
Abstract
Caveolin-1 is a palmitoylated protein involved in the formation of plasma membrane subdomains termed caveolae, intracellular cholesterol transport, and assembly and regulation of signaling molecules in caveolae. Caveolin-1 interacts via a consensus binding motif with several signaling proteins, including H-Ras. Ras oncogene products function as molecular switches in several signal transduction pathways regulating cell growth and differentiation. Post-translational modifications, including palmitoylation, are critical for the membrane targeting and function of H-Ras. Subcellular localization regulates the signaling pathways engaged by H-Ras activation. We show here that H-Ras is localized at the plasma membrane in caveolin-1-expressing cells but not in caveolin-1-deficient cells. Since palmitoylation is required for trafficking of H-Ras from the endomembrane system to the plasma membrane, we tested whether the altered localization of H-Ras in caveolin-1-null cells is due to decreased H-Ras palmitoylation. Although the palmitoylation profiles of cultured embryo fibroblasts isolated from wild type and caveolin-1 gene-disrupted mice differed, suggesting that caveolin-1, or caveolae, play a role in the palmitate incorporation of a subset of palmitoylated proteins, the palmitoylation of H-Ras was not decreased in caveolin-1-null cells. We conclude that the altered localization of H-Ras in caveolin-1-deficient cells is palmitoylation-independent. This article shows two important new mechanisms by which loss of caveolin-1 expression may perturb intracellular signaling, namely the mislocalization of signaling proteins and alterations in protein palmitoylation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Caveolae are ~70nm invaginations of the plasma membrane enriched in glycosphingolipids, cholesterol and lipid-anchored membrane proteins. These specialized subdomains serve signaling and trafficking functions (Anderson et al. 1992; Schlegel et al. 2000; Mineo and Anderson 2001). The biogenesis and functions of caveolae are believed to require caveolin-1, the major protein component of caveolae (Fra et al. 1995; Drab et al. 2001; Yu et al. 2006). Caveolin-1 is an integral membrane protein which participates in multiple protein/protein interactions. Via a consensus binding motif (the scaffolding domain, amino acids 82–101), caveolin-1 interacts with (and generally inhibits) signaling proteins such as endothelial nitric oxide synthase, heterotrimeric G-protein α-subunits, protein tyrosine kinases, and H-Ras (Li et al. 1995; Li et al. 1996; Garcia-Cardena et al. 1997).
Ras family GTPases function as molecular switches regulating cell growth and differentiation. In order for Ras to signal, it must associate with the cytoplasmic leaflet of cellular membranes, including the plasma membrane, but also endosomes, the Golgi apparatus and the endoplasmic reticulum (ER; Chiu et al. 2002; Bivona and Philips 2003). Three Ras isoforms, H-, K- and N-Ras are ubiquitously expressed in mammalian cells and signal differently despite their high degree of homology. It is currently believed that this specificity derives from compartmentalization in different cellular domains (Prior et al. 2001). All Ras proteins are synthesized on soluble ribosomes, and undergo post translational modifications initiated by a CAAX motif present at the C-terminus of all Ras proteins: farnesylation by a cytosolic farnesyltransferase is followed by proteolytic removal of the AAX amino acids and methylesterification by enzymes located in the ER. CAAX processing is required for membrane association (Hancock et al. 1990). In addition, N-Ras and H-Ras are palmitoylated in the early secretory pathway on one or two cysteines, respectively (Chiu et al. 2002; Smotrys and Linder 2004).
Palmitoylation is the addition of palmitate (C16:0) to cysteine residues through thioester linkage. Protein palmitoylation and depalmitoylation reactions are considered to be primarily enzymatic and catalyzed by membrane-bound palmitoyl acyltransferases (Dunphy et al. 1996; Das et al. 1997) and by cytoplasmic and lysosomal forms of palmitoyl thioesterases (Camp and Hofmann 1993; Duncan and Gilman 1998; Yeh et al. 1999), respectively. Palmitoyl acyltransferase activities have been purified from tissues and cell lines, but their molecular identity has only recently been unveiled. In yeast, Erf2/Erf4 complex stimulates palmitoylation of Ras2 (similar to mammalian H-Ras and N-Ras) (Lobo et al. 2002). In humans, H- and N-Ras are palmitoylated by a DHHC9/GCP16 complex localized in the Golgi complex (Swarthout et al. 2005).
