handling and experimental procedures were abided by the Ethics Committee of Shanghai East Hospital, Tongji University School of Medicine (Authorization number: 2019tjdx62) and in accordance with the ARRIVE guidelines (https://www.nc3rs.org.uk/arrive-guidelines).
Statistical analysis
Data were presented as mean ± standard deviation (SD) for three biological replicates for all experiments, and statistical analyses were performed using GraphPad Prism software v7.0. Gaussian distribution data were analyzed by two-tailed Student’s t test, while non-Gaussian distribution data by Mann–Whitney of nonparametric test, and one-way ANOVA with a post-hoc Turkey test was used for multiple comparisons, and P < 0.05 was considered to indicate a statistically significant difference.
Results
CSNK2B expression levels were frequently increased in CRC tissues
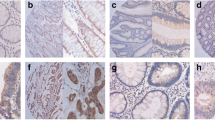
To explore the role of CSNK2B in CRC, we firstly analyzed CSNK2B mRNA expression utilizing 4 previously published gene expression profiles from GEO database (GSE6988, GSE8671 GSE20842 and GSE20916). As shown in Fig. 1a, increased CSNK2B mRNA level was observed in all CRC tissues compared with normal tissues. Furthermore, CSNK2B protein expression was evaluated by IHC method on a CRC tissue microarray containing 50 CRC tissues and paired adjacent normal tissues. Consistently, CSNK2B protein level in CRC tissues was also significantly higher than in normal tissues (Fig. 1b, P < 0.0001). Therefore, these data suggested an overall up-regulation of CSNK2B mRNA and protein levels in CRC tissues, hinting us that CSNK2B is possibly involved in the development of CRC.

CSNK2B expression was significantly up-regulated in CRC tissues. a Human CRC gene expression data from public GEO database (GSE6988, GSE8671 GSE20842 and GSE20916) were analyzed to compare CSNK2B mRNA level between tumor and normal tissues. b CSNK2B protein level in CRC was detected by IHC analysis on a tissue microarray containing 50 CRC tissues and paired adjacent normal tissues. Representative images (left panel, magnification: ×40) and column diagram (right panel) were shown
Overexpression of CSNK2B accelerated CRC cell proliferation in vitro
We next investigated whether CSNK2B plays a role in CRC progression. Endogenous CSNK2B expression in different CRC cell lines was analyzed using western blot analysis. As shown in Fig. 2a, CSNK2B was present in all of the analyzed cell lines. To assess the function of CSNK2B in CRC cell proliferation, RKO and HCT116 with a relatively low level of CSNK2B were transiently transfected with CSNK2B-expressing plasmids (Fig. 2b), and CCK-8 cell proliferation assays were performed. The growth curves suggested that ectopic expression of CSNK2B significantly facilitated CRC cell growth rates (Fig. 2c). Besides, we generated stable CSNK2B-overrexpressing RKO and HCT116 cells with lentivirus-mediated transduction and carried out colony formation assays. As expected, the number of colonies formed from cells with overexpression of CSNK2B was remarkably higher than those from the control cells (Fig. 2d). In addition, EdU incorporation assay was performed to verify the effects of CSNK2B on CRC cell proliferation. Similarly, CSNK2B-overexpressing RKO and HCT116 cells exhibited a higher percentage of EdU-positvie cells, which further supported the above observations (Fig. 2e). Hence, these findings demonstrated that increased CSNK2B expression strongly enhanced CRC cell proliferation abilities.

Overexpression of CSNK2B promoted CRC cell proliferation in vitro. a CSNK2B protein expression in different CRC cell lines was evaluated by western blot analysis. b RKO and HCT116 cells were transfected with Flag-tagged CSNK2B-expressing plasmids, total proteins were extracted after 24 h and western blot analysis was performed. c CCK-8 cell proliferation assays were employed at 24 h post-transfection of CSNK2B-expressing plasmids into RKO and HCT116 cells. d RKO and HCT116 cells with stable CSNK2B overexpression were used to perform colony formation assays. Representative images (left panel) and quantification (right panel) of colonies were shown. e Representative images (left panel) and quantification (right panel) of EdU-positive RKO cells were shown. *P < 0.05, **P < 0.01
Negative regulation of cell growth by CSNK2B knockdown in vitro
Subsequently, whether down-regulated CSNK2B expression could affect proliferation capacities of the CRC cells were explored. DLD1 and SW620 cells with a relatively higher CSNK2B expression level were transfected with siRNAs against CSNK2B (siCSNK2B-1 and siCSNK2B-2), and immunoblots confirmed the knockdown efficiencies (Fig. 3a). In contrast to overexpression, blockade of CSNK2B markedly retarded cell growth rates comparing with the corresponding control groups (Fig. 3b). Moreover, DLD-1 and SW620 with stable knockdown of CSNK2B using lentiviral infection of shRNA were established, and concordant results were observed in colony formation assays (Fig. 3c). Moreover, CSNK2B-knockdown cells exhibited a significantly lower percentage of EdU-positive cells in EdU incorporation assays (Fig. 3d). Taken together, these in vitro observations identified that CSNK2B plays an oncogenic role in CRC cell proliteration in vitro.

