
Abstract
The extracellular matrix molecule tenascin-C is highly expressed during embryonic development, tissue repair and in pathological situations such as chronic inflammation and cancer. Tenascin-C interacts with several other extracellular matrix molecules and cell-surface receptors, thus affecting tissue architecture, tissue resilience and cell responses. Tenascin-C modulates cell migration, proliferation and cellular signaling through induction of pro-inflammatory cytokines and oncogenic signaling molecules amongst other mechanisms. Given the causal role of inflammation in cancer progression, common mechanisms might be controlled by tenascin-C during both events. Drugs targeting the expression or function of tenascin-C or the tenascin-C protein itself are currently being developed and some drugs have already reached advanced clinical trials. This generates hope that increased knowledge about tenascin-C will further improve management of diseases with high tenascin-C expression such as chronic inflammation, heart failure, artheriosclerosis and cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Today it is well accepted that the microenvironment plays an essential role in inflammatory diseases (Schafer and Werner 2008) and cancer (Marx 2008). In particular in cancer a normal tissue architecture has a tumor suppressive function (Bissell and Labarge 2005; Bissell and Radisky 2001). Chronic inflammation can cause cancer and thus, similar mechanisms involving the role of the microenvironment might underlie both pathologies. The microenvironment is composed of a complex extracellular matrix (ECM) and the embedded cells. The information encoded by the ECM can be of a mechanical as well as of a signaling nature. In this review we will summarize current knowledge about the roles of the ECM molecule tenascin-C during inflammation and tumorigenesis, its mechanistic basis and how this knowledge could be used to combat tenascin-C-associated pathologies such as chronic inflammation and cancer. Moreover, we will also elaborate on the functions of tenascin-C as an architectural molecule and highlight evidence for its direct signaling nature.
Structure and expression pattern of tenascin-C
The presence of tenascin-C was discovered more than 20 years ago in gliomas, in muscle tissue and in the nervous system, hence the different names for this molecule: myotendinous antigen, glial/mesenchymal extracellular matrix protein (GMEM), cytotactin, J1 220/200, neuronectin and hexabrachion (reviewed in Chiquet-Ehrismann and Chiquet 2003; Chiquet-Ehrismann et al. 1994). Tenascin-C is the founding member of a family of extracellular matrix glycoproteins comprising tenascin-X (termed tenascin-Y in the chicken), -R and -W in addition to tenascin-C. Its name, coined by Ruth Chiquet-Ehrismann (Chiquet-Ehrismann et al. 1986), represents a combination of the Latin verbs “tenere“ and “nasci“ (to be born, to grow, to develop), which provided the roots of the English words “tendon“ and “nascent“, and reflect the location and developmental expression of the protein observed at that time.
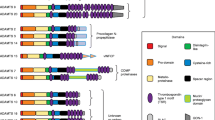
The human tenascin-C gene locus of 97`680 bp (Gherzi et al. 1995) is located on chromosome 9q33. The tenascin-C gene was first determined to comprise 28 exons separated by 27 introns (Gherzi et al. 1995). Subsequently, two additional exons, AD1 (Sriramarao and Bourdon 1993) and AD2 (Mighell et al. 1997) were identified, thus resulting in a total number of 30 exons. The first exon is untranslated and translation starts in exon 2. The transcript is 8150 bp long encoding a protein of a maximal putative length of 2385 amino acids (Hancox et al. 2009; Jones et al. 1989; Pas et al. 2006) (Fig. 1). Tenascin-C exhibits a modular organization consisting of an N-terminal region containing a chaperone-like sequence that forms coiled coil structures and interchain disulfide bonds that are essential for subunit oligomerization into hexamers. Human tenascin-C comprises 14.5 epidermal growth factor (EGF)-like repeats, 30–50 amino acids in length, which contain six cysteine residues involved in intrachain disulfide bonds. Up to 17 fibronectin type III domains (FNIII) are present that are about 90 amino acids in length and that are composed of seven antiparallel β-strands arranged in two sheets. The number of fibronectin type III domains is generated by alternative splicing, but the underlying mechanisms are little understood, although there is evidence that the proliferative state of a cell (Borsi et al. 1994), extracellular pH (Borsi et al. 1996), TGFβ1 (Zhao and Young 1995) and the splicing factor sam68 (Moritz et al. 2008) are involved. At least nine different FNIII domains are differentially included or excluded by RNA splicing. This can generate a considerable diversity in normal tissue such as in the nervous system (Joester and Faissner 2001), teeth (Sahlberg et al. 2001), human skin (Latijnhouwers et al. 1996), human fetal membranes (Bell et al. 1999), avian optic tectum (Tucker 1998), corneas (Ljubimov et al. 1998), gamma irradiated tissue (Geffrotin et al. 1998), tissue chronically infected with hepatitis C (El-Karef et al. 2007), lungs affected by asthma (Matsuda et al. 2005) and, in cancer tissues (Adams et al. 2002; Carnemolla et al. 1999; Derr et al. 1997; Dueck et al. 1999; Mighell et al. 1997; Richter et al. 2009). The different tenascin-C splice forms may cause distinct but yet unknown cell responses. The C-terminal fibrinogen globular domain (FBG) resembling the β- and γ-chains of fibrinogen, 210 amino acids in length, forms intrachain disulfide bonds (Fig. 1). The tenascin-C protein displays 23 potential glycosylation sites, two in the assembly domain, two in the EGF repeats, 18 in the FNIII repeats and one in the FBG domain. Erickson and coworkers observed that tenascin-C purified from human glioma cells is indeed glycosylated, probably even to a higher extent than fibronectin (Taylor et al. 1989). Glycosylation and alternative splicing affect the molecular weight of the tenascin-C protein and could explain the wide range between 190 and 330 kDa of the tenascin-C monomer.

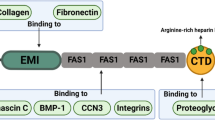
Tenascin-C structure and binding partners a The exon/intron structure of the human tenascin-C gene on chromosome 9q33 and the organization into the different protein domains is depicted schematically. Information on the exon/intron organization is based on Hancox et al. (2009) and analysis of sequence entries in the EMBL sequence data bank. Organization of tenascin-C into an assembly domain, EGF-L, constant (grey) and alternatively spliced in FNIII repeats (black) and the C-terminal FBG. Tenascin-C-interacting molecules are shown at the bottom of the figure. CALEB, chicken acidic leucine-rich EGF like domain containing brain protein; EGFR, epidermal growth factor receptor; NaN, sodium channel subunit β2; RPTPβ, receptor protein tyrosine phosphatase β/ζ (Orend and Chiquet-Ehrismann, 2006). b The appearance of purified tenascin-C protein as a hexamer upon electron microscopy. Taken from (Orend and Chiquet-Ehrismann, 2006). c Tenascin-C present in matrix tubes of paraffin preserved human colorectal carcinoma tissue is visualized by immunohistochemistry
Tenascin-C has a tightly regulated pattern of expression which has been reviewed in detail elsewhere (Tucker and Chiquet-Ehrismann 2009). In addition to the high levels expressed in the tumor stroma, it is significantly up-regulated in response to tissue injury (Orend and Chiquet-Ehrismann 2006). Tenascin-C expression can be induced by various pro- and anti-inflammatory cytokines (see below) and by growth factors that are mostly secreted by stromal cells. In addition, hypoxia, reactive oxygen species, and mechanical stress, which are also present in inflamed and tumor tissue, induce tenascin-C expression. In contrast, glucocorticoids (Cella et al. 2000) and GATA-6 (Ghatnekar and Trojanowska 2008) suppress tenascin-C expression. Signaling causing activation of transcription factors such as TCF/LEF, NfkB, c-Jun, Ets, SP1, and Prx-1 is involved in tenascin-C gene transcription (Orend and Chiquet-Ehrismann 2006). Recently, Notch1 and Notch2 signaling (Sivasankaran et al. 2009) and HNF-4α (Ishikawa et al. 2008) were also described to induce tenascin-C transcription in gliomas and mammary epithelial NMuNG cells, respectively. In addition, a combination of signals induced by ectopic expression of c-myc and VEGFA in the mammary epithelium also induced tenascin-C (Calvo et al. 2008). Tenascin-C RNA levels can also be downregulated by miR-335, and low expression levels of miR-335 (allowing high tenascin-C expression) appear to be causally linked to lung metastasis in breast cancer (Tavazoie et al. 2008). Tenascin-C is cleaved by matrix metalloproteases and serine proteases, thus potentially releasing cryptic sites that may create adhesive sites for cell adhesion receptors (Table 1). Indeed, MMP2 cleavage of tenascin-C generated a cryptic adhesive site within the FNIII A2 domain (Saito et al. 2007; Watanabe et al. 2000). A 22mer peptide of this domain, termed TNIIIA2, comprising an adhesive YTITIRG sequence that is homologous to a similar sequence in the FBG domain, bound to the heparan sulfated side chains of syndecan-4 and thus caused clustering and activation of β1 integrins and cell spreading (Saito et al. 2007). Cleavage of tenascin-C by MMP2 also caused removal of the alternativley spliced in FNIII repeats A3/A4 and B-D (Siri et al. 1995). In vitro cleavage of tenascin-C by MMP1, 3 and 7 resembled tenascin-C cleavage products of human melanoma cells (Imai et al. 1994). Cell contact with tenascin-C also induced expression of matrix metalloproteases (Tremble et al. 1994), thus presenting a positive feedback loop between induction of metalloproteases by tenascin-C and cleavage of tenascin-C by these enzymes. It is most likely that hexameric, monomeric and protease-cleaved tenascin-C exhibit distinct functions, although the full extent of these functions is not currently clear.
Tenascin-C binds to ECM molecules such as fibronectin, perlecan, aggrecan, versican, brevican (Fig. 1) and presumably many more ECM molecules that remain to be identified. Its promiscuous binding to a variety of ECM molecules and its hexameric organization may provide tenascin-C with crosslinking functions, thus potentially modulating the architecture and resilience of tissues. Cells can interact with tenascin-C via cell-surface receptors including integrins α2β1,α7β1, α9β1 and αvβ3, syndecans 1 and 4, annexin II, and epidermal growth factor receptor (EGFR) amongst others (Fig. 1, Table 1). These data are extensively reviewed elsewhere (Orend and Chiquet-Ehrismann 2006). Thus tenascin-C is equipped not only to modulate the cellular environment, but also to influence the behaviour of cells. However, the resulting cell responses and their potential in vivo relevance are still poorly understood at a mechanistic level.
Overview of the response to tissue injury
The response to tissue injury is a highly orchestrated process. It proceeds through three distinct but overlapping phases; inflammation, tissue rebuilding and tissue remodelling (for more detailed reviews see (Clark 1993; Midwood et al. 2004b)). Inflammation is a critical and immediate response to tissue injury and infection. Pattern recognition receptors (PRRs) recognize both pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) and in response stimulate the expression of pro-inflammatory genes (Gordon 2002). These include cytokines and chemokines, which attract immune cells including neutrophils, myeloid cells and lymphocytes to the site of injury or infection. These cells mediate removal of the injurious stimuli and dead or damaged tissue. They also initiate the healing process by inducing the proliferation of other cell types, including fibroblasts and endothelial cells (EC), and stimulate their migration to the site of injury (Clark 1993; Midwood et al. 2004b).
Immediately upon injury, a provisional ECM consisting predominantly of fibrin and plasma fibronectin is synthesized. Deposition of this matrix temporarily fills the wound bed and prevents excessive blood loss. Assembly of new ECM upon this scaffold is vital in order to replace lost and damaged tissue and restore the architecture of tissue. Upon activation, cells such as fibroblasts migrate into the provisional matrix where they assemble fibronectin-rich granulation tissue. This in turn forms a scaffold for the subsequent deposition of collagen types I and III and other ECM molecules. EC re-vascularize the new tissue and fibroblasts differentiate into myofibroblasts, which contract the ECM to bring the margins of the wound together. The resultant scar tissue is then remodeled and collagen cross-linked over the following years, although it fails to exactly recapitulate the cellularity, vascularity, and tensile strength of the original tissue (Clark 1993; Midwood et al. 2004b).
Regulation of each of these phases is vital: over-enthusiastic inflammation can lead to damage of healthy tissues, too little matrix deposition results in chronic non-healing wounds, whereas too much is the hallmark of fibrotic disease, including pulmonary fibrosis and liver cirrhosis; excessive myofibroblast contraction mediates hypertrophic scarring commonly associated with burns (Clark 1993; Midwood et al. 2004b).
The role of tenascin-C in tissue injury
A. Inflammation
Tenascin-C expression is induced at sites of inflammation, apparently regardless of the location or type of causative insult. Expression is observed upon both tissue injury, for example during incisional wounding or photo-damage to the skin (Betz et al. 1993; Filsell et al. 1999), tendon rupture (Riley et al. 1996) and asbestos-induced lung damage (Kaarteenaho-Wiik et al. 2000). Tenascin-C is also induced by infections, for example in the lungs of tuberculosis patients (Kaarteenaho-Wiik et al. 2000), upon infection with staphylococcus aureus (Paallysaho et al. 1993) and in the cervical epithelium of patients with human papilloma virus (Tiitta et al. 1992). This pattern of expression suggests that tenascin-C comprises part of a common, global inflammatory response.
Tenascin-C expression is induced rapidly upon tissue injury. In normal human skin, low levels of tenascin-C expression are restricted to the dermal-epidermal junction, eccrine glands and to large blood vessels (Lightner et al. 1989; Schalkwijk et al. 1991b). The appearance of tenascin-C mRNA and protein often precedes obvious signs of tissue damage or inflammation induced in human skin by UVB (Seite et al. 2000), gamma irradiation (Geffrotin et al. 1998; van der Vleuten et al. 1996), excisional wounds and punch biopsies (Betz et al. 1993; Latijnhouwers et al. 1996). Early induction of tenascin-C expression is also observed during hapten-induced allergic reaction in mice (Kusubata et al. 1999) and in incisional wounds in rats (Mackie et al. 1988) and mice (Fassler et al. 1996). During the acute inflammatory phase, tenascin-C expression is concentrated in areas of increased immune cell infiltration. It co-localizes with polymorphonuclear lymphocyte cell infiltration in the inflamed human dermis upon application of leukotriene-B4 (Seyger et al. 1997), in areas of mononuclear cell infiltrates, in degenerate tendons resulting from rotator cuff rupture (Riley et al. 1996), at sites of leukocyte accumulation in patients with myocarditis (Tsukada et al. 2009) and during experimental liver inflammation in mice (El-Karef et al. 2007).