Since post-translational modification of Ras proteins is critical for their function, the enzymes involved in these steps are considered potential targets for anti cancer therapy. Palmitoylation is necessary for the localization of N-Ras and H-Ras at the plasma membrane (Hancock et al. 1990). Palmitoylation-deficient N- and H-Ras mutants accumulate predominantly in the ER and to some extent in the Golgi (Choy et al. 1999; Apolloni et al. 2000). ER localization of H-Ras is also seen when cells are treated with a palmitoylation inhibitor (Michaelson et al. 2002). Recent studies have shown that Ras is actively signaling not only from the plasma membrane but also on intracellular membranes, with different signaling pathways engaged from different membranes (Chiu et al. 2002). Insight into the cellular factors that regulate Ras palmitoylation is therefore essential to our understanding of the regulation of Ras function.
The relationship between caveolin-1 and Ras is complex. Activation of the Ras-MAP kinase pathway transcriptionally down-regulates caveolin-1 promoter activity (Engelman et al. 1999) and cellular transformation by constitutively active, oncogenic H-Ras (G12V) leads to reduced cellular levels of caveolin-1 and absence of caveolae (Koleske et al. 1995). Reexpression of caveolin-1 in H-Ras (G12V)-transformed NIH 3T3 cells abrogates their anchorage-independent growth in soft agar (Engelman et al. 1997). On the other hand, a fraction of H-Ras is localized in caveolae (Song et al. 1996; Mineo et al. 1996; Hancock 2003) and caveolae disruption in mammalian cells blocks H-Ras-mediated Raf activation (Roy et al. 1999). In this work, we determined whether H-Ras localization was altered in the absence of caveolin-1 expression: we compared the subcellular distribution of GFP-H-Ras in fibroblasts isolated from wild type or caveolin-1-null mice. The plasma membrane localization of GFP-H-Ras was lost in caveolin-1-deficient cells. This was similar to the localization of palmitoylation-deficient H-Ras in wild type cells. We therefore determined whether the absence of caveolin-1 influenced the palmitoylation of H-Ras and other proteins. The palmitoylation profiles of cultured embryo fibroblasts isolated from wild type and caveolin-1 gene-disrupted mice differed, suggesting that caveolin-1, or caveolae, play a role in the palmitate incorporation of a subset of palmitoylated proteins. However the palmitoylation of H-Ras was not decreased in caveolin-1-null cells, ruling out the possibility that the altered localization of H-Ras in caveolin-1-deficient cells is palmitoylation-dependent.
Materials and methods
Materials
[9,10–3H]palmitic acid (35Ci/mmol) was from Perkin Elmer Life Sciences (Boston, MA, USA). Agarose-conjugated rat monoclonal anti H-Ras antibody and its blocking peptide were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Rabbit anti-caveolin-1 was obtained from BD Biosciences (San Jose, CA). Biotinylated goat anti-rabbit antibody, Texas red-avidin, and Vectashield mounting medium were from Vector laboratories (Burlingame, CA). Fatty acid-free bovine serum albumin (BSA) was from Roche (Indianapolis, IN, USA). Geneticin was from Gibco (Carlsbad, CA, USA). Other assay reagents were from Sigma (St. Louis, MO, USA).
Cell culture
Mouse embryo fibroblasts (MEF) isolated from caveolin-1+/+ and caveolin-1−/− animals were a generous gift from Dr. Philippe Frank (Thomas Jefferson University, Philadelphia, PA, USA). Fibroblasts isolated from adult caveolin-1+/+, caveolin-1+/− or caveolin-1−/− mice were a generous gift from Dr. Richard G.W. Anderson (University of Texas Southwestern Medical Center, Dallas, TX, USA). Cells were maintained in DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% (v/v) fetal bovine serum, 100IU/ml penicillin, 100μg/ml streptomycin and 2.5μg/ml amphotericin B in a humidified air/CO2 (19:1) atmosphere.
Immunoblot analyses
Proteins were separated by SDS-PAGE and transferred to nitrocellulose. Immunoblot analysis was performed using rabbit polyclonal or mouse monoclonal primary antibodies and peroxidase-conjugated secondary antibody. Blots were developed by chemiluminescense using ECL and hyperfilm-ECL (Amersham, Arlington Heights, IL, USA).
Live cell confocal fluorescence microscopy
The plasmid encoding N-terminally tagged GFP-H-Ras (Choy et al. 1999) was a generous gift from Dr Mark Philips (New York University School of Medicine, New York, NY, USA). Wild type and caveolin-1-null MEF were transfected using superfect (Qiagen) according to the manufacturer’s instructions, selected in geneticin-containing medium, and pooled after two weeks of selection. Live cells were observed using a Leica TCS-SP2 AOBS confocal microscope (Leica Microsystems, GmbH, Wetzlar, Germany) with an HC PL Apo 63X, 1.4 NA, oil immersion objective lens on an inverted Leica DMIRB microscope equipped for confocal fluorescence imaging with a heated stage and environmental CO2 chamber.