Knockdown of CSNK2B retarded CRC cell viability in vitro. a DLD1 and SW620 cells were transfected with siRNAs against CSNK2B, respectively. 48 h after transfection, cells were lysed and immunoblots were performed. b Cell growth curves were measured using CCK-8 assays after 24 h post-transfection of the indicated siRNAs. c Representative images (left panel) and quantification (right panel) of colonies formed from DLD1 and SW620 cells with stable knockdown of CSNK2B were exhibited. d Relative proliferation rates of the indicated cells were assessed by EdU staining, and representative images (left panel) and counting analysis (right panel) were shown. *P < 0.05, **P < 0.01
CSNK2B promoted CRC tumor growth in vivo
With the aim of verifying the role of CSNK2B in CRC tumorigenesis in vivo, we established a subcutaneous xenograft mice model by inoculation of the CRC cells with stable overexpression or knockdown of CSNK2B. As shown in Fig. 4a, increased CSNK2B expression significantly accelerated tumor growth of HCT116 cells. In sharp contrast, CSNK2B-knockdown tumors exhibited a markedly tumor growth rate than the control cells (Fig. 4b). Thus, these above results demonstrated that CSNK2B contributes to CRC tumorigenesis in vivo.

CRC tumor growth was positively regulated by CSNK2B. a–b A subcutaneous xenograft model of nude mice was established using HCT116 cells with stable CSNK2B overexpression (a) or SW620 cells with stable CSNK2B knockdown (b) (n = 6). Xenograft tumor images (left panel), tumor volume curves (middle panel) and tumor weight quantification (right panel) were shown. **P < 0.01
The mTOR/p70S6K signaling mediated the regulation of CRC proliferation by CSNK2B
In order to further explore potential mechanisms by which CSNK2B affected tumor proliferation, we compared the differential expression of key proteins in crucial tumor-associated pathways including mTOR (Murugan 2019), STAT3 (Garg et al. 2020), FAK (Worthmuller and Ruegg 2020), p53 (Huang 2020), cyclin D1 (Bates and Peters 1995), CDK4 (Teh and Aplin 2019) and NRF2 (Liu et al. 2020) by immunoblots after knockdown of CSNK2B in DLD1 cells. Interestingly, down-regulation of CSNK2B induced a decrease of mTOR expression, while the other proteins showed no significant changes (Fig. 5a). It is well established that mTOR is a key member of the phosphatidylinositol 3-kinase-associated kinase superfamily and plays an important role in regulating mammalian cell proliferation and survival (Bracho-Valdes et al. 2011; Sun 2020). More importantly, substantial evidence has elucidated that mTOR acts as an oncogene in various cancers through activation its downstream molecule such as p70S6K and 4E-BP1 (Sridharan and Basu 2020). Thus we subsequently examined the relationship between CSNK2B and the activation of the mTOR signaling pathway. As expected, decreased phosphorylation levels of mTOR, p70S6K and 4E-BP1 were observed in DLD-1 cells with knockdown of CSNK2B, whereas overexpression of CSNK2B led to opposite results (Fig. 5b, c), suggesting that CSNK2B can positively regulate the activity of the mTOR signaling. Therefore, we hypothesized CSNK2B promoted CRC cell proliferation dominantly by activation of the mTOR pathway. To verify our hypothesis, CRC cells with stable overexpression of CSNK2B were treated with rapamycin, an inhibitor of mTOR (Edwards and Wandless 2007). CCK-8 assays showed that the promoting effects of CSNK2B on cell proliferation were largely reversed by rapamycin treatment (Fig. 5c), and similar results were observed in colony formation assays (Fig. 5d). Thus, the above findings confirmed that CSNK2B promoted CRC cell proliferation via activating the mTOR signaling pathway.