Tenascin-C expression is particularly well documented in lung inflammation. It associates with inflammatory foci in inflamed bronchi of smokers (Amin et al. 2003) and in patients with interstitial lung diseases such as interstitial pneumonia and sarcoidosis (Kaarteenaho-Wiik et al. 1998; Kuhn and Mason 1995). Expression is also increased in the subepithelial ECM of patients with bronchial asthma, a disease characterized by chronic airway inflammation, where it correlates with the grade of bronchial infiltration of mast cells and eosinophils (Amin et al. 2000; Karjalainen et al. 2000). Matsuda et al. (2005) demonstrated that a single nucleotide polymorphism in the alternatively spliced FNIII-D domain of tenascin-C is associated with a high risk of developing asthma. Molecular modeling of the resultant amino acid substitution (Leu1677Ile) demonstrated that steric hindrance by a isoleucine side chain may hinder hydrophobic interactions at the outer edge of the beta sheet, causing domain instability. The authors propose that this change may alter FNIII-D domain elasticity and result in changes in the integrity or stiffness of the asthmatic airway (Matsuda et al. 2005). Tenascin-C-induced PDGFRβ signaling that causes smooth muscle cell expansion contributes to airway obstructions in the lung. This mechanism might also be relevant in Aspergillus fumigatus-induced asthma in the mouse and in pulmonary hypertension in humans since a high tenascin-C expression correlated with high expression of PDGFRβ and SMC proliferation in the diseased lung (Cohen et al. 2009).
Nakahara et al. (2006) subsequently demonstrated that tenascin-C-null mice are protected from experimentally induced asthma due to a dampened Th2 cell response (Nakahara et al. 2006). Th2 lymphocyte activation plays a fundamental role in the pathogenesis of asthma (Busse and Lemanske 2001). Lower levels of cytokines secreted by Th2 CD4+ T lymphocytes including IL-5, IL-4, IL-13, were detected in the bronchiolar lavage fluid of tenascin-C-null mice. Subsequent stimulation of IgE synthesis by B lymphocytes, recruitment and prolonged survival of eosinophils and airway activation were also attenuated in null mice. In addition, lower levels of MCP-1 and MMP-9 induction were observed in tenascin-C-null mice (Nakahara et al. 2006). Together these data indicate that the role of tenascin-C in the pathogenesis of bronchial asthma may lie in mediating lymphocyte trafficking to the inflamed bronchial mucosa and/or activation of T cells.
A role for tenascin-C in stimulating lymphocyte migration is suggested by data that demonstrate that tenascin-C supports primary human peripheral blood and tonsillar lymphocyte adhesion and rolling (Clark et al. 1997). In addition, tenascin-C-null mice exhibit reduced lymphocyte infiltration and lower levels of IFN, TNF and IL-4 mRNA upon concanavalin A-induced liver injury in mice (El-Karef et al. 2007). However, tenascin-C appears to exhibit a cell-type specific effect on migration; it inhibits monocyte chemotaxis in vitro (Loike et al. 2001) and tenascin-C-null mice exhibit increased migration of monocytes and macrophages in mammary tumor stroma (Talts et al. 1999).
There is also evidence that tenascin-C can directly regulate the activation of lymphocytes. Addition of exogenous tenascin-C to mouse spleen lymphocytes cultured on fibronectin substrates, stimulates secretion of IL5, IL13, IFN and IgE (Nakahara et al. 2006) and tenascin-C also induces calcium mobilization in Jurkat T lymphocytes (Kuhn and Mason 1995). Whilst these data suggest a role for tenascin-C in promoting T cell responses there is also evidence that it limits their inflammatory function (Kuznetsova and Roberts 2004). Tenascin-C inhibits the activation of human peripheral blood T cells induced by natural antigens (Ruegg et al. 1989), by immobilization on CD3 and fibronectin (Hemesath et al. 1994) and by anti-CD28, ICAM-1, or laminin (Hibino et al. 1998). It also significantly inhibits anti-CD3 and mitogen-induced proliferation and IFNγ production in human peripheral blood lymphocytes and lymphocytes isolated from lung tumors (Parekh et al. 2005). The alternatively spliced domains FNIII-A1-3 mediate the suppression of anti CD3/fibronectin-induced proliferation, and TNF and IFNγ secretion (Puente Navazo et al. 2001), although the molecular mechanism remains unclear. It is unlikely that tenascin-C affects protein kinase C signaling pathways required for T cell activation, as it does not prevent T cell activation induced by the protein kinase C agonist PMA (Parekh et al. 2005). It may therefore interfere with other pathways, for example, the activation of Rho-induced proliferation (Woodside et al. 1998), given the ability of tenascin-C to suppress Rho activation in fibroblasts (Wenk et al. 2000). Consistent with a role in suppressing the immune response, tenascin-C-deficient mice develop progressive inflammation and glomerular damage in experimental venom-induced glomerulonephritis (Nakao et al. 1998). These mice also develop severe dermatitis in response to 2,4-dinitrofluorobenzene and exhibit prolonged infiltration of polymorphonuclear cells (Koyama et al. 1998).
In addition to a role for tenascin-C in modulating T cell behavior to influence the adaptive immune response, we have demonstrated that tenascin-C contributes to driving innate immunity. Tenascin-C stimulates the production of pro-inflammatory cytokines TNFα, IL-6 and IL-8 in primary human macrophages and synovial fibroblasts, via activation of Toll-like receptor 4 (TLR4) mediated signaling pathways (Midwood et al. 2009). It can also stimulate cytokine and MMP synthesis in murine synovial fibroblasts via activation of α9 integrins (Kanayama et al. 2009). Tenascin-C induces inflammation in vivo when injected into the joints of mice, and tenascin-C-null mice exhibit accelerated resolution of joint inflammation and are protected from persistent inflammation and erosive joint disease (Midwood et al. 2009). Together these data demonstrate that tenascin-C can influence many aspects of immunity and that its precise role at sites of inflammation is defined by context: for example the site and mode of tissue injury, or the participating cell types (Table 2).
Re-epithelialization
In the first 24 h following dermal wounding, keratinocytes proliferate and migrate through the fibrin clot. This mediates early wound closure that is key to preventing further microbial invasion (Clark 1993; Midwood et al. 2004b). In addition to the expression of tenascin-C in areas of immune cell infiltration during inflammation, it is also observed within 24 h at the dermal/epidermal junction underneath migrating and proliferating keratinocytes (Betz et al. 1993; Mackie et al. 1988). In situ hybridization demonstrates that keratinocytes are the major source of this tenascin-C (Latijnhouwers et al. 1997). Strong expression of tenascin-C is also observed underneath the hyperproliferative epidermis in patients with acne (Knaggs et al. 1994), psoriasis (Gerritsen et al. 1997) and blistering diseases (Schenk et al. 1995). However it is absent from keratinocyte pathologies that do not proceed via hyperproliferative pathways, including X-linked recessive ichthyosis, autosomal dominant ichthyosis vulgaris and non-erythrodermic lamellar ichthyosis (Schalkwijk et al. 1991a). During development of the chick cornea, keratinocytes emigrate from tenascin-C rich areas during embryonic stage 27 (Kaplony et al. 1991). Furthermore, corneal suture wounds in tenascin-C-null mice are devoid of migrating keratinocytes (Matsuda et al. 1999). These data suggest that tenascin-C may play a role in regulating epithelial cell functions, such as promoting their proliferation and migration.
Immortalized human epithelial corneal cells secrete a tenascin-C- and laminin-rich plaque that is essential for early adhesion events (Katz et al. 2006), and isolated primary human epidermal keratinocytes maintain a rounded morphology on tenascin-C substrates (Latijnhouwers et al. 1997), suggesting that tenascin-C may promote adhesion and migration during re-epitheliazation. Moreover, in studying the development of the prostate, (Ishii et al. 2008) identified tenascin-C as a morphogenic signal that drives epithelial differentiation and function by up-regulating androgen receptor expression (Ishii et al. 2008). Thus tenascin-C may also promote epithelial cytodifferentiation during wound repair.
B. Tissue rebuilding
Towards the end of the inflammatory phase of tissue repair, tissue rebuilding begins. In incisional or abrasion wounds to the dermis, tenascin-C expression peaks at this point. It increases at wound edges of the dermis associated with activated fibroblasts, and then extends into connective tissue and is observed throughout the dermis. High levels are also found in the new granulation tissue (Betz et al. 1993; Fassler et al. 1996; Latijnhouwers et al. 1996; Mackie et al. 1988). Persistent tenascin-C expression correlates with excessive matrix deposition in patients with fibrotic diseases including chronic liver disease (Van Eyken et al. 1990; Yamada et al. 1992) and several fibrotic lung disorders such as pulmonary fibrosis (Kaarteenaho-Wiik et al. 1998). Tissue tenascin-C levels are increasingly used as a predictive marker for fibrosis (Lieber et al. 2008; Paivaniemi et al. 2008). Tenascin-C-null mice exhibit thinner stromata during corneal suture wounds with reduced fibronectin deposition (Matsuda et al. 1999). Similarly, lower levels of fibronectin were detected in the granulation tissue during healing of dermal excisional wounds in tenascin-C-null mice (Forsberg et al. 1996), and only small and immature granulation tissue was observed during experimental kidney injury (Nakao et al. 1998). Together these data suggest a role for tenascin-C in promoting tissue rebuilding during wound healing (Fig. 2).

Tenascin-C in tissue repair The interaction of tenascin-C with distinct cell types throughout tissue repair and the respective cell responses are shown during the different phases of the response to tissue damage
Cell proliferation and migration
The proliferation of cells in healthy tissue adjacent to the wound and their migration into the wound site is essential for tissue rebuilding. Upon dermal injury, fibroblast infiltration into the provisional matrix-filled wound bed occurs around 4 days after injury and is the key rate-limiting step of tissue rebuilding (McClain et al. 1996).
Tenascin-C has diverse effects on cell migration in vitro that reflect differences in cell type, species, and the assay used in each study (Trebaul et al. 2007). For example, human corneal fibroblasts migrate on tenascin-C substrates but soluble tenascin-C added to these cells on collagen substrates inhibits migration (Schmidinger et al. 2003). Tenascin-C induces a migratory phenotype in mouse NIH3T3 fibroblasts cultured on a fibrin-fibronectin matrix; cells remain rounded and do not form actin stress fibers or focal adhesions but instead form extended actin-rich filopodia. These changes in morphology are mediated by inhibition of FAK and RhoA activity (Midwood and Schwarzbauer 2002; Wenk et al. 2000). Tenascin-C also induces a migratory phenotype in human, rat, and mouse fibroblasts, and in glioma cells plated on fibronectin substrata. Again, FAK activation was inhibited by tenascin-C, but here down-regulation of tropomyosin-1 and RhoA, rather than inhibition of RhoA was proposed to decrease actin stabilization (Lange et al. 2007; Ruiz et al. 2004). In both cases, tenascin-C acts by blocking the interaction of fibronectin with cell-surface syndecan-4 (Huang et al. 2001; Midwood et al. 2004a) by binding directly to FNIII13 in the HepII domain of fibronectin (Huang et al. 2001). Tenascin-C also possesses heparin-binding activity (Marton et al. 1989) raising the possibility that it may additionally compete for binding to syndecan HSPG side chains. Indeed, one of the heparin-binding sites has been mapped to the FBG domain of tenascin-C (Aukhil et al. 1993; Fischer et al. 1995). The FBG posseses no fibronectin binding activity (Chung et al. 1995), but it can promote migration of human fibroblasts within 3D fibrin-fibronectin gels (Trebaul et al. 2007). These data suggest that heparin binding may also be sufficient to mediate changes in cell morphology required for migration.
The epidermal growth factor-like (EGF-L) domain of tenascin-C also plays a role in regulating fibroblast proliferation and migration. The EGF-L repeats, Ten1-2, Ten11-13 and Ten14 induce EGF receptor (EGFR) phosphorylation and stimulate MAP kinase activation and mitogenesis in the NR6 fibroblast cell line (Swindle et al. 2001). Ten14 was also shown to bind to EGFR with low affinity and to induce migration through activation of PLCγ and m-calpain (Iyer et al. 2008). Tenascin-C activation of EGFR also stimulates Rac activity in myofibroblasts which, via inhibition of Rho activation, promotes their invasion in collagen gels (De Wever et al. 2004). In direct contrast to other EGFR ligands, which induce migration and proliferation at similar concentrations, tenascin-C EGF-L repeats induce mitogenesis at high micromolar levels and migration at lower levels (Swindle et al. 2001). The Kd of Ten14 for EGFR is 74 μM, 3 orders of magnitude weaker than EGF. This is due to lack of a C terminal loop in Ten14 that mediates high-affinity interactions of EGF. The resultant rapid off rate is proposed to be preferential for cell motility at low concentrations (Tran et al. 2004; Tran et al. 2005). Thus, different domains of tenascin-C can affect cell migration or proliferation by different mechanisms, including modulation of cell adhesion to the ECM or via direct interactions with cell-surface growth factor receptors.
ECM synthesis
Deposition of new ECM is key to restoring tissue architecture and functionality (Clark 1993). Tenascin-C may promote matrix deposition or fibrosis by increasing pro-collagen synthesis. In a model of immune-mediated hepatitis, collagen deposition was reduced in tenascin-C-null mice. This occurred at the level of gene transcription with a 50% reduction in collagen I mRNA induction and failure to induce any collagen type III transcripts (El-Karef et al. 2007). These studies do not rule out a role for tenascin-C in direct transcriptional control of collagen genes, but they do offer several other potential mechanisms of action. In this model, weekly injections of concanavalin A induce local inflammation, which causes continuous cellular injury leading to increased collagen deposition and eventual fibrosis. Tenascin-C-null mice exhibit attenuated liver inflammation with reduced numbers of infiltrating lymphocytes concomitant with lower levels of cytokines such as IFNγ, TNF and IL-4 mRNA. These mice also demonstrate low numbers of activated hepatic stellate cells and myofibroblasts, the cellular source of procollagens, as well as reduced induction of transforming growth factor (TGF)-β1 mRNA, a growth factor that stimulates collagen transcription (El-Karef et al. 2007). Thus the failure of mice to induce collagen transcription in the absence of tenascin-C could simply result as a consequence of an attenuated inflammatory response that fails to stimulate subsequent recruitment of collagen-synthesizing cells and TGFβ1 expression. Conversely tenascin-C could have direct effects on each stage of the progression of hepatitis to fibrosis (Table 2, Fig. 2).