Analysis of plasma membrane caveolin expression
Analysis of plasma membrane caveolin expression in MEF was performed using customized Image-Pro Plus macros (v6.2, Media Cybernetics, Silver Spring, MD, USA). Briefly, grayscale images of transfected cells were processed using median, spectral (high-pass), and morphological “closing” filters to enhance cell boundaries. Within the resultant images, regions of interests (ROIs) were created around cells using a semi-automated tracing tool. Each ROI was saved and loaded sequentially into a blank image, filled with pixels at a gray-level intensity of 255, and “eroded” using an 11 × 11 kernel (four passes). The subtraction of the original and eroded ROI’s produced a binary plasma membrane mask with a uniform thickness of 1.35μm. This membrane mask and the original ROI (cell mask) were applied to the unprocessed GFP images to determine overall fluorescence intensity and total fluorescent area (100–255 gray-level range) in the plasma membrane and entire cell, respectively.
Detection of caveolin-1 by immunofluorescence microscopy
Cells were transiently transfected with wild type or palmitoylation-deficient GFP-H-Ras (GFP-H-rasC181S,C184S; Choy et al. 1999). After recovery, cells were washed with phosphate-buffered saline (PBS), fixed with 3.7% formaldehyde in PBS for 20min, and then washed three times at room temperature. The cells were permeabilized with 0.1% Triton X-100 for 10min and washed. To reduce non-specific binding of antibody, cells were pre-incubated with 3% goat serum (Gibco, Grand Island, NY, USA) in PBS for 20min. Cells were then incubated with primary antibody (1μg/ml) in a solution containing 1.5% goat serum in PBS for 60min. After three washes with PBS and a 5-min incubation with 3% non-immune goat serum, the cells were incubated with biotinylated goat anti-rabbit antibody 1:1,000 in 1.5% goat serum for 45min followed by Texas red-avidin for 10min. After washing, cells were placed in Vectashield mounting medium. No reactivity was observed when the primary antibody was substituted with non-immune rabbit IgG (1μg/ml). Cells were visualized using an HCX Plan Apo 63X, 1.4 NA, oil immersion objective lens on a Leica TCS-SP2 AOBS confocal microscope (Leica Microsystems, GmbH, Wetzlar, Germany).
Determination of palmitoylation profiles by metabolic labeling with [3H]palmitate
Confluent cells in 60mm dishes were washed with serum-free DMEM and incubated for 2h in the same medium. Cells were labeled for 30min by incubation with 2ml 250μCi/ml [3H]palmitic acid in serum-free medium containing 3.5mg/ml fatty acid-free BSA.
Cells were washed twice in phosphate-buffered saline and lysed for 90min at 4°C in 0.4ml of immunoprecipitation buffer containing 50mM Hepes, pH7.4, 150mM NaCl, 1mM EDTA, 2.5mM MgCl2, 0.5% sodium deoxycholate, 1% (v/v) Nonidet P40, 20μg/ml aprotinin and 5μg/ml leupeptin. The lysate was collected by scraping and cleared by centrifugation at 16,000×g for 3min. The lysates were added with Laemmli buffer and separated by SDS-PAGE [4–15% or 11% (w/v)].
For two dimensional gel electrophoresis, cells were lysed in buffer (25mM Tris, pH7.5, 2.5mM MgCl2, 0.5% SDS) added with 100μg/ml DNase I and 100μg/ml RNase A for 15min. Samples were heated at 96°C for 5min. One mg of total protein was precipitated in acetone (80%, v/v) overnight. The precipitate was resuspended in a rehydration buffer (7M urea, 2M thiourea, 1% dithiothreitol, 1% CHAPS, 1% Triton X-100, and 1% ampholytes (Bio-Rad Bio-Lyte 3/10). Rehydration of IPG strips and isoelectric focusing was performed using a Bio-Rad Protean IEF Cell. The focused IPG strips were equilibrated in SDS under reducing [6M urea, 2% (w/v) SDS, 20% (v/v) glycerol 100mM dithiothreitol] and then alkylating [6M urea, 2% (w/v) SDS, 20% (v/v) glycerol, 100mM iodoacetamide) conditions for 15min. The focused IPG strips were subjected to SDS-PAGE [12% (w/v)]. Gels were then processed for fluorography.