The mTOR/p70S6K signaling pathway mediated the positive effects of CSNK2B on CRC cell proliferation. a DLD1 cells were transfected with siCSNK2B-1 and siCSNK2B-2 for 48 h, then total proteins were extracted and western blot analysis was performed to detect the levels of the indicated genes. b–c CRC cells transfected with CSNK2B-expressing plasmids or siCSNK2B-1/2 were subjected to immunoblots to verify the expression changes of the indicated genes, images (b) and densitometric quantification (c) were shown. RKO cells with stable CSNK2B overexpression were treated with rapamycin (20 nM) or DMSO, CCK-8 assays (c) and colony formation assays (d) were performed to explore whether mTOR inhibition could reverse the effects of CSNK2B overexpression on CRC cell viability. *P < 0.05, **P < 0.01
Discussion
Although CSNK2B has been implicated in the progression of several human malignancies, its expression patterns and biological function in CRC are extensively unknown. In the current work, we for the first time identified CSNK2B as an oncogene in CRC. By analyzing public gene expression data from GEO database, we observed a significant up-regulation of CSNK2B mRNA level in CRC tissues comparing with normal tissues, and further IHC analysis showed that CSNK2B protein level is up-regulated in CRC. Meanwhile, considering that there is a discrepancy of CSNK2B gene expression between different datasets, it is necessary to collect clinical and pathological data to further verify the effectiveness of the above findings and explore whether CSNK2B expression is associated with clinicopathological parameters and prognosis of CRC patients in future studies.
Subsequently, we conducted a range of functional experiments to investigate the role of CSNK2B in proliferation ability of CRC cells. Both in vitro and in vivo experiments indicated that CSNK2B potentiated CRC cell proliferation and tumorigenesis. By mechanistic studies, we found that the effects of CSNK2B on CRC cell proliferation were dominantly mediated by abnormal activation of the mTOR signaling pathway, which thus contributing to CRC progression.
Currently, mTOR is commonly recognized as a key regulator of cell proliferation and survival by stimulating two major downstream targets p70S6K and 4E-BP1 (Bracho-Valdes et al. 2011; Sridharan and Basu 2020). Abnormal activation of the mTOR signaling pathway is correlated with the progression of different tumor types including CRC (Ghanaatgar-Kasbi et al. 2019; Lu et al. 2020; Murugan 2019). To date, the regulatory relationship between CSNK2B and the mTOR signaling has not been reported. Herein, we reported that up-regulated CSNK2B expression can result in activation of mTOR and its downstream effectors p70S6K and 4E-BP1, and inhibition of mTOR activity by rapamycin treatment significantly abolished the positive effects of CSNK2B on CRC cell viability. Thus, inhibition of the CSNK2B-mTOR signaling may be effective strategy to suppress CRC progression. For example, 4,5,6,7-tetrabromobenzotriazole (TBB), a selective CK2 inhibitor, has been reported to directly inhibit CSNK2B activity in vitro (Stengel et al. 2020), suggesting that it will be interesting to test whether TBB treatment can inhibit the biological functions of CSNK2B in CRC cells. Still, how CSNK2B activates the mTOR signaling pathway and its correlation with CK2 protein, remain to be further addressed.
In summary, we identified CSNK2B as an oncogene in CRC, which promotes tumor cell proliferation in vitro and in vivo by activating the mTOR signaling pathway. Our findings indicated that CSNK2B might be a potential biomarker and therapeutic target for the diagnosis and treatment against CRC.
References
Aparicio J et al (2020) Metastatic colorectal cancer. First line therapy for unresectable disease. J Clin Med 9:73–82. https://doi.org/10.3390/jcm9123889
Bates S, Peters G (1995) Cyclin D1 as a cellular proto-oncogene. Semin Cancer Biol 6:73–82. https://doi.org/10.1006/scbi.1995.0010
Bracho-Valdes I, Moreno-Alvarez P, Valencia-Martinez I, Robles-Molina E, Chavez-Vargas L, Vazquez-Prado J (2011) mTORC1- and mTORC2-interacting proteins keep their multifunctional partners focused. IUBMB Life 63:896–914. https://doi.org/10.1002/iub.558
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. https://doi.org/10.3322/caac.21492
Edwards SR, Wandless TJ (2007) The rapamycin-binding domain of the protein kinase mammalian target of rapamycin is a destabilizing domain. J Biol Chem 282:13395–13401. https://doi.org/10.1074/jbc.M700498200