Indeed, tenascin-C can affect stromal cell activity and growth factor synthesis independently of its effects on inflammation. Tenascin-C-null mice are unable to resolve renal inflammation in response to snake venom-induced glomerulonephritis (Nakao et al. 1998). In wild-type mice destruction of the kidney mesangial structure caused by inflammation is reversible and heals spontaneously. However, renal damage induced in tenascin-C-null mice persists and can be fatal. In this model, renal fibrosis is not likely to be attributable to reduced inflammation in the absence of tenascin-C, as equal numbers of infiltrating immune cells were observed in wild-type and null mice. Instead, lower numbers of mesangial cells that play a central role in regeneration of tissue were observed. Furthermore, induction of PDGF was reduced and induction of TGFβ1, collagen type VI and fibronectin delayed (Nakao et al. 1998). The proliferation of mesangial cells isolated from wild-type mice in response to PDGF and TGFβ1 can be blocked by an antibody to the EGF-L repeats of tenascin-C. Conversely, cells isolated from tenascin-C-null mice do not proliferate in response to PDGF or TGFβ (Nakao et al. 1998), suggesting that tenascin-C modulates cellular response to growth factors. These findings are consistent with data showing that proliferation of mouse 3T3 fibroblasts induced by EGF was enhanced when tenascin-C was added to the culture medium. This effect was mediated by the constitutively expressed FNIII domains, but not by the alternatively spliced domains (End et al. 1992). The precise details of the molecular mechanism(s) by which tenascin-C regulates the cellular response to growth factors are not known. Tenascin-C may bind directly to growth factors in order to increase local concentrations at the cell-surface, in a manner similar to heparin (Goodger et al. 2008). It may increase growth factor receptor availability at the cell-surface, either by directly affecting receptor expression, as it appears to do for androgen receptors (Ishii et al. 2008) and endothelin receptor type A (EDNRA) (Lange et al. 2007), or by stabilizing receptor clustering upon ligation, potentially via modulation of syndecan-4/FGF/FGFR complex formation (Alexopoulou et al. 2007). Since tenascin-C competed with syndecan-4 binding to fibronectin, it is possible that syndecan-4 is exclusively available as a coreceptor for growth factor receptors in the presence of tenascin-C. In summary, in addition to its role in regulating the inflammatory response, there is strong evidence that tenascin-C may also play a direct role in the control of growth factor signaling that promotes ECM synthesis.
ECM assembly
Tenascin-C interacts directly with many ECM molecules including fibronectin (Orend and Chiquet-Ehrismann 2006) (Fig. 1). It can bind to soluble plasma fibronectin in vitro (Chiquet-Ehrismann et al. 1991; Chung et al. 1995; Ingham et al. 2004) as well to the fibronectin fibrils of cultured cells (Chiquet and Fambrough 1984; Chung et al. 1995; Ramos et al. 1998). Tenascin-C co-localizes to fibronectin fibrils and to regions of fibronectin deposition and assembly in vivo (Chiquet and Fambrough 1984; Kusubata et al. 1999; Riou et al. 1990; Whitby et al. 1991). Fibronectin and tenascin-C matrix assembly appear to be interdependent (Ramos et al. 1998) and the recombinant tenascin-C protein 70Ten, which contains the FNIII like repeats and the FBG domain of tenascin-C inhibits fibronectin matrix assembly (Midwood and Schwarzbauer 2002). It was demonstrated, using in vitro solid phase binding assays, that tenascin-C can bind to fibrillar collagen types I — VI and IX, but not denatured collagen (Faissner et al. 1990). However, despite colocalization of tenascin-C and collagen in several tissues, little evidence exists to show any functional relevance of this interaction. This is in contrast to tenascin-X a family member whose deficiency results in Ehlers Danlos syndrome due to an inability to properly regulate dermal collagen fiber maturation (Chiquet-Ehrismann 2004).
Tenascin-C also interacts with ECM-resident proteoglycans, including members of the lectican family; versican, neurocan, aggrecan and brevican (Day et al. 2004). The FNIII3-5 domain binds with high affinity to the highly conserved G3 domain in aggrecan, versican and neurocan but exhibits weaker binding to brevican (Day et al. 2004). Electron microscopy shows that purified hexameric tenascin-C crosslinks aggrecan-hyaluronan complexes, suggesting a role in mediating the structural organization of the ECM (Lundell et al. 2004). This interaction also occurs with FNIII3-5 of tenascin-R and crystalization of these domains in complex with the aggrecan C-type lectin domain revealed a direct protein-protein interaction between the lectin domain and FNIII4 (Lundell et al. 2004). A role for tenascin-C in promoting proteoglycan assembly is supported by the fact that neointima formation during the intimal hyperplasia that follows longitudinal aortotomy in rats is reduced in tenascin-C-null mice, with significantly lower levels of alcian blue-positive proteoglycan deposition (Yamamoto et al. 2005). Together these data suggest a role for tenascin-C in promoting ECM synthesis during tissue repair by regulating cell migration and proliferation, tissue levels of pro-fibrotic growth factors and cell response to these factors, but they also indicate a potential role as a structural template that mediates assembly of ECM components.
Vascular remodeling
New tissue deposited during repair is called granulation tissue because the formation of new blood vessels gives it a granular appearance. Revascularization is vital to maintain the viability of any new tissue. Tenascin-C expression is highly associated with sites of vascular remodeling during dermal tissue repair (Betz et al. 1993; Fassler et al. 1996; Latijnhouwers et al. 1996; Mackie et al. 1988). In the central nervous system, tenascin-C is expressed by migrating EC and the kinetics of its expression are linked with maximal angiogenic activity during infectious, ischemic, and traumatic processes (Zagzag and Capo 2002). Tenascin-C expression is also highly associated with angiogenesis in a wide range of disease states, including cancer, diabetes, aortic aneurysm (Castellon et al. 2002; Jallo et al. 1997; Paik et al. 2004), artheriosclerosis (Fischer 2007), ulcerative colitis (Dueck et al. 1999), inflammatory bowel disease (Geboes et al. 2001), Crohns disease (Riedl et al. 2001) and vasculitis (Gindre et al. 1995).
During tissue neovascularization, EC undergo pro-angiogenic activation, disrupting their ECM interactions and assuming a migratory morphology (Carmeliet 2000; Ingber 2002). In vitro, tenascin-C is specifically expressed by sprouting and cord forming bovine aortic EC but not by non-sprouting, i.e. resting cells (Canfield and Schor 1995; Schenk et al. 1999). This angiogenic phenotype was inhibited when cells were grown in the presence of anti—tenascin-C antibodies, suggesting that the transition from a resting to a sprouting phenotype may be promoted by tenascin-C (Canfield and Schor 1995). And in fact, soluble tenascin-C reduces focal adhesions in EC (Chung et al. 1996; Murphy-Ullrich et al. 1991) and enhances EC migration (Chung et al. 1996).
Studies using a cardiac allograft model demonstrate that tenascin-C is required for vascularization in mice in vivo. Upon subdermal transplantation of wild-type cardiac tissue into a syngenic host, a fibrin clot forms around the allograft and both the clot and cardiac tissue become vascularized, resulting in engraftment of viable cardiac tissue. Clot formation is unaffected in tenascin-C-null mice; however, these mice fail to form any vascular channels and no engraftment of cardiac tissue is observed. In wild-type mice the donor EC were observed to engraft at sites of tenascin-C expression suggesting that tenascin-C acts to promote EC homing and incorporation. Indeed, in vitro, rat cardiac microvascular EC adhered to tenascin-C substrata, but spreading and monolayer formation were delayed compared to that in cells adherent to fibronectin or collagen. Futhermore, migration of these cells into a collagen gel was enhanced when cultured on tenascin-C (Ballard et al. 2006). These data support a role for tenascin-C in the early stages of angiogenesis by modulating EC adhesiveness, and thereby to promote migration.
In addition to changes in endothelial-ECM interactions, a number of humoral mediators, including basic fibroblast growth factor (bFGF) and vascular endothelial growth factor A (VEGFA), are required for angiogenesis (Nomi et al. 2006). Tenascin-C can regulate the EC response to growth factors as it does for fibroblasts. Resting EC can be activated by addition of exogenous FBG domain of tenascin-C in the presence of bFGF (Schenk et al. 1999). Furthermore, soluble tenascin-C was found to facilitate the mitogenic response of EC to growth factors such as bFGF. These effects were mediated by the alternatively spliced region of tenascin-C (Murphy-Ullrich et al. 1991) (Table 1). Likewise, enhancement of EC proliferation required a combination of both bFGF and tenascin-C. This effect was mediated by binding of the alternatively spliced domains of tenascin-C to cell-surface annexin II (Chung et al. 1996). Together, data suggest that modulation of tissue levels of humoral factors such as VEGFA, regulation of cell response to key growth factors, and direct effects on cell morphology and adhesion are mechanisms used by tenascin-C to guide blood vessel formation during repair.
C. Tissue remodelling
The final stages of tissue repair include differentiation of fibroblasts into highly contractile α-smooth muscle-rich myofibroblasts that mediate wound contraction. This change is followed by cell death by apoptosis and collagen fibril remodeling (Clark 1993). Tenascin-C expression is not detected after repair is complete; tenascin-C levels return to normal after wound contraction and the protein is not present in scar tissue (Betz et al. 1993; Fassler et al. 1996; Mackie et al. 1988). Tenascin-C mRNA is down-regulated and the protein is cleared from the tissue suggesting that it is not likely to play a role in long term ECM remodeling. However, mice that lack tenascin-C exhibit delayed infiltration of myofibroblasts upon myocardial infarction and the alternatively spliced FNIII repeats and FBG domain stimulated migration, differentiation and collagen gel contraction by cultured cardiac fibroblasts (Tamaoki et al. 2005). These data suggest that tenascin-C may promote wound closure at the end of repair. Conversely, tenascin-C prevents fibroblast contraction of a 3D fibrin-fibronectin matrix (Midwood and Schwarzbauer 2002), suggesting that during early repair stages it may delay contraction as a mechanism of limiting excessive wound closure until sufficient rebuilding has occurred. In addition, tenascin-C may modulate cell apoptosis. Tenascin-C-null mice show earlier mesangial cell apoptosis than wild-type mice upon kidney injury (Nakao et al. 1998) and the EGF-L repeats of tenascin-C are pro-apoptotic when added to cultured smooth muscle cells (Wallner et al. 2004) (Fig. 2).
Summary: tenascin-C in tissue repair
Tenascin-C influences many physiological processes during tissue repair (Table 2). The pleiotrophic nature of this ECM molecule was recently highlighted. Using microarray analysis of gene expression during the repair of striated leg muscle from wild-type and tenascin-C-null mice, expression of full length tenascin-C was required for the regulation of 155 genes that contributed to processes including de-adhesion, inflammation, angiogenesis, energy metabolism, fibrinolysis and differentiation (Fluck et al. 2008).
Tenascin-C exhibits archetypal matricellular properties during the complex progression of events that comprise tissue repair (Table 2, Fig. 2). Whilst little is currently known of the molecular mechanisms by which tenascin-C affects the adaptive immune response, its context-specific function here is already apparent. It promotes the innate immune response via activation of key immune-family receptors and its anti-adhesive capabilities promote the migration of keratinocytes, fibroblasts and EC. Competitive binding of tenascin-C to both ECM proteins and their counterpart cell-surface receptors mediates its ability to modulate cell-ECM interactions. The capacity of tenascin-C to interact with a wide range of ECM molecules may also enable it to contribute to the structural organization of the ECM. In addition, tenascin-C can promote migration and proliferation by direct activation of cell-surface growth factor receptors and cellular differentiation by up-regulating androgen receptor and EDNRA expression. Tenascin-C also appears to regulate cellular responses to growth factors on several levels. It may control tissue levels of growth factors by regulating their synthesis, by direct binding to increase local concentrations, or it may affect receptor expression, localization and clustering. These molecular details remain to be uncovered, as do the mechanisms by which tenascin-C mediates apoptosis.
Much of the mechanistic data from the last 5 years has derived from studies using mice with a targeted disruption of the tenascin-C gene causing the absence of the tenascin-C protein in the tenascin-C-null mice. These mice have a grossly normal phenotype that originally implied a redundancy of tenascin-C. Indeed, recent data have shown that other ECM proteins exhibit shared functions with tenascin-C. Fibulin-1 and tenascin-C posses a common mechanism for modulating fibronectin-syndecan interactions (Williams and Schwarzbauer 2009), thrombospondin-1 can suppress T lymphocyte proliferation as effectively as tenascin-C (Puente Navazo et al. 2001) and osteopontin can substitute for tenascin-C during vascular remodeling (Cowan et al. 2000). Reduction in fibronectin levels found in suture and excsional wounds of tenascin-C null mice (Forsberg et al. 1996; Matsuda et al. 1999) present another potential adaptation mechanism. However, closer inspection of the response of tenascin-C-null mice to tissue injury has demonstrated that this matricellular protein is absolutely required for many aspects of the repair response.
Overview of the role of tenascin-C in tumorigenesis
The tumor microenvironment includes several cell types besides the cancer cells. Soluble and insoluble factors are secreted by these cells. But it is still little acknowledged that the insoluble part of the tumor bed, the “stroma“ (greek for “mat to lie on“), that is represented by the ECM, is as instrumental in tumor initiation and cancer progression as the cells and soluble factors. The tumor ECM is very complex and heterogenous amongst different cancer tissues. Moreover its composition is still poorly understood. The past literature on the role of tenascin-C in cancer has been extensively reviewed elsewhere (Chiquet-Ehrismann and Chiquet 2003; Jones and Jones 2000; Orend 2005; Orend and Chiquet-Ehrismann 2006; Vollmer 1997) (Fig. 3). Therefore, here we will focus on more recent data and will only briefly mention already reviewed data.

Tenascin-C in cancer The interaction of tenascin-C with distinct cell types within a tumor and the respective cell responses are shown. Tenascin-C both inhibits and promotes certain actions, including cell proliferation, migration, invasion, angiogenesis and metastasis. Since tenascin-C induced secretion of pro-inflammatory cytokines through binding to TLR4 in rheumathoid arthritis (Midwood et al., 2009), it is possible that a similar mechanism plays a role in cytokine induction in cancer metastasis
Tenascin-C has distinct effects on normal, tumor and tumor-associated cells based on as yet poorly understood cell-type specific responses toward this ECM molecule (Orend and Chiquet-Ehrismann 2000). Tenascin-C is part of the stroma of most solid cancers, and plays a role in enhancing proliferation, invasion and angiogenesis during tumorigenesis and metastasis. Moreover, data support the possibility that tenascin-C contributes to cancer formation via its potential interference with genomic stability, by blocking immunosurveillance, and by providing a favorable niche for normal and tumor stem cells to survive and expand. Finally, its high expression correlates with a poor prognosis for disease-free survival in patients with several cancers such as glioma and lung cancer (Orend and Chiquet-Ehrismann 2006), but the molecular basis for this tumorigenesis-promoting effect of tenascin-C is poorly understood.
A. Cell adhesion modulation by tenascin-C in cancer
Tenascin-C contains adhesive and anti-adhesive sequences that coexist in the native molecule. These opposing activities arise as a consequence of the binding of tenascin-C to ECM components and cell-surface receptors, and by proteolytic cleavage releasing cryptic adhesive sites. One mechanism that triggers tenascin-C-specific cell rounding on fibronectin involves inhibition of syndecan-4 (Huang et al. 2001; Midwood et al. 2004a). This interference prevents activation of small GTPase RhoA (Wenk et al. 2000), paxillin and focal adhesion kinase (FAK) and blocks expression of the actin stress fiber-stabilizing tumor suppressor-like molecule tropomyosin-1 in fibroblasts and glioblastoma cells (Lange et al. 2008). Activation of oncogenic Wnt, EDNRA, and MAPK signaling, induced by tenascin-C (Ruiz et al. 2004), and elimination of G1 cell-cycle transition control (Huang et al. 2001; Orend et al. 2003) could contribute to enhanced tumor cell proliferation by tenascin-C. The inhibition of cell proliferation of fibroblasts by tenascin-C might also have an impact on the function of these cells, since this inhibition might turn on a secretory program that potentially transforms a quiescent microenvironment into a tumorigenesis-stimulating “tumor bed“. For further information the reader is referred to Chiquet-Ehrismann and Chiquet (2003), Orend (2005), Orend and Chiquet-Ehrismann (2006).