Immunoprecipitation of palmitoylated proteins
Cell lysates in immunoprecipitation buffer were added with SDS (0.1% final concentration). To immunoprecipitate endogenous H-Ras, the mixture was incubated with 5μg agarose-conjugated rat monoclonal anti H-Ras antibody for 18h. Control cell lysates were incubated with 5μg agarose-conjugated rat monoclonal anti H-Ras antibody and 12.5μg/ml competing peptide. For immunoprecipitation of GFP-H-ras, the lysates were incubated with 1μg rabbit polyclonal anti-H-Ras for 18h, then with Protein A-Sepharose for 60min. Control cell lysates were immunoprecipitated with rabbit non-immune IgG. Immunoprecipitated complexes were collected by low-speed centrifugation, washed three times with lysis buffer and then analyzed by SDS/PAGE [11 or 15% (w/v) polyacrylamide]. Incorporation of the radiolabel was detected by fluorography after a 6–12-week exposure.
Results
The subcellular localization of H-Ras is altered in caveolin-1-deficient cells
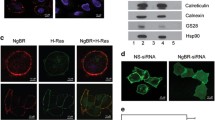
To examine the effect of caveolin-1 expression on the subcellular localization of H-Ras, we made use of a plasmid which contains wild type H-Ras inserted into the pEGFP-C3 vector, and encodes GFP-H-Ras (Choy et al. 1999). Wild type and caveolin-1-null immortalized MEF (Razani et al. 2001; Sotgia et al. 2002) were transfected, selected in geneticin-containing medium, and pooled after 2weeks of selection. The expressed protein was detected by green fluorescence in live transfected cells (Fig. 1). As expected, in wild type MEF the localization of H-Ras included the plasma membrane and a perinuclear compartment (Fig. 1a–c). In contrast, the plasma membrane localization of H-Ras was lost in caveolin-1-null MEF, which exhibited green fluorescence only in the perinuclear region (Fig. 1d–f). Quantitative analysis confirmed the statistically significant decrease of the mean plasma membrane fluorescence intensity (Fig. 1g) as well as the membrane/total cell fluorescence intensity ratio (not shown).

Effect of caveolin-1 deficiency on subcellular localization of H-Ras. Cav-1+/+ (a–c) and Cav-1−/− (d–f) mouse embryo fibroblasts were transfected with a plasmid encoding GFP-H-Ras. Live cells were imaged using confocal fluorescence microscopy and the exogenous protein detected by the green fluorescence of GFP. Bar represents 20 μm in a, b, d and f; 10 μm in c and e. Bar graph shows plasma membrane mean fluorescence intensity (g). Error bars represent SEM. **p < 0.01
The localization of H-Ras in caveolin-1-deficient cells mimics the localization of palmitoylation-deficient H-Ras
Endoplasmic reticulum and Golgi localization of palmitoylation-deficient GFP-H-Ras has previously been shown by Choy et al. who demonstrated that palmitoylation is required for trafficking of endomembrane-associated Ras molecules out of the endomembrane system (Choy et al. 1999). We transfected wild type and caveolin-1-deficient MEF with wild type GFP-H-Ras, and wild type MEF with palmitoylation-deficient GFP-H-Ras. After recovery, cells were fixed and caveolin-1 expression and localization assessed by immunofluorescence confocal microscopy. As seen in live cells, the plasma membrane localization of GFP-H-Ras was lost in caveolin-1-null cells (Fig. 2). Similarly, the palmitoylation-deficient mutant of H-Ras was confined in the perinuclear region of wild type MEF, raising the possibility that the loss of plasma membrane localization of H-Ras in caveolin-1 null cells may be due to a loss of H-Ras palmitoylation. The overlay of caveolin-1 staining and wild type GFP-H-Ras fluorescence further showed that while both proteins partially localize at the plasma membrane, their distribution did not substantially overlap (Fig. 2).

Subcellular localization of caveolin-1 and wild type or palmitoylation-deficient H-Ras. Cav-1+/+ mouse embryo fibroblasts were transfected with a plasmid encoding wild type or palmitoylation-deficient GFP-H-Ras as indicated. Cav-1−/− MEF were transfected with a plasmid encoding wild type GFP-H-Ras. Fixed cells were labeled using rabbit anti-human caveolin-1 as primary antibody followed by biotinylated goat anti-rabbit antibody and Texas red-avidin. GFP-H-Ras was detected by the green fluorescence of GFP. The overlay of GFP-H-Ras and cellular caveolin-1 is shown in the merged image. Plasma membrane localization is indicated by white arrows. Localization in a perinuclear endomembrane compartment is indicated by open arrows. Bar represents 20 μm
Caveolin-1 expression affects the palmitoylation profile of mouse embryo fibroblasts
Since the distribution of wild type H-Ras in caveolin-1-deficient cells resembled that of palmitoylation-deficient H-Ras in caveolin-1-expressing cells, we considered the possibility that absence of caveolin-1 might alter protein palmitoylation.