Filhol O, Giacosa S, Wallez Y, Cochet C (2015) Protein kinase CK2 in breast cancer: the CK2beta regulatory subunit takes center stage in epithelial plasticity. Cell Mol Life Sci CMLS 72:3305–3322. https://doi.org/10.1007/s00018-015-1929-8
Garg M et al (2020) The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Med Res Rev. https://doi.org/10.1002/med.21761
Ghanaatgar-Kasbi S, Amerizadeh F, Rahmani F, Hassanian SM, Khazaei M, Ferns GA, Avan A (2019) AMP-kinase inhibitor dorsomorphin reduces the proliferation and migration behavior of colorectal cancer cells by targeting the AKT/mTOR pathway. IUBMB Life 71:1929–1936. https://doi.org/10.1002/iub.2136
Guerra B, Issinger OG (1999) Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis 20:391–408. https://doi.org/10.1002/(SICI)1522-2683(19990201)20:2%3c391::AID-ELPS391%3e3.0.CO;2-N
Heller-Harrison RA, Meisner H, Czech MP (1989) Cloning and characterization of a cDNA encoding the beta subunit of human casein kinase II. Biochemistry 28:9053–9058. https://doi.org/10.1021/bi00449a014
Huang J (2020) Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol Ther 220:107720. https://doi.org/10.1016/j.pharmthera.2020.107720
Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369:1–15. https://doi.org/10.1042/BJ20021469
Liu Y, Lang F, Yang C (2020) NRF2 in human neoplasm: Cancer biology and potential therapeutic target. Pharmacol Ther 369:107664. https://doi.org/10.1016/j.pharmthera.2020.107664
Lu X, Paliogiannis P, Calvisi DF, Chen X (2020) Role of the mammalian target of rapamycin pathway in liver cancer: from molecular genetics to targeted therapies. Hepatology. https://doi.org/10.1002/hep.31310
Murugan AK (2019) mTOR: role in cancer, metastasis and drug resistance. Semin Cancer Biol 59:92–111. https://doi.org/10.1016/j.semcancer.2019.07.003
Pallares J et al (2009) CK2beta is expressed in endometrial carcinoma and has a role in apoptosis resistance and cell proliferation. Am J Pathol 174:287–296. https://doi.org/10.2353/ajpath.2009.080552
Shuford RA, Cairns AL, Moaven O (2020) Precision approaches in the management of colorectal cancer: current evidence and latest advancements towards individualizing the treatment. Cancers 12:3481. https://doi.org/10.3390/cancers12113481
Siegel RL et al (2020) Colorectal cancer statistics, 2020. CA Cancer J Clin 70:145–164. https://doi.org/10.3322/caac.21601
Silva-Pavez E, Tapia JC (2020) Protein kinase CK2 in cancer energetics. Front Oncol 10:893. https://doi.org/10.3389/fonc.2020.00893
Sridharan S, Basu A (2020) Distinct roles of mTOR targets S6K1 and S6K2 in breast cancer. Int J Mol Sci 21:1199. https://doi.org/10.3390/ijms21041199
Stengel ST et al (2020) Activating transcription factor 6 mediates inflammatory signals in intestinal epithelial cells upon endoplasmic reticulum stress. Gastroenterology 159:1357–1374.e1310. https://doi.org/10.1053/j.gastro.2020.06.088
Sun SY (2020) mTOR-targeted cancer therapy: great target but disappointing clinical outcomes, why? Front Med. https://doi.org/10.1007/s11684-020-0812-7
Teh JLF, Aplin AE (2019) Arrested developments: CDK4/6 inhibitor resistance and alterations in the tumor immune microenvironment. Clin Cancer Res Off J Am Assoc Cancer Res 25:921–927. https://doi.org/10.1158/1078-0432.CCR-18-1967
Wang W et al (2019) Huaier suppresses breast cancer progression via linc00339/miR-4656/CSNK2B signaling pathway. Front Oncol 9:1195. https://doi.org/10.3389/fonc.2019.01195
Worthmuller J, Ruegg C (2020) The crosstalk between FAK and Wnt signaling pathways in cancer and its therapeutic implication. Int J Mol Sci 21:9107. https://doi.org/10.3390/ijms21239107
Xiao Y et al (2020) Tumor necrosis factor alpha-induced protein 1 as a novel tumor suppressor through selective downregulation of CSNK2B blocks nuclear factor-kappaB activation in hepatocellular carcinoma. EBioMedicine 51:102603. https://doi.org/10.1016/j.ebiom.2019.102603
Funding
This work was supported by the National Natural Science Foundation of China (No. 81972290) and the Joint Project of Pudong new area Health and Family Planning Commission (No. PW2018D-06) and the Outstanding Clinical Discipline Project of Shanghai Pudong (No. PWYgy2018-02).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have declared that no competing interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, S., Hu, Q., Fan, K. et al. CSNK2B contributes to colorectal cancer cell proliferation by activating the mTOR signaling. J. Cell Commun. Signal. 15, 383–392 (2021). https://doi.org/10.1007/s12079-021-00619-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-021-00619-1