B. Tenascin-C in angiogenesis and matrix tube formation
As mentioned above, tenascin-C is expressed during formation of new blood vessels in the adult as, e.g. in granulation tissue of wounds after myocardial infarction, in arthritis, and in neoplastic diseases. In human gliomas, tenascin-C expression correlates with the degree of tumor neovascularization (Herold-Mende et al. 2002). Tenascin-C may promote angiogenesis through several mechanisms, such as by serving as a chemoattractant for EC, and by initiating EC differentiation, survival and proliferation. These events appear to involve integrin αvβ3, FAK, Prx1 and VEGFA amongst other, not yet identified, molecules (Orend and Chiquet-Ehrismann 2006). In particular, regulation of VEGFA expression by tenascin-C could be relevant to tenascin-C-associated tumor angiogenesis, since xenografted melanoma cells were largely impaired in triggering a new vasculature in tenascin-C-null mice, which correlated with reduced VEGFA levels in the circulation (Tanaka et al. 2004).
In the tissue of malignant melanomas (Kaariainen et al. 2006), breast (Degen et al. 2007) and colorectal carcinomas (Degen et al. 2008), tenascin-C is frequently not homogenously expressed, but accumulates in matrix tracks. In search of metastasis-determining factors by differential expression screening, a combined high expression of tenascin-C and fibronectin, amongst other factors, turned out to discriminate between metastatic and non-metastatic melanomas. Moreover, both molecules were found to colocalize with laminin and procollagen in tubular channels that were hollow, contained tumor cells, and were not lined by blood endothelial or lymph endothelial cells (Kaariainen et al. 2006). In metastatic breast cancers from MMTV-VEGF/c-myc transgenic mice, a panel of ECM molecules, including tenascin-C, several laminins, and collagens were highly expressed, which was in contrast with non-metastatic c-myc-induced tumors that displayed largely reduced or no expression of these molecules (Calvo et al. 2008). It is intriguing that a combined high expression of distinct ECM molecules, including tenascin-C and its presentation in tubular matrix channels, is linked to metastasis.
It is likely that cancer cells use the tenascin-C-containing matrix tubes to disseminate. This assumption is supported by data derived from coculture experiments of fibroblasts together with squamous carcinoma cells (Gaggioli et al. 2007). Fibroblasts scouted their way through matrigel by degrading the ECM at the front, leaving tubes behind that they filled with fibronectin and tenascin-C. Subsequently, the squamous carcinoma cells invaded the matrigel by using these matrix tubes. The invasive behaviour of the squamous carcinoma cells was dependent on the fibroblasts (Gaggioli et al. 2007). A similar observation was originally made by De Wever et al. (2004) who observed that carcinoma-associated fibroblasts or fibroblasts that were “educated“ by TGFβ1 to become myofibroblasts secreted tenascin-C into the collagen gels, and thus prepared the ground for colorectal carcinoma cells to be able to invade in a c-MET- and EGFR-dependent manner. Their invasive behaviour was due to activation of Rac and the concomitant inhibition of RhoA (De Wever et al. 2004). Tenascin-C was instrumental for invasion of the colorectal carcinoma cells since invasion was inhibited with an anti-tenascin-C antibody. Tumor-associated macrophages appear to use collagen tracks within a tumor to guide cancer cells to blood vessels (Iyengar et al. 2003; Lin et al. 2006; Pollard 2008). Whether these collagen tracks contain tenascin-C and potentially represent the described matrix tubes is not known. The existence of vessel-like structures distinct from blood and lymphatic vessels, linked to metastasis, has been known for a long time and was coined “vasculogenic mimicry“ (Hendrix et al. 2003). Whether the observed tenascin-C containing matrix tubes indeed represent the same phenomenon remains to be determined.
In melanomas the tenascin-C-rich matrix tubes were connected to blood vessels since they contained erythrocytes (Kaariainen et al. 2006). Recently, data from Lammert and colleagues suggest that the evolutionary development of endothelium-lined blood vessels in vertebrates preceeded laminin-based matrix tracks in invertebrates (Kucera et al. 2009). In the invertebrate fish amphioxus, laminin filled tubes are laid down as a scaffold in which a hole is drilled by cells of unknown origin to generate a coelum that allows blood to circulate. A similar scenario can be recapitulated in cocultures of EC with a macrophage cell line, in which the EC deposited laminin. Subsequently, the macrophages created a coelum by partially digesting and clearing the ECM to generate space. The observed tenascin-C matrix tubes in melanomas and other cancers may offer an alternative route for dissemination of tumor cells through their continuum with blood vessels. They also potentially provide a scaffold to support growth of vessels. This possibility is interesting considering that the tumor vasculature is mostly distorted and far from providing a functional circulation system, but nevertheless promotes cancer cell dissemination. Anti-angiogenic therapeutic approaches, despite efficient killing of the EC, fail in the long-run, and even promote tumor progression, which results in increased and earlier tumor metastasis (Paez-Ribes et al. 2009; Stockmann et al. 2008). Tenascin-C matrix tubes would not be affected by anti-angiogenic drugs. Fusenig and coworkers demonstrated that expression of tenascin-C in xenografts of squamous carcinoma cells was unchanged upon elimination of the EC with a VEGFA-targeting antibody (Vosseler et al. 2005).
Tenascin-C-associated matrix tubes are also present in a normal setting in so called reticular fibres of secondary lymphoid tissues. They combine characteristics of basement membranes and interstitial or fibrillar matrices, resulting in scaffolds that are strong and flexible, and in certain organs, such as the thymus, form conduit networks for rapid fluid transport (Lokmic et al. 2008). In the thymus, the conduits exhibit a collagen core, a wrapping with laminin and an outer lining of tenascin-C. In addition, fibrillins 1 and 2 and procollagen II were detected within these conduits (Drumea-Mirancea et al. 2006). Whether tenascin-C is required for the formation of the conduits is not known. We speculate that a program that encodes the establishment of “thymic conduits“ has been turned on inappropriately in cancer resulting in the described tenascin-C-containing matrix tubes. It will be important to understand how these matrix tubes are created, and which signals induce their emergence. It will also be necessary to determine what exact role tenascin-C plays in the formation of the matrix tubes in the thymus and in cancer.
C. Tenascin-C and mechanical stress
The tenascin-C-rich matrix tubes might also have an impact on local tissue stiffness, which is an increasingly important issue in cancer (Boyd et al. 2009). Tenascin-C might affect tissue resilience and is itself regulated by mechanical stress. Mechanical stress induced tenascin-C transcription in an ILK- and MAPK-dependent manner that did not require protein synthesis. It triggered nuclear translocation of the SRF (serum response factor) transcriptional coreceptor MAL (Maier et al. 2008). Conversely, cell adhesion to composite matrices containing tenascin-C triggered cell rounding, which could be considered as a relaxed form of the cell and its actin cytoskeleton. Cell rounding by tenascin-C was linked to inhibition of membrane recruitment of RhoA (Wenk et al. 2000), reduced RhoA protein stability, and reduced expression of actin stress fiber-stabilizing high molecular weight tropomyosins 1 – 3 (Lange et al. 2007). Thus, mechanical stress induces tenascin-C, tenascin-C may enhance tissue tension and local stiffness, and cells may counteract this tension through reduced adhesion (Chiquet et al. 2009).
D. Role of tenascin-C in tumor progression
Multiple oncogenic signaling molecules are induced by tenascin-C (Ruiz et al. 2004) that potentially are relevant in tenascin-C-associated metastasis. In particular, a fibronectin/tenascin-C substratum induced activation of Wnt signaling in glioblastoma cells by blocking expression of the soluble Wnt signaling inhibitor Dickkopf 1 (DKK1) (Ruiz et al. 2004). There is accumulating evidence for a role of DKK1 as an instrumental factor in tumor progression by regulating Wnt signaling in several cell types, which include tumor cells (Pendas-Franco et al. 2008) and vasculogenic progenitors (Aicher et al. 2008). Repression of DKK1 may also be a prerequisite for tumor cells to become responsive to epithelial-to-mesenchymal transition (EMT)-inducing signals (DiMeo et al. 2009). Tenascin-C does not only induce Wnt signaling but is itself also induced by Wnt signaling. In the lung Wnt7b induced tenascin-C (Cohen et al. 2009). In another study it was demonstrated that Wnt signaling transactivated the tenascin-C promoter in dependence of its TCF binding sites. This presents an amplification mechanism causing high tenascin-C expression and persistent Wnt signaling activation. The potential relevance of this interrelationship between tenascin-C and Wnt signaling in cancer is supported by the presence of nuclear β-catenin in human colorectal carcinomas at the invasive tumor front, where cells are embedded into a tenascin-C-rich matrix (Beiter et al. 2005).
Tenascin-C induces several molecules that are involved in EMT. At least six (Wnt, EDNRA, PDGFRα, notch, TGFβ and EGFR) of the eight pathways that induce EMT (Polyak and Weinberg 2009) are regulated by tenascin-C. Tenascin-C may not only activate Wnt signaling but may also affect TGFβ signaling, since follistatin, a negative regulator, was repressed by tenascin-C in glioblastoma cells (Ruiz et al. 2004). Moreover, RNA levels of platelet derived growth factor α, EDNRA and the notch signaling target Hes-1 were also increased under the same conditions (Ruiz et al. 2004). In fibroblasts, tenascin-C activated EGFR signaling and cell migration through binding of the EGF-like repeats of tenascin-C to the EGFR (Iyer et al. 2008). In synovial fibroblasts, tenascin-C induced expression of MMP3 (Tremble et al. 1994). A potential role of tenascin-C in EMT is also supported by a study in which MCF7 cells experienced an EMT-like morphological change on a tenascin-C substratum, that was associated with induction of the adapter molecule 14-3-3 tau (Martin et al. 2003). Despite the lack of a formal proof of a causal role of tenascin-C in full EMT so far, which might be an issue of appropriate model systems, it is intriguing to notice that tenascin-C activates multiple oncogenic signaling molecules and pathways that induce EMT under other conditions.
E. Role of tenascin-C in metastasis
The first experimental evidence for an instructive role of tenascin-C in metastasis was recently provided by data obtained from two murine tumorigenesis models. It was demonstrated that xenografts of breast carcinoma cells, in which tenascin-C expression was suppressed by siRNA, were largely impaired in establishing metastasis in the lung, which was in contrast with the parental cells that strongly induced lung metastasis (Calvo et al. 2008; Tavazoie et al. 2008). Tenascin-C was identified as a downstream target of miR-335 which also regulated expression of the transcription factor sox-4 and, suppression of sox-4 in breast cancer cells by siRNA upregulated tenascin-C expression and lung metastasis (Tavazoie et al. 2008). Sox-4 was also identified as target gene to be induced by tenascin-C in glioblastoma cells (Ruiz et al. 2004), which suggests that expression of these two molecules could be interdependent and crucial for metastasis formation.
Summary: tenascin-C in cancer
Tenascin-C performs multiple functions that depend on its presence as a structural component and on its interactions with cells by means of specific binding to cell-surface receptors (Table 3, Fig. 3). Many cells interact with tenascin-C and the resulting responses are context dependent. Tenascin-C activates oncogenic signaling and by that might promote tumor progression which represents its signaling nature. A role of tenascin-C as architectual molecule is demonstrated by its interaction with multiple ECM molecules and its presence in neuronal and neural crest cell guiding cues, underneath basement membranes, in reticular fibers, thymic conduits and in matrix channels of melanomas. Ectopic expression of tenascin-C in matrix tubes that are physically linked to blood vessels in metastastic cancers might not only have a huge impact on the global mechanical properties of the tissue, and thereby affect cellular signaling, but might also function as cues for tumor cell dissemination and metastasis.
Improved diagnosis and treatment of tenascin-C associated pathologies
Expression of tenascin-C has already some application in disease diagnosis. Tissue tenascin-C levels are used as predictive marker for fibrosis (Lieber et al. 2008; Paivaniemi et al. 2008) and inflammation in myocarditis (Tsukada et al. 2009), and high tenascin-C serum levels in patients with inflammatory bowel disease correlate with disease severity (Riedl et al. 2001). Moreover, in breast cancer patients treated with tamoxifen the high expression of tenascin-C correlated with a reduced drug responsiveness (Helleman et al. 2008).
Tenascin-C expression correlates with adverse effects in pulmonary hypertension, cardiovascular diseases (Fischer 2007), arterial grafting (Sawada et al. 2007), artheriosclerosis (Kenji et al. 2004; von Lukowicz et al. 2007), chronic inflammation and cancer. Inhibition of tenascin-C expression at the transcriptional level would be the first choice to prevent tenascin-C actions. Unfortunately, this approach is not easy to accomplish since many factors and conditions that trigger tenascin-C expression are not specific for tenascin-C alone but would affect many other genes with unwanted side-effects. In cardiovascular diseases, tenascin-C promotes survival of vascular smooth muscle cells and hyperproliferation which is linked to heart failure (Hedin et al. 1991; Jones et al. 1997; Milting et al. 2008). Inhibition of phosphodiesterase III by cilostazol may present a suitable approach since this drug reduces tenascin-C expression in vascular smooth muscle cells and prevents neointimal hyperplasia (Fujinaga et al. 2004; Yamamoto et al. 2007). Inhibition of angiotensin II, which is a potent inducer of tenascin-C, with drugs such as angiotensin II type 1 receptor (AT-1) antagonists and angiotensin converting enzyme (ACE) inhibitors potentially present another means to block vascular tenascin-C expression in hypertensive patients (Fischer 2007). In a glioblastoma patient double stranded anti-tenascin-C RNA was used to reduce tenascin-C expression (Zukiel et al. 2006). Tenascin-C expression was also shown to be blocked with a tenascin-C specific antisense/ribozyme, which prevented progression of the vascular disease (Cowan et al. 2000).
More mechanistic insight into tenascin-C actions in diseased tissues could provide information that may be useful in disease treatment. In this sense it needs to be determined whether blocking TLR4 in rheumatoid arthritis and cancer, and restoration of syndecan-4 function, or blocking tenascin-C-induced oncogenic signaling in tumors are suitable strategies to counteract tenascin-C and potentially ameliorate or erradicate the pathological situation.
Given its high expression in diseased tissues, a promising approach is the targeting of tenascin-C with anti-tenascin-C-directed antibodies coupled to cytotoxic agents. A bispecific CD95/anti-tenascin-C antibody that activated the Fas/Apo-1 death receptor efficiently caused glioma cell lysis that was strongly enhanced in combination with topoisomerase inhibitors or doxorubicin (Herrmann et al. 2008). Similarly, recruitment of the immune system by attaching IL2 to an anti-tenascin-C antibody (G11) (Marlind et al. 2008) or coupling cytotoxic radioactivity to the B8C6 anti-tenascin-C antibody represent other potentially efficient strategies (Reardon et al. 2008). DNA based aptamers that specifically recognize tenascin-C may also be suited for targeting tenascin-C (Hicke et al. 2006). Tenascin-C targeting antibodies are in advanced clinical trials for use in tumor imaging and therapy (Akabani et al. 2005; Silacci et al. 2006). These applications would require high amounts of tenascin-C specific antibodies which could be achieved by expression of a single chain antibody that recognizes human tenascin-C in the plant nicotiana bentamiana (Villani et al. 2009).
Parallels between tenascin-C-associated inflammation, the repair process and tumorigenesis
Clinical and epidemiologic studies suggest a strong association between inflammation and cancer (Coussens and Werb 2002; Hussain et al. 2003; Lin and Karin 2007; Shacter and Weitzman 2002). Inflammation appears to orchestrate the tumor microenvironment and is a critical condition for both tumor promotion and tumor progression (Balkwill et al. 2005). The inflammatory milieu of tumors is characterized by cells such as resident and recruited macrophages, dendritic cells, T cells and NK cells (Luster et al. 2005). These infiltrated immune cells can produce pro-inflammatory mediators that weaken anti-tumor immunity (Ben-Baruch 2006; Smyth et al. 2004). Most pro-inflammatory cytokines (e.g. IL6, IL8, TNFα) produced by either host immune cells or tumor cells can themselves promote tumor development. By contrast, pro-apoptotic (e.g. TRAIL) and anti-inflammatory (e.g. IL10) cytokines usually interfere with tumor development (Lin and Karin 2007). A persistent inflammatory microenvironment can induce tumor promotion, and accelerate tumor progression, invasion, angiognesesis and often metastasis (Coussens and Werb 2002; Dobrovolskaia and Kozlov 2005; Karin and Greten 2005).
Activation of TLR signaling presents a mechanistic link between inflammation and cancer. TLR signaling can enhance tumor development by means of various mechanisms. In a mouse model of transplanted metastatic cancer cells, activation of TLR4 by intraperitoneal injection of bacterial LPS stimulated the growth of lung metastasis, which was linked to the production of several different inflammatory cytokines (Harmey et al. 2002; Luo et al. 2004; Pidgeon et al. 1999). In this scenario activation of TLR4 by tenascin-C might play a similarly important role. Tenascin-C maintained chronic inflammation in rheumatoid arthritic joints through binding and activation of TLR4, which subsequently stimulated secretion of pro-inflammatory cytokines IL6, IL8 and TNFα (Midwood et al. 2009). Whether TLR4 activation by tenascin-C promotes tumor progression is an intriguing possibility that needs to be addressed in the future. Other ECM molecules such as biglycan and versican also activated TLRs, with biglycan binding to TLR2 and TLR4 (Schaefer et al. 2005), and versican binding to TLR2 (Kim et al. 2009). Activation of TLR2 by versican in myeloid cells induced secretion of TNFα and IL6, which caused metastatic spread of murine LLC lung carcinoma cells to the lung (Kim et al. 2009). These data suggest that activation of TLRs by soluble and insoluble ECM ligands can induce secretion of pro-inflammatory cytokines that would promote tumor progression and metastasis (Table 2).
An extensive literature supports similarities between wound healing and cancer. Tumors are even considered as wounds that exhibit an abnormal healing process (Schafer and Werner 2008). Several components of the tumor stroma are common to those of the granulation tissue in healing skin wounds, which suggests that epithelial tumors potentially promote the formation of their stroma by activating the wound-healing response of the host. However, in contrast to healing wounds, the process is not self-limiting in cancer tissue, and this leads to uncontrolled cell proliferation, invasion and metastasis. In particular, a matrix of crosslinked fibrin and fibronectin containing tenascin-C is present during wound healing (Midwood et al. 2004b), and ectopic expression of fibronectin, fibrin and tenascin-C is a hallmark of cancer (Kaspar et al. 2006; Konstantopoulos and Thomas 2009; McMahon and Kwaan 2008; Orend and Chiquet-Ehrismann 2006; Rybak et al. 2007). Wound healing and tumor vessels are initially immature and leaky, and VEGFA controls angiogenesis in wounds and tumors. Tenascin-C regulates VEGFA levels by an unknown mechanism (Tanaka et al. 2004), and suture wounds in skin and retina heal poorly in tenascin-C-null mice (Forsberg et al. 1996; Matsuda et al. 1999). Wound re-epithelialization is an event that is reminiscent of EMT, which is also a hallmark of cancer. Tenascin-C might be involved in EMT in both wounds and tumors, since tenascin-C not only induces EMT-like morphologically changes in culture, and induces genes that trigger EMT, but is also expressed in wounds and cancer tissue at sites, where EMT occurs.
Our increasing insight into the mechanisms involved in the functions of tenascin-C combined with the existing strategies for targeting this protein, generate optimism that some life-threatening and painful diseases in which tenascin-C is highly expressed will be better managed in the not so distant future by applying this knowledge for treatment and/or diagnosis.
Abbreviations
- ACE:
-
angiotensin converting enzyme
- AT-1:
-
angiotensin II type 1 receptor
- bFGF:
-
basic fibroblast growth factor
- CALEB:
-
chicken acidic leucine-rich EGF like domain containing brain protein
- c-Met:
-
mesenchymal-epithelial transition factor
- DAMP:
-
damage associated molecular pattern
- DKK1:
-
Dickkopf 1
- EC:
-
endothelial cell
- ECM:
-
extracellular matrix
- EDNRA:
-
endothelin receptor type A
- EGF:
-
epidermal growth factor
- EGF-L:
-
epidermal growth factor like repeat
- EGFR:
-
epidermal growth factor receptor
- EMT:
-
epithelial-to-mesenchymal transition
- FAK:
-
focal adhesion kinase
- FBG:
-
fibrinogen like globe
- FGFR:
-
fibroblast growth factor receptor
- FNIII:
-
fibronectin type III like repeat
- GMEM:
-
glial/mesenchymal extracellular matrix protein
- HepII:
-
Heparin binding domain II
- HNF-4α:
-
hepatocyte nuclear factor 4α
- HSPG:
-
heparan sulfate proteoglycan
- ICAM-1:
-
Inter cellular adhesion molecule 1
- IL:
-
interleukin
- IFN:
-
interferon
- IgE:
-
Immunoglobulin E
- ILK:
-
integrin linked kinase
- LEF/TCF:
-
lymphoid enhancer-binding factor, T cell factor
- LPS:
-
lipopolysaccharide
- MAL:
-
myocardin related transcription factor
- MAPK:
-
mitogen activated protein kinase
- MCP1:
-
monocyte chemotactic protein-1
- miR:
-
micro RNA
- MMP:
-
matrix metalloprotease
- NFκB:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- NK cell:
-
natural killer cell
- PAMP:
-
pathogen associated molecular pattern
- PDGFRα/β:
-
platelet dervied growth factor receptor-α/-β
- PMA:
-
phorbol 12myristate 13acetate
- PLC:
-
phospholipase C
- PRR:
-
pattern recognition receptor
- Prx-1:
-
peroxiredoxin -1
- RPTPβ:
-
receptor protein tyrosine phosphatase β
- siRNA:
-
small interfering RNA
- SMC:
-
smooth muscle cell
- SRF:
-
serum response factor
- TCF:
-
T-cell factor
- TGF:
-
transforming growth factor
- Th2:
-
T helper cell
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor
- TRAIL:
-
tumour necrosis factor related apoptosis inducing ligand
- VEGFA:
-
vascular endothelial growth factor
References
Adams M, Jones JL, Walker RA, Pringle JH, Bell SC (2002) Changes in tenascin-C isoform expression in invasive and preinvasive breast disease. Cancer Res 62:3289–3297
Aicher A, Kollet O, Heeschen C, Liebner S, Urbich C, Ihling C, Orlandi A, Lapidot T, Zeiher AM, Dimmeler S (2008) The Wnt antagonist Dickkopf-1 mobilizes vasculogenic progenitor cells via activation of the bone marrow endosteal stem cell niche. Circ Res 103:796–803
Akabani G, Reardon DA, Coleman RE, Wong TZ, Metzler SD, Bowsher JE, Barboriak DP, Provenzale JM, Greer KL, DeLong D, Friedman HS, Friedman AH, Zhao XG, Pegram CN, McLendon RE, Bigner DD, Zalutsky MR (2005) Dosimetry and radiographic analysis of 131I-labeled anti-tenascin 81C6 murine monoclonal antibody in newly diagnosed patients with malignant gliomas: a phase II study. J Nucl Med. 46:1042–1051
Alexopoulou AN, Multhaupt HA, Couchman JR (2007) Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol 39:505–528
Amin K, Ekberg-Jansson A, Lofdahl CG, Venge P (2003) Relationship between inflammatory cells and structural changes in the lungs of asymptomatic and never smokers: a biopsy study. Thorax 58:135–142
Amin K, Ludviksdottir D, Janson C, Nettelbladt O, Bjornsson E, Roomans GM, Boman G, Seveus L, Venge P (2000) Inflammation and structural changes in the airways of patients with atopic and nonatopic asthma BHR Group. Am J Respir Crit Care Med 162:2295–2301
Aukhil I, Joshi P, Yan Y, Erickson HP (1993) Cell- and heparin-binding domains of the hexabrachion arm identified by tenascin expression proteins. J Biol Chem. 268:2542–2553
Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 7:211–217
Ballard VL, Sharma A, Duignan I, Holm JM, Chin A, Choi R, Hajjar KA, Wong SC, Edelberg JM (2006) Vascular tenascin-C regulates cardiac endothelial phenotype and neovascularization. Faseb J. 20:717–719
Beiter K, Hiendlmeyer E, Brabletz T, Hlubek F, Haynl A, Knoll C, Kirchner T, Jung A (2005) beta-Catenin regulates the expression of tenascin-C in human colorectal tumors. Oncogene. 24:8200–8204
Bell SC, Pringle JH, Taylor DJ, Malak TM (1999) Alternatively spliced tenascin-C mRNA isoforms in human fetal membranes. Mol Hum Reprod. 5:1066–1076
Ben-Baruch A (2006) Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol 16:38–52
Betz P, Nerlich A, Tubel J, Penning R, Eisenmenger W (1993) Localization of tenascin in human skin wounds-an immunohistochemical study. Int J Legal Med 105:325–328
Bissell MJ, Labarge MA (2005) Context tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 7:17–23
Bissell MJ, Radisky D (2001) Putting tumours in context. Nat Rev Cancer. 1:46–54
Borsi L, Allemanni G, Gaggero B, Zardi L (1996) Extracellular pH controls pre-mRNA alternative splicing of tenascin-C in normal, but not in malignantly transformed, cells. Int J Cancer 66:632–635
Borsi L, Balza E, Castellani P, Carnemolla B, Ponassi M, Querze G, Zardi L (1994) Cell-cycle dependent alternative splicing of the tenascin primary transcript. Cell Adhes Commun 1:307–317
Boyd NL, Martin S, Chavez A, Gunasekara A, Salleh O, Melnichouk M, Yaffe C, Friedenreich S, Minkin, Bronskill M (2009) Breast-tissue composition and other risk factors for breast cancer in young women: a cross-sectional study. Lancet Oncol 10:569–580
Busse WW, Lemanske RF Jr (2001) Asthma. N Engl J Med. 344:350–362
Calvo A, Catena R, Noble MS, Carbott D, Gil-Bazo I, Gonzalez-Moreno O, Huh JI, Sharp R, Qiu TH, Anver MR, Merlino G, Dickson RB, Johnson MD, Green JE (2008) Identification of VEGF-regulated genes associated with increased lung metastatic potential: functional involvement of tenascin-C in tumor growth and lung metastasis. Oncogene 27:5373–5386
Canfield AE, Schor AM (1995) Evidence that tenascin and thrombospondin-1 modulate sprouting of endothelial cells. J Cell Sci. 108(Pt 2):797–809
Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6:389–395
Carnemolla B, Castellani P, Ponassi M, Borsi L, Urbini S, Nicolo G, Dorcaratto A, Viale G, Winter G, Neri D, Zardi L (1999) Identification of a glioblastoma-associated tenascin-C isoform by a high affinity recombinant antibody. Am J Pathol 154:1345–1352
Castellon R, Caballero S, Hamdi HK, Atilano SR, Aoki AM, Tarnuzzer RW, Kenney MC, Grant MB, Ljubimov AV (2002) Effects of tenascin-C on normal and diabetic retinal endothelial cells in culture. Invest Ophthalmol Vis Sci 43:2758–2766
Cella N, Chiquet-Ehrismann R, Hynes NE (2000) Lactogenic hormones and tenascin-C regulate C/EBPalpha and beta in mammary epithelial cells. J Cell Biochem. 76:394–403
Chiquet M, Fambrough DM (1984) Chick myotendinous antigen I. A monoclonal antibody as a marker for tendon and muscle morphogenesis. J Cell Biol. 98:1926–1936
Chiquet M, Gelman L, Lutz R, Maier S (2009) From mechanotransduction to extracellular matrix gene expression in fibroblasts. Biochim Biophys Acta 1793:911–920
Chiquet-Ehrismann R (2004) Tenascins. Int J Biochem Cell Biol 36:986–990
Chiquet-Ehrismann R, Chiquet M (2003) Tenascins: regulation and putative functions during pathological stress. J Pathol. 200:488–499
Chiquet-Ehrismann R, Hagios C, Matsumoto K (1994) The tenascin gene family. Perspect Dev Neurobiol 2:3–7
Chiquet-Ehrismann R, Mackie EJ, Pearson CA, Sakakura T (1986) Tenascin: an extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell 47:131–139
Chiquet-Ehrismann R, Matsuoka Y, Hofer U, Spring J, Bernasconi C, Chiquet M (1991) Tenascin variants: differential binding to fibronectin and distinct distribution in cell cultures and tissues. Cell Regul. 2:927–938
Chung CY, Erickson HP (1994) Cell surface annexin II is a high affinity receptor for the alternatively spliced segment of tenascin-C. J Cell Biol. 126:539–548
Chung CY, Erickson HP (1997) Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-C. J Cell Sci. 110:1413–1419
Chung CY, Murphy-Ullrich JE, Erickson HP (1996) Mitogenesis, cell migration, and loss of focal adhesions induced by tenascin-C interacting with its cell surface receptor, annexin II. Mol Biol Cell 7:883–892
Chung CY, Zardi L, Erickson HP (1995) Binding of tenascin-C to soluble fibronectin and matrix fibrils. J Biol Chem. 270:29012–29017
Clark RA (1993) Biology of dermal wound repair. Dermatol Clin 11:647–666
Clark RA, Erickson HP, Springer TA (1997) Tenascin supports lymphocyte rolling. J Cell Biol. 137:755–765
Cohen ED, Ihida-Stansbury K, Lu MM, Panettieri RA, Jones PL, Morrisey EE (2009) Wnt signaling regulates smooth muscle precursor development in the mouse lung via a tenascin C/PDGFR pathway. J Clin Invest. 119:2538–2549
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420:860–867
Cowan KN, Jones PL, Rabinovitch M (2000) Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. 105:21–34
Crossin KL (1991) Cytotactin binding: inhibition of stimulated proliferation and intracellular alkalinization in fibroblasts. Proc Natl Acad Sci U S A. 88:11403–11407
Day JM, Olin AI, Murdoch AD, Canfield A, Sasaki T, Timpl R, Hardingham TE, Aspberg A (2004) Alternative splicing in the aggrecan G3 domain influences binding interactions with tenascin-C and other extracellular matrix proteins. J Biol Chem. 279:12511–12518
De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, Mareel M (2004) Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. Faseb J. 18:1016–1018
Degen M, Brellier F, Kain R, Ruiz C, Terracciano L, Orend G, Chiquet-Ehrismann R (2007) Tenascin-W is a novel marker for activated tumor stroma in low-grade human breast cancer and influences cell behavior. Cancer Res 67:9169–9179
Degen M, Brellier F, Schenk S, Driscoll R, Zaman K, Stupp R, Tornillo L, Terracciano L, Chiquet-Ehrismann R, Ruegg C, Seelentag W (2008) Tenascin-W, a new marker of cancer stroma, is elevated in sera of colon and breast cancer patients. Int J Cancer 122:2454–2461
Denda S, Müller U, Crossin KL, Erickson HP, Reichardt LF (1998) Utilization of a soluble integrin-alkaline phosphatase chimera to characterize integrin alpha 8 beta 1 receptor interactions with tenascin: murine alpha 8 beta 1 binds to the RGD site in tenascin-C fragments, but not to native tenascin-C. Biochemistry 37:5464–5474
Derr LB, Chiquet-Ehrismann R, Gandour-Edwards R, Spence J, Tucker RP (1997) The expression of tenascin-C with the AD1 variable repeat in embryonic tissues, cell lines and tumors in various vertebrate species. Differentiation. 62:71–82
DiMeo TA, Anderson K, Phadke P, Feng C, Perou CM, Naber S, Kuperwasser C (2009) A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res 69:5364–5373
Dobrovolskaia MA, Kozlov SV (2005) Inflammation and cancer: when NF-kappaB amalgamates the perilous partnership. Curr Cancer Drug Targets. 5:325–344
Drumea-Mirancea M, Wessels JT, Muller CA, Essl M, Eble JA, Tolosa E, Koch M, Reinhardt DP, Sixt M, Sorokin L, Stierhof YD, Schwarz H, Klein G (2006) Characterization of a conduit system containing laminin-5 in the human thymus: a potential transport system for small molecules. J Cell Sci. 119:1396–1405
Dueck M, Riedl S, Hinz U, Tandara A, Moller P, Herfarth C, Faissner A (1999) Detection of tenascin-C isoforms in colorectal mucosa, ulcerative colitis, carcinomas and liver metastases. Int J Cancer 82:477–483
El-Karef A, Yoshida T, Gabazza EC, Nishioka T, Inada H, Sakakura T, Imanaka-Yoshida K (2007) Deficiency of tenascin-C attenuates liver fibrosis in immune-mediated chronic hepatitis in mice. J Pathol. 211:86–94
End P, Panayotou G, Entwistle A, Waterfield MD, Chiquet M (1992) Tenascin: a modulator of cell growth. Eur J Biochem 209:1041–1051
Faissner A (1997) The tenascin gene family in axon growth and guidance. Cell Tissue Res 290:331–341
Faissner A, Kruse J, Kuhn K, Schachner M (1990) Binding of the J1 adhesion molecules to extracellular matrix constituents. J Neurochem. 54:1004–1015
Fassler R, Sasaki T, Timpl R, Chu ML, Werner S (1996) Differential regulation of fibulin, tenascin-C, and nidogen expression during wound healing of normal and glucocorticoid-treated mice. Exp Cell Res 222:111–116
Filsell W, Rudman S, Jenkins G, Green MR (1999) Coordinate upregulation of tenascin C expression with degree of photodamage in human skin. Br J Dermatol 140:592–599
Fischer D, Chiquet-Ehrismann R, Bernasconi C, Chiquet M (1995) A single heparin binding region within the fibrinogen-like domain is functional in chick tenascin-C. J Biol Chem. 270:3378–3384
Fischer D, Tucker RP, Chiquet-Ehrismann R, Adams JC (1997) Cell-adhesive responses to tenascin-C splice variants involve formation of fascin microspikes. Mol Biol Cell 8:2055–2075
Fischer JW (2007) Tenascin-C: a key molecule in graft stenosis. Cardiovasc Res 74:335–336
Fluck M, Mund SI, Schittny JC, Klossner S, Durieux AC, Giraud MN (2008) Mechano-regulated tenascin-C orchestrates muscle repair. Proc Natl Acad Sci U S A. 105:13662–13667
Forsberg E, Hirsch E, Frohlich L, Meyer M, Ekblom P, Aszodi A, Werner S, Fassler R (1996) Skin wounds and severed nerves heal normally in mice lacking tenascin-C. Proc Natl Acad Sci U S A. 93:6594–6599
Fujinaga K, Onoda K, Yamamoto K, Imanaka-Yoshida K, Takao M, Shimono T, Shimpo H, Yoshida T, Yada I (2004) Locally applied cilostazol suppresses neointimal hyperplasia by inhibiting tenascin-C synthesis and smooth muscle cell proliferation in free artery grafts. J Thorac Cardiovasc Surg. 128:357–363
Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E (2007) Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9:1392–1400
Geboes K, El-Zine MY, Dalle I, El-Haddad S, Rutgeerts P, Van Eyken P (2001) Tenascin and strictures in inflammatory bowel disease: an immunohistochemical study. Int J Surg Pathol. 9:281–286
Geffrotin C, Tricaud Y, Crechet F, Castelli M, Lefaix JL, Vaiman M (1998) Unlike tenascin-X, tenascin-C is highly up-regulated in pig cutaneous and underlying muscle tissue developing fibrosis after necrosis induced by very high-dose gamma radiation. Radiat Res 149:472–481
Gerritsen MJ, Elbers ME, de Jong EM, van de Kerkhof PC (1997) Recruitment of cycling epidermal cells and expression of filaggrin, involucrin and tenascin in the margin of the active psoriatic plaque, in the uninvolved skin of psoriatic patients and in the normal healthy skin. J Dermatol Sci. 14:179–188
Ghatnekar A, Trojanowska M (2008) GATA-6 is a novel transcriptional repressor of the human Tenascin-C gene expression in fibroblasts. Biochim Biophys Acta 1779:145–151
Gherzi R, Carnemolla B, Siri A, Ponassi M, Balza E, Zardi L (1995) Human tenascin gene. Structure of the 5′-region, identification, and characterization of the transcription regulatory sequences. J Biol Chem 270:3429–3434
Giese A, Loo MA, Norman SA, Treasurywala S, Berens ME (1996) Contrasting migratory response of astrocytoma cells to tenascin mediated by different integrins. J Cell Sci. 109(Pt 8):2161–2168
Gindre D, Peyrol S, Raccurt M, Sommer P, Loire R, Grimaud JA, Cordier JF (1995) Fibrosing vasculitis in Wegener’s granulomatosis: ultrastructural and immunohistochemical analysis of the vascular lesions. Virchows Arch 427:385–393
Goodger SJ, Robinson CJ, Murphy KJ, Gasiunas N, Harmer NJ, Blundell TL, Pye DA, Gallagher JT (2008) Evidence that heparin saccharides promote FGF2 mitogenesis through two distinct mechanisms. J Biol Chem. 283:13001–13008
Gordon S (2002) Pattern recognition receptors: doubling up for the innate immune response. Cell 111:927–930
Gotz M, Bolz J, Joester A, Faissner A (1997) Tenascin-C synthesis and influence on axonal growth during rat cortical development. Eur J NeuroSci 9:496–506
Hancox RA, Allen MD, Holliday DL, Edwards DR, Pennington CJ, Guttery DS, Shaw JA, Walker RA, Pringle JH, Jones JL (2009) Tumor-associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase-dependent and independent mechanisms. Breast Cancer Res 11:R24
Harmey JH, Bucana CD, Lu W, Byrne AM, McDonnell S, Lynch C, Bouchier-Hayes D, Dong Z (2002) Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int J Cancer 101:415–422
Hauzenberger D, Olivier P, Gundersen D, Ruegg C (1999) Tenascin-C inhibits beta1 integrin-dependent T lymphocyte adhesion to fibronectin through the binding of its fnIII 1-5 repeats to fibronectin. Eur J Immunol 29:1435–1447
Hedin U, Holm J, Hansson GK (1991) Induction of tenascin in rat arterial injury Relationship to altered smooth muscle cell phenotype. Am J Pathol 139:649–656
Helleman J, Jansen MP, Ruigrok-Ritstier K, van Staveren IL, Look MP, Meijer-van Gelder ME, Sieuwerts AM, Klijn JG, Sleijfer S, Foekens JA, Berns EM (2008) Association of an extracellular matrix gene cluster with breast cancer prognosis and endocrine therapy response. Clin Cancer Res. 14:5555–5564
Hemesath TJ, Marton LS, Stefansson K (1994) Inhibition of T cell activation by the extracellular matrix protein tenascin. J Immunol. 152:5199–5207
Hendrix MJ, Seftor EA, Hess AR, Seftor RE (2003) Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 3:411–421
Herold-Mende C, Mueller MM, Bonsanto MM, Schmitt HP, Kunze S, Steiner HH (2002) Clinical impact and functional aspects of tenascin-C expression during glioma progression. Int J Cancer 98:362–369
Herrmann T, Grosse-Hovest L, Otz T, Krammer PH, Rammensee HG, Jung G (2008) Construction of optimized bispecific antibodies for selective activation of the death receptor CD95. Cancer Res 68:1221–1227
Hibino S, Kato K, Kudoh S, Yagita H, Okumura K (1998) Tenascin suppresses CD3-mediated T cell activation. Biochem Biophys Res Commun 250:119–124
Hicke BJ, Stephens AW, Gould T, Chang YF, Lynott CK, Heil J, Borkowski S, Hilger CS, Cook G, Warren S, Schmidt PG (2006) Tumor targeting by an aptamer. J Nucl Med. 47:668–678
Huang W, Chiquet-Ehrismann R, Moyano JV, Garcia-Pardo A, Orend G (2001) Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res 61:8586–8594
Hussain SP, Hofseth LJ, Harris CC (2003) Radical causes of cancer. Nat Rev Cancer. 3:276–285
Imai K, Kusakabe M, Sakakura T, Nakanishi I, Okada Y (1994) Susceptibility of tenascin to degradation by matrix metalloproteinases and serine proteinases. FEBS Lett 352:216–218
Ingber DE (2002) Mechanical signaling and the cellular response to extracellular matrix in angiogenesis and cardiovascular physiology. Circ Res 91:877–887
Ingham KC, Brew SA, Erickson HP (2004) Localization of a cryptic binding site for tenascin on fibronectin. J Biol Chem. 279:28132–28135
Ishii K, Imanaka-Yoshida K, Yoshida T, Sugimura Y (2008) Role of stromal tenascin-C in mouse prostatic development and epithelial cell differentiation. Dev Biol 324:310–319
Ishikawa F, Nose K, Shibanuma M (2008) Downregulation of hepatocyte nuclear factor-4alpha and its role in regulation of gene expression by TGF-beta in mammary epithelial cells. Exp Cell Res 314:2131–2140
Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C, Flanagan L, Tenniswood MP, Guha C, Lisanti MP, Pestell RG, Scherer PE (2003) Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene. 22:6408–6423
Iyer AK, Tran KT, Borysenko CW, Cascio M, Camacho CJ, Blair HC, Bahar I, Wells A (2007) Tenascin cytotactin epidermal growth factor-like repeat binds epidermal growth factor receptor with low affinity. J Cell Physiol. 211:748–758
Iyer AK, Tran KT, Griffith L, Wells A (2008) Cell surface restriction of EGFR by a tenascin cytotactin-encoded EGF-like repeat is preferential for motility-related signaling. J Cell Physiol. 214:504–512
Jallo GI, Friedlander DR, Kelly PJ, Wisoff JH, Grumet M, Zagzag D (1997) Tenascin-C expression in the cyst wall and fluid of human brain tumors correlates with angiogenesis. Neurosurgery. 41:1052–1059
Joester A, Faissner A (2001) The structure and function of tenascins in the nervous system. Matrix Biol 20:13–22
Jones FS, Hoffman S, Cunningham BA, Edelman GM (1989) A detailed structural model of cytotactin: protein homologies, alternative RNA splicing, and binding regions. Proc Natl Acad Sci U S A. 86:1905–1909
Jones PL, Boudreau N, Myers CA, Erickson HP, Bissell MJ (1995) Tenascin-C inhibits extracellular matrix-dependent gene expression in mammary epithelial cells Localization of active regions using recombinant tenascin fragments. J Cell Sci. 108(Pt 2):519–527
Jones PL, Crack J, Rabinovitch M (1997) Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the alpha v beta 3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol. 139:279–293
Jones PL, Jones FS (2000) Tenascin-C in development and disease: gene regulation and cell function. Matrix Biol 19:581–596
Julian J, Chiquet-Ehrismann R, Erickson HP, Carson DD (1994) Tenascin is induced at implantation sites in the mouse uterus and interferes with epithelial cell adhesion. Development. 120:661–671
Kaariainen E, Nummela P, Soikkeli J, Yin M, Lukk M, Jahkola T, Virolainen S, Ora A, Ukkonen E, Saksela O, Holtta E (2006) Switch to an invasive growth phase in melanoma is associated with tenascin-C, fibronectin, and procollagen-I forming specific channel structures for invasion. J Pathol. 210:181–191
Kaarteenaho-Wiik R, Lakari E, Soini Y, Pollanen R, Kinnula VL, Paakko P (2000) Tenascin expression and distribution in pleural inflammatory and fibrotic diseases. J Histochem Cytochem. 48:1257–1268
Kaarteenaho-Wiik R, Mertaniemi P, Sajanti E, Soini Y, Paakko P (1998) Tenascin is increased in epithelial lining fluid in fibrotic lung disorders. Lung 176:371–380
Kalembeyi I, Inada H, Nishiura R, Imanaka-Yoshida K, Sakakura T, Yoshida T (2003) Tenascin-C upregulates matrix metalloproteinase-9 in breast cancer cells: direct and synergistic effects with transforming growth factor beta1. Int J Cancer 105:53–60
Kanayama M, Kurotaki D, Morimoto J, Asano T, Matsui Y, Nakayama Y, Saito Y, Ito K, Kimura C, Iwasaki N, Suzuki K, Harada T, Li HM, Uehara J, Miyazaki T, Minami A, Kon S, Uede T (2009) Alpha9 integrin and its ligands constitute critical joint microenvironments for development of autoimmune arthritis. J Immunol. 182:8015–8025
Kaplony A, Zimmermann DR, Fischer RW, Imhof BA, Odermatt BF, Winterhalter KH, Vaughan L (1991) Tenascin Mr 220, 000 isoform expression correlates with corneal cell migration. Development. 112:605–614
Karin M, Greten FR (2005) NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 5:749–759
Karjalainen EM, Laitinen A, Sue-Chu M, Altraja A, Bjermer L, Laitinen LA (2000) Evidence of airway inflammation and remodeling in ski athletes with and without bronchial hyperresponsiveness to methacholine. Am J Respir Crit Care Med 161:2086–2091
Kaspar M, Zardi L, Neri D (2006) Fibronectin as target for tumor therapy. Int J Cancer 118:1331–1339
Katz S, Hukkanen M, Lounatmaa K, Rousselle P, Tervo T, Virtanen I (2006) Cooperation of isoforms of laminin-332 and tenascin-CL during early adhesion and spreading of immortalized human corneal epithelial cells. Exp Eye Res 83:1412–1422
Kenji K, Hironori U, Hideya Y, Michinori I, Yasuhiko H, Nobuoki K (2004) Tenascin-C is associated with coronary plaque instability in patients with acute coronary syndromes. Circ J. 68:198–203
Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457:102–106
Knaggs HE, Layton AM, Morris C, Wood EJ, Holland DB, Cunliffe WJ (1994) Investigation of the expression of the extracellular matrix glycoproteins tenascin and fibronectin during acne vulgaris. Br J Dermatol 130:576–582
Konstantopoulos K, Thomas SN (2009) Cancer Cells in Transit: The Vascular Interactions of Tumor Cells. Annu Rev Biomed Eng 11:177–202
Koyama Y, Kusubata M, Yoshiki A, Hiraiwa N, Ohashi T, Irie S, Kusakabe M (1998) Effect of tenascin-C deficiency on chemically induced dermatitis in the mouse. J Invest Dermatol. 111:930–935
Kucera T, Strilic B, Regener K, Schubert M, Laudet V, Lammert E (2009) Ancestral vascular lumen formation via basal cell surfaces. PLoS ONE 4:e4132
Kuhn C, Mason RJ (1995) Immunolocalization of SPARC, tenascin, and thrombospondin in pulmonary fibrosis. Am J Pathol 147:1759–1769
Kusubata M, Hirota A, Ebihara T, Kuwaba K, Matsubara Y, Sasaki T, Kusakabe M, Tsukada T, Irie S, Koyama Y (1999) Spatiotemporal changes of fibronectin, tenascin-C, fibulin-1, and fibulin-2 in the skin during the development of chronic contact dermatitis. J Invest Dermatol. 113:906–912
Kuznetsova SA, Roberts DD (2004) Functional regulation of T lymphocytes by modulatory extracellular matrix proteins. Int J Biochem Cell Biol 36:1126–1134
Lange K, Kammerer M, Hegi ME, Grotegut S, Dittmann A, Huang W, Fluri E, Yip GW, Gotte M, Ruiz C, Orend G (2007) Endothelin receptor type B counteracts tenascin-C-induced endothelin receptor type A-dependent focal adhesion and actin stress fiber disorganization. Cancer Res 67:6163–6173
Lange K, Kammerer M, Saupe F, Hegi ME, Grotegut S, Fluri E, Orend G (2008) Combined lysophosphatidic acid/platelet-derived growth factor signaling triggers glioma cell migration in a tenascin-C microenvironment. Cancer Res 68:6942–6952
Latijnhouwers M, Bergers M, Ponec M, Dijkman H, Andriessen M, Schalkwijk J (1997) Human epidermal keratinocytes are a source of tenascin-C during wound healing. J Invest Dermatol. 108:776–783
Latijnhouwers MA, Bergers M, Van Bergen BH, Spruijt KI, Andriessen MP, Schalkwijk J (1996) Tenascin expression during wound healing in human skin. J Pathol. 178:30–35
Lieber CS, Weiss DG, Paronetto F (2008) Value of fibrosis markers for staging liver fibrosis in patients with precirrhotic alcoholic liver disease. Alcohol Clin Exp Res 32:1031–1039
Lightner VA, Gumkowski F, Bigner DD, Erickson HP (1989) Tenascin/hexabrachion in human skin: biochemical identification and localization by light and electron microscopy. J Cell Biol. 108:2483–2493
Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW (2006) Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66:11238–11246
Lin WW, Karin M (2007) A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 117:1175–1183
Ljubimov AV, Saghizadeh M, Spirin KS, Khin HL, Lewin SL, Zardi L, Bourdon MA, Kenney MC (1998) Expression of tenascin-C splice variants in normal and bullous keratopathy human corneas. Invest Ophthalmol Vis Sci 39:1135–1142
Loike JD, Cao L, Budhu S, Hoffman S, Silverstein SC (2001) Blockade of alpha 5 beta 1 integrins reverses the inhibitory effect of tenascin on chemotaxis of human monocytes and polymorphonuclear leukocytes through three-dimensional gels of extracellular matrix proteins. J Immunol. 166:7534–7542
Lokmic Z, Lammermann T, Sixt M, Cardell S, Hallmann R, Sorokin L (2008) The extracellular matrix of the spleen as a potential organizer of immune cell compartments. Semin Immunol 20:4–13
Lundell A, Olin AI, Morgelin M, al-Karadaghi S, Aspberg A, Logan DT (2004) Structural basis for interactions between tenascins and lectican C-type lectin domains: evidence for a crosslinking role for tenascins. Structure 12:1495–1506
Luo JL, Maeda S, Hsu LC, Yagita H, Karin M (2004) Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 6:297–305
Luster AD, Alon R, von Andrian UH (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 6:1182–1190
Mackie EJ, Halfter W, Liverani D (1988) Induction of tenascin in healing wounds. J Cell Biol. 107:2757–2767
Maier S, Lutz R, Gelman L, Sarasa-Renedo A, Schenk S, Grashoff C, Chiquet M (2008) Tenascin-C induction by cyclic strain requires integrin-linked kinase. Biochim Biophys Acta 1783:1150–1162
Marlind J, Kaspar M, Trachsel E, Sommavilla R, Hindle S, Bacci C, Giovannoni L, Neri D (2008) Antibody-mediated delivery of interleukin-2 to the stroma of breast cancer strongly enhances the potency of chemotherapy. Clin Cancer Res. 14:6515–6524
Martin D, Brown-Luedi M, Chiquet-Ehrismann R (2003) Tenascin-C signaling through induction of 14-3-3 tau. J Cell Biol. 160:171–175
Marton LS, Gulcher JR, Stefansson K (1989) Binding of hexabrachions to heparin and DNA. J Biol Chem. 264:13145–13149
Marx J (2008) Cancer biology. All in the stroma: cancer’s Cosa Nostra. Science 320:38–41
Matsuda A, Hirota T, Akahoshi M, Shimizu M, Tamari M, Miyatake A, Takahashi A, Nakashima K, Takahashi N, Obara K, Yuyama N, Doi S, Kamogawa Y, Enomoto T, Ohshima K, Tsunoda T, Miyatake S, Fujita K, Kusakabe M, Izuhara K, Nakamura Y, Hopkin J, Shirakawa T (2005) Coding SNP in tenascin-C Fn-III-D domain associates with adult asthma. Hum Mol Genet 14:2779–2786
Matsuda A, Yoshiki A, Tagawa Y, Matsuda H, Kusakabe M (1999) Corneal wound healing in tenascin knockout mouse. Invest Ophthalmol Vis Sci 40:1071–1080
McClain SA, Simon M, Jones E, Nandi A, Gailit JO, Tonnesen MG, Newman D, Clark RA (1996) Mesenchymal cell activation is the rate-limiting step of granulation tissue induction. Am J Pathol 149:1257–1270
McMahon B, Kwaan HC (2008) The plasminogen activator system and cancer. Pathophysiol Haemost Thromb. 36:184–194
Mercado ML, Nur-e-Kamal A, Liu HY, Gross SR, Movahed R, Meiners S (2004) Neurite outgrowth by the alternatively spliced region of human tenascin-C is mediated by neuronal alpha7beta1 integrin. J Neurosci. 24:238–247
Michele M, Faissner A (2009) Tenascin-C stimulates contactin-dependent neurite outgrowth via activation of phospholipase C. Mol Cell Neurosci 41:397–408
Midwood KS, Schwarzbauer JE (2002) Tenascin-C modulates matrix contraction via focal adhesion kinase- and Rho-mediated signaling pathways. Mol Biol Cell 13:3601–3613
Midwood KS, Valenick LV, Hsia HC, Schwarzbauer JE (2004a) Coregulation of fibronectin signaling and matrix contraction by tenascin-C and syndecan-4. Mol Biol Cell 15:5670–5677
Midwood KS, Williams LV, Schwarzbauer JE (2004b) Tissue repair and the dynamics of the extracellular matrix. Int J Biochem Cell Biol 36:1031–1037
Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F, Foxwell B (2009) Tenascin-C is an endogenous activator of toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 15:774–780
Mighell AJ, Thompson J, Hume WJ, Markham AF, Robinson PA (1997) Human tenascin-C: identification of a novel type III repeat in oral cancer and of novel splice variants in normal, malignant and reactive oral mucosae. Int J Cancer 72:236–240
Milting H, Ellinghaus P, Seewald M, Cakar H, Bohms B, Kassner A, Korfer R, Klein M, Krahn T, Kruska L, El Banayosy A, Kramer F (2008) Plasma biomarkers of myocardial fibrosis and remodeling in terminal heart failure patients supported by mechanical circulatory support devices. J Heart Lung Transplant. 27:589–596
Moritz S, Lehmann S, Faissner A, von Holst A (2008) An induction gene trap screen in neural stem cells reveals an instructive function of the niche and identifies the splicing regulator sam68 as a tenascin-C-regulated target gene. Stem Cells. 26:2321–2331
Murphy-Ullrich JE, Lightner VA, Aukhil I, Yan YZ, Erickson HP, Hook M (1991) Focal adhesion integrity is downregulated by the alternatively spliced domain of human tenascin. J Cell Biol. 115:1127–1136
Nakahara H, Gabazza EC, Fujimoto H, Nishii Y, D’Alessandro-Gabazza CN, Bruno NE, Takagi T, Hayashi T, Maruyama J, Maruyama K, Imanaka-Yoshida K, Suzuki K, Yoshida T, Adachi Y, Taguchi O (2006) Deficiency of tenascin C attenuates allergen-induced bronchial asthma in the mouse. Eur J Immunol 36:3334–3345
Nakao N, Hiraiwa N, Yoshiki A, Ike F, Kusakabe M (1998) Tenascin-C promotes healing of Habu-snake venom-induced glomerulonephritis: studies in knockout congenic mice and in culture. Am J Pathol 152:1237–1245
Nomi M, Miyake H, Sugita Y, Fujisawa M, Soker S (2006) Role of growth factors and endothelial cells in therapeutic angiogenesis and tissue engineering. Curr Stem Cell Res Ther. 1:333–343
Orend G (2005) Potential oncogenic action of tenascin-C in tumorigenesis. Int J Biochem Cell Biol 37:1066–1083
Orend G, Chiquet-Ehrismann R (2000) Adhesion modulation by antiadhesive molecules of the extracellular matrix. Exp Cell Res 261:104–110
Orend G, Chiquet-Ehrismann R (2006) Tenascin-C induced signaling in cancer. Cancer Lett 244:143–163
Orend G, Huang W, Olayioye MA, Hynes NE, Chiquet-Ehrismann R (2003) Tenascin-C blocks cell-cycle progression of anchorage-dependent fibroblasts on fibronectin through inhibition of syndecan-4. Oncogene. 22:3917–3926
Paallysaho T, Tervo K, Kivela T, Virtanen I, Tarkkanen A, Tervo T (1993) Cellular fibronectin and tenascin in an orbital nylon prosthesis removed because of infection caused by Staphylococcus aureus. Graefes Arch Clin Exp Ophthalmol 231:61–65
Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 15:220–231
Paik DC, Fu C, Bhattacharya J, Tilson MD (2004) Ongoing angiogenesis in blood vessels of the abdominal aortic aneurysm. Exp Mol Med. 36:524–533
Paivaniemi OE, Maasilta PK, Alho HS, Vainikka TL, Salminen US (2008) Epithelial tenascin predicts obliterative airway disease. J Heart Lung Transplant. 27:400–407
Parekh K, Ramachandran S, Cooper J, Bigner D, Patterson A, Mohanakumar T (2005) Tenascin-C, over expressed in lung cancer down regulates effector functions of tumor infiltrating lymphocytes. Lung Cancer. 47:17–29
Pas J, Wyszko E, Rolle K, Rychlewski L, Nowak S, Zukiel R, Barciszewski J (2006) Analysis of structure and function of tenascin-C. Int J Biochem Cell Biol 38:1594–1602
Pendas-Franco N, Aguilera O, Pereira F, Gonzalez-Sancho JM, Munoz A (2008) Vitamin D and Wnt/beta-catenin pathway in colon cancer: role and regulation of DICKKOPF genes. Anticancer Res 28:2613–2623
Phillips GR, Krushel LA, Crossin KL (1998) Domains of tenascin involved in glioma migration. J Cell Sci. 111(Pt 8):1095–1104
Pidgeon GP, Harmey JH, Kay E, Da Costa M, Redmond HP, Bouchier-Hayes DJ (1999) The role of endotoxin/lipopolysaccharide in surgically induced tumour growth in a murine model of metastatic disease. Br J Cancer 81:1311–1317
Pollard JW (2008) Macrophages define the invasive microenvironment in breast cancer. J Leukoc Biol. 84:623–630
Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 9:265–273
Prieto AL, Andersson-Fisone C, Crossin KL (1992) Characterization of multiple adhesive and counteradhesive domains in the extracellular matrix protein cytotactin. J Cell Biol. 119:663–678
Prieto AL, Edelman GM, Crossin KL (1993) Multiple integrins mediate cell attachment to cytotactin/tenascin. Proc Natl Acad Sci U S A. 90:10154–10158
Probstmeier R, Pesheva P (1999) Tenascin-C inhibits beta1 integrin-dependent cell adhesion and neurite outgrowth on fibronectin by a disialoganglioside-mediated signaling mechanism. Glycobiology. 9:101–114
Puente Navazo MD, Valmori D, Ruegg C (2001) The alternatively spliced domain TnFnIII A1A2 of the extracellular matrix protein tenascin-C suppresses activation-induced T lymphocyte proliferation and cytokine production. J Immunol. 167:6431–6440
Ramos DM, Chen B, Regezi J, Zardi L, Pytela R (1998) Tenascin-C matrix assembly in oral squamous cell carcinoma. Int J Cancer 75:680–687
Reardon DA, Zalutsky MR, Akabani G, Coleman RE, Friedman AH, Herndon JE 2nd, McLendon RE, Pegram CN, Quinn JA, Rich JN, Vredenburgh JJ, Desjardins A, Guruangan S, Boulton S, Raynor RH, Dowell JM, Wong TZ, Zhao XG, Friedman HS, Bigner DD (2008) A pilot study: 131I-antitenascin monoclonal antibody 81c6 to deliver a 44-Gy resection cavity boost. Neuro Oncol. 10:182–189
Richter P, Tost M, Franz M, Altendorf-Hofmann A, Junker K, Borsi L, Neri D, Kosmehl H, Wunderlich H, Berndt A (2009) B and C domain containing tenascin-C: urinary markers for invasiveness of urothelial carcinoma of the urinary bladder? J Cancer Res Clin Oncol 135:1351–1358
Riedl S, Tandara A, Reinshagen M, Hinz U, Faissner A, Bodenmuller H, Buhr HJ, Herfarth C, Moller P (2001) Serum tenascin-C is an indicator of inflammatory bowel disease activity. Int J Colorectal Dis 16:285–291
Riley GP, Harrall RL, Cawston TE, Hazleman BL, Mackie EJ (1996) Tenascin-C and human tendon degeneration. Am J Pathol 149:933–943
Riou JF, Shi DL, Chiquet M, Boucaut JC (1990) Exogenous tenascin inhibits mesodermal cell migration during amphibian gastrulation. Dev Biol 137:305–317
Ruegg CR, Chiquet-Ehrismann R, Alkan SS (1989) Tenascin, an extracellular matrix protein, exerts immunomodulatory activities. Proc Natl Acad Sci U S A. 86:7437–7441
Ruiz C, Huang W, Hegi ME, Lange K, Hamou MF, Fluri E, Oakeley EJ, Chiquet-Ehrismann R, Orend G (2004) Growth promoting signaling by tenascin-C. Cancer Res 64:7377–7385
Rybak JN, Roesli C, Kaspar M, Villa A, Neri D (2007) The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res 67:10948–10957
Sahlberg C, Aukhil I, Thesleff I (2001) Tenascin-C in developing mouse teeth: expression of splice variants and stimulation by TGFbeta and FGF. Eur J Oral Sci 109:114–124
Saito Y, Imazeki H, Miura S, Yoshimura T, Okutsu H, Harada Y, Ohwaki T, Nagao O, Kamiya S, Hayashi R, Kodama H, Handa H, Yoshida T, Fukai F (2007) A peptide derived from tenascin-C induces beta1 integrin activation through syndecan-4. J Biol Chem. 282:34929–34937
Sawada Y, Onoda K, Imanaka-Yoshida K, Maruyama J, Yamamoto K, Yoshida T, Shimpo H (2007) Tenascin-C synthesized in both donor grafts and recipients accelerates artery graft stenosis. Cardiovasc Res 74:366–376
Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Gotte M, Malle E, Schaefer RM, Grone HJ (2005) The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 115:2223–2233
Schafer M, Werner S (2008) Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 9:628–638
Schalkwijk J, Steijlen PM, van Vlijmen-Willems IM, Oosterling B, Mackie EJ, Verstraeten AA (1991a) Tenascin expression in human dermis is related to epidermal proliferation. Am J Pathol 139:1143–1150
Schalkwijk J, Van Vlijmen I, Oosterling B, Perret C, Koopman R, Van den Born J, Mackie EJ (1991b) Tenascin expression in hyperproliferative skin diseases. Br J Dermatol 124:13–20
Schenk S, Bruckner-Tuderman L, Chiquet-Ehrismann R (1995) Dermo-epidermal separation is associated with induced tenascin expression in human skin. Br J Dermatol 133:13–22
Schenk S, Chiquet-Ehrismann R, Battegay EJ (1999) The fibrinogen globe of tenascin-C promotes basic fibroblast growth factor-induced endothelial cell elongation. Mol Biol Cell 10:2933–2943
Schmidinger G, Hanselmayer G, Pieh S, Lackner B, Kaminski S, Ruhswurm I, Skorpik C (2003) Effect of tenascin and fibronectin on the migration of human corneal fibroblasts. J Cataract Refract Surg. 29:354–360
Schnapp LM, Hatch N, Ramos DM, Klimanskaya IV, Sheppard D, Pytela R (1995) The human integrin alpha 8 beta 1 functions as a receptor for tenascin, fibronectin, and vitronectin. J Biol Chem. 270:23196–23202
Seiffert M, Beck SC, Schermutzki F, Muller CA, Erickson HP, Klein G (1998) Mitogenic and adhesive effects of tenascin-C on human hematopoietic cells are mediated by various functional domains. Matrix Biol 17:47–63
Seite S, Colige A, Piquemal-Vivenot P, Montastier C, Fourtanier A, Lapiere C, Nusgens B (2000) A full-UV spectrum absorbing daily use cream protects human skin against biological changes occurring in photoaging. Photodermatol Photoimmunol Photomed 16:147–155
Seyger MM, van Pelt JP, van den Born J, Latijnhouwers MA, de Jong EM (1997) Epicutaneous application of leukotriene B4 induces patterns of tenascin and a heparan sulfate proteoglycan epitope that are typical for psoriatic lesions. Arch Dermatol Res 289:331–336
Shacter E, Weitzman SA (2002) Chronic inflammation and cancer. Oncology (Williston Park) 16:217–226 229; discussion 230–2
Silacci M, Brack SS, Spath N, Buck A, Hillinger S, Arni S, Weder W, Zardi L, Neri D (2006) Human monoclonal antibodies to domain C of tenascin-C selectively target solid tumors in vivo. Protein Eng Des Sel. 19:471–478
Siri A, Knauper V, Veirana N, Caocci F, Murphy G, Zardi L (1995) Different susceptibility of small and large human tenascin-C isoforms to degradation by matrix metalloproteinases. J Biol Chem. 270:8650–8654
Sivasankaran B, Degen M, Ghaffari A, Hegi ME, Hamou MF, Ionescu MC, Zweifel C, Tolnay M, Wasner M, Mergenthaler S, Miserez AR, Kiss R, Lino MM, Merlo A, Chiquet-Ehrismann R, Boulay JL (2009) Tenascin-C is a novel RBPJkappa-induced target gene for Notch signaling in gliomas. Cancer Res 69:458–465
Smyth MJ, Cretney E, Kershaw MH, Hayakawa Y (2004) Cytokines in cancer immunity and immunotherapy. Immunol Rev 202:275–293
Spring J, Beck K, Chiquet-Ehrismann R (1989) Two contrary functions of tenascin: dissection of the active sites by recombinant tenascin fragments. Cell 59:325–334
Sriramarao P, Bourdon MA (1993) A novel tenascin type III repeat is part of a complex of tenascin mRNA alternative splices. Nucleic Acids Res 21:163–168
Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA, Johnson RS (2008) Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 456:814–818
Swindle CS, Tran KT, Johnson TD, Banerjee P, Mayes AM, Griffith L, Wells A (2001) Epidermal growth factor (EGF)-like repeats of human tenascin-C as ligands for EGF receptor. J Cell Biol. 154:459–468
Talts JF, Wirl G, Dictor M, Muller WJ, Fassler R (1999) Tenascin-C modulates tumor stroma and monocyte/macrophage recruitment but not tumor growth or metastasis in a mouse strain with spontaneous mammary cancer. J Cell Sci. 112:1855–1864
Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T (2005) Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol 167:71–80
Tanaka K, Hiraiwa N, Hashimoto H, Yamazaki Y, Kusakabe M (2004) Tenascin-C regulates angiogenesis in tumor through the regulation of vascular endothelial growth factor expression. Int J Cancer 108:31–40
Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massague J (2008) Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451:147–152
Taylor HC, Lightner VA, Beyer WF Jr, McCaslin D, Briscoe G, Erickson HP (1989) Biochemical and structural studies of tenascin/hexabrachion proteins. J Cell Biochem. 41:71–90
Tiitta O, Wahlstrom T, Paavonen J, Linnala A, Sharma S, Gould VE, Virtanen I (1992) Enhanced tenascin expression in cervical and vulvar koilocytotic lesions. Am J Pathol 141:907–913
Tran KT, Griffith L, Wells A (2004) Extracellular matrix signaling through growth factor receptors during wound healing. Wound Repair Regen. 12:262–268
Tran KT, Lamb P, Deng JS (2005) Matrikines and matricryptins: Implications for cutaneous cancers and skin repair. J Dermatol Sci. 40:11–20
Trebaul A, Chan EK, Midwood KS (2007) Regulation of fibroblast migration by tenascin-C. Biochem Soc Trans 35:695–697
Tremble P, Chiquet-Ehrismann R, Werb Z (1994) The extracellular matrix ligands fibronectin and tenascin collaborate in regulating collagenase gene expression in fibroblasts. Mol Biol Cell 5:439–453
Tsukada B, Terasaki F, Shimomura H, Otsuka K, Otsuka K, Katashima T, Fujita S, Imanaka-Yoshida K, Yoshida T, Hiroe M, Kitaura Y (2009). High prevalence of chronic myocarditis in dilated cardiomyopathy referred for left ventriculoplasty: expression of tenascin C as a possible marker for inflammation. Hum Pathol 40:1015–1022
Tucker RP (1998) Quantitative in situ localization of tenascin-C alternatively spliced transcripts in the avian optic tectum. Mol Vis. 4:18
Tucker RP (2001) Abnormal neural crest cell migration after the in vivo knockdown of tenascin-C expression with morpholino antisense oligonucleotides. Dev Dyn 222:115–119
Tucker RP, Chiquet-Ehrismann R (2009) The regulation of tenascin expression by tissue microenvironments. Biochim Biophys Acta 1793:888–892
van der Vleuten CJ, Snijders CG, de Jong EM, van de Kerkhof PC (1996) Effects of calcipotriol and clobetasol-17-propionate on UVB-irradiated human skin: an immunohistochemical study. Skin Pharmacol 9:355–365
Van Eyken P, Sciot R, Desmet VJ (1990) Expression of the novel extracellular matrix component tenascin in normal and diseased human liver. An immunohistochemical study. J Hepatol. 11:43–52
Villani ME, Morgun B, Brunetti P, Marusic C, Lombardi R, Pisoni I, Bacci C, Desiderio A, Benvenuto E, Donini M (2009) Plant pharming of a full-sized, tumour-targeting antibody using different expression strategies. Plant Biotechnol J 7:59–72
Vollmer G (1997) Biologic and oncologic implications of tenascin-C/hexabrachion proteins. Crit Rev Oncol Hematol 25:187–210
von Lukowicz T, Silacci M, Wyss MT, Trachsel E, Lohmann C, Buck A, Luscher TF, Neri D, Matter CM (2007) Human antibody against C domain of tenascin-C visualizes murine atherosclerotic plaques ex vivo. J Nucl Med. 48:582–587
Vosseler S, Mirancea N, Bohlen P, Mueller MM, Fusenig NE (2005) Angiogenesis inhibition by vascular endothelial growth factor receptor-2 blockade reduces stromal matrix metalloproteinase expression, normalizes stromal tissue, and reverts epithelial tumor phenotype in surface heterotransplants. Cancer Res 65:1294–1305
Wallner K, Li C, Shah PK, Wu KJ, Schwartz SM, Sharifi BG (2004) EGF-Like domain of tenascin-C is proapoptotic for cultured smooth muscle cells. Arterioscler Thromb Vasc Biol 24:1416–1421
Watanabe K, Takahashi H, Habu Y, Kamiya-Kubushiro N, Kamiya S, Nakamura H, Yajima H, Ishii T, Katayama T, Miyazaki K, Fukai F (2000) Interaction with heparin and matrix metalloproteinase 2 cleavage expose a cryptic anti-adhesive site of fibronectin. Biochemistry. 39:7138–7144
Wenk MB, Midwood KS, Schwarzbauer JE (2000) Tenascin-C suppresses Rho activation [see comments]. J Cell Biol. 150:913–920
Whitby DJ, Longaker MT, Harrison MR, Adzick NS, Ferguson MW (1991) Rapid epithelialisation of fetal wounds is associated with the early deposition of tenascin. J Cell Sci. 99(Pt 3):583–586
Williams SA, Schwarzbauer JE (2009) A shared mechanism of adhesion modulation for tenascin-C and fibulin-1. Mol Biol Cell 20:1141–1149
Woodside DG, Wooten DK, McIntyre BW (1998) Adenosine diphosphate (ADP)-ribosylation of the guanosine triphosphatase (GTPase) rho in resting peripheral blood human T lymphocytes results in pseudopodial extension and the inhibition of T cell activation. J Exp Med. 188:1211–1221
Yamada S, Ichida T, Matsuda Y, Miyazaki Y, Hatano T, Hata K, Asakura H, Hirota N, Geerts A, Wisse E (1992) Tenascin expression in human chronic liver disease and in hepatocellular carcinoma. Liver. 12:10–16
Yamamoto K, Onoda K, Sawada Y, Fujinaga K, Imanaka-Yoshida K, Shimpo H, Yoshida T, Yada I (2005) Tenascin-C is an essential factor for neointimal hyperplasia after aortotomy in mice. Cardiovasc Res 65:737–742
Yamamoto K, Onoda K, Sawada Y, Fujinaga K, Imanaka-Yoshida K, Yoshida T, Shimpo H (2007) Locally applied cilostazol suppresses neointimal hyperplasia and medial thickening in a vein graft model. Ann Thorac Cardiovasc Surg. 13:322–330
Yokosaki Y, Monis H, Chen J, Sheppard D (1996) Differential effects of the integrins alpha9beta1, alphavbeta3, and alphavbeta6 on cell proliferative responses to tenascin Roles of the beta subunit extracellular and cytoplasmic domains. J Biol Chem. 271:24144–24150
Yokoyama K, Erickson HP, Ikeda Y, Takada Y (2000) Identification of amino acid sequences in fibrinogen gamma -chain and tenascin C C-terminal domains critical for binding to integrin alpha vbeta 3. J Biol Chem. 275:16891–16898
Zagzag D, Capo V (2002) Angiogenesis in the central nervous system: a role for vascular endothelial growth factor/vascular permeability factor and tenascin-C. Common molecular effectors in cerebral neoplastic and non-neoplastic “angiogenic diseases”. Histol Histopathol 17:301–321
Zhao Y, Young SL (1995) TGF-beta regulates expression of tenascin alternative-splicing isoforms in fetal rat lung. Am J Physiol 268:L173–L180
Zukiel R, Nowak S, Wyszko E, Rolle K, Gawronska I, Barciszewska MZ, Barciszewski J (2006) Suppression of human brain tumor with interference RNA specific for tenascin-C. Cancer Biol Ther. 5:1002–1007
Acknowledgement
We thank Isabelle Gasser, who is supported by a grant from the Region Alsace, France, for providing a picture of tenascin-C expression in human colorectal carcinoma tissue. Table 1 is based on work by Pia Schulz zur Wiesch that was generated during the course of her master thesis at the University of Basel, Switzerland. G.O. is generously supported by Inserm, an INCa/Inserm grant and by a Contrat d`interface with the hospital Hautepierre. K.M. is supported by the Arthritis Research Campaign and the Medical Research Council, UK.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Midwood, K.S., Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 3, 287–310 (2009). https://doi.org/10.1007/s12079-009-0075-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-009-0075-1