To determine whether the loss of caveolin-1 expression affected protein palmitoylation, we compared the palmitoylation profile of caveolin-1+/+ and caveolin-1−/− MEF. Cells were subjected to [3H]palmitate labeling, and total cell lysates were analyzed by non reducing SDS-PAGE and fluorography. In caveolin-1-deficient cells, several proteins showed decreased or increased palmitate incorporation (Fig. 3). Cellular uptake of palmitic acid seemed unaffected by the absence of caveolin-1 since for most proteins, palmitoylation was identical in caveolin-1-expressing and-deficient cells. These results suggested that caveolin-1, or caveolae, play a role in the palmitate incorporation of a subset of palmitoylated proteins.

Effect of caveolin-1 deficiency on the palmitoylation profile of mouse embryo fibroblasts. a Cav-1+/+ (WT) and Cav-1−/− (KO) cells were incubated with [3H]palmitic acid for 20 min. Cell lysates were separated by SDS-PAGE and analyzed by fluorography after Coomassie Blue staining. The position of proteins with increased or decreased palmitoylation in KO cells compared to WT cells is indicated by arrows. b The profile of each lane of the Coomassie Blue-stained gel and the fluorograph was analyzed using Image J and plotted with corresponding molecular weight
To achieve a better resolution of the differentially labeled bands, we performed [3H]palmitate labeling of caveolin-1+/+ and caveolin-1−/− cells followed by total cell lysate analysis by two-dimensional gel electrophoresis and fluorography (Fig. 4). The resolution offered by two-dimensional gel electrophoresis allowed the visualization of several proteins between pI 4–7 that were differentially palmitoylated in caveolin-1+/+ and caveolin-1−/− cells (Fig. 4c–d), confirming that the loss of caveolin-1 expression affects the palmitoylation profile of cultured cells.

Two-dimensional analysis of the palmitoylation profile of caveolin-1-expressing and caveolin-1-deficient mouse embryo fibroblasts. Cav-1+/+ and Cav-1−/− cells were incubated with [3H]palmitic acid. Cell lysates were analyzed by two-dimensional gel electrophoresis and fluorography after staining with Coomassie Blue. a Coomassie Blue-stained gel of Cav-1+/+ cell lysate. b Coomassie Blue-stained gel of Cav-1−/− cell lysate. c, d Fluorographs of the gels shown in a and b, respectively. Proteins with increased palmitoylation are shown by white circles on the fluorographs (c, d)
H-Ras palmitoylation is not decreased in caveolin-1-null cells
To determine whether palmitoylation of H-Ras was decreased in the absence of caveolin-1 expression, confluent adult fibroblasts isolated from caveolin-1+/+, caveolin-1+/− and caveolin-1−/− mice were incubated with [3H]palmitate and cell lysates were added with agarose-conjugated rat monoclonal anti H-Ras antibody. Control samples were added with blocking peptide. [3H]palmitate-labeled protein was detected by fluorography. Palmitate incorporation into H-Ras was unaffected by variations in caveolin-1 expression levels (Fig. 5a). To confirm this finding, we similarly labeled MEF isolated from caveolin-1+/+ and caveolin-1−/− mice. [3H]palmitate labeling of the cells followed by immunoprecipitation and fluorography confirmed that palmitate incorporation into H-Ras was not reduced in caveolin-1-null cells compared to wild type cells (not shown).

Effect of caveolin-1 expression on H-Ras palmitoylation. (a) Cav-1+/+, Cav-1+/− and Cav-1−/− adult mouse fibroblasts were incubated with [3H]palmitic acid. Immunoprecipitation of H-Ras from the cell lysates was done using agarose-conjugated rat monoclonal anti H-Ras antibody. Control samples were added with blocking peptide. The immunoprecipitated material was subjected to SDS-PAGE and fluorography. Using duplicate samples, the immunoprecipitated material was subjected to SDS-PAGE and Western Blotting using rabbit polyclonal anti H-Ras antibody. (b) Cav-1+/+ (WT) and Cav-1−/− (KO) mouse embryo fibroblasts were transfected with a plasmid encoding GFP-H-Ras. After recovery, cells were incubated with [3H]palmitic acid and immunoprecipitation of GFP-H-ras from the cell lysates was done using rabbit anti H-Ras antibody (RAS) or non immune rabbit purified IgG (IgG) as control. The immunoprecipitated material was subjected to SDS-PAGE and fluorography. Using duplicate samples, the immunoprecipitated material was subjected to SDS-PAGE and Western Blotting using rabbit polyclonal anti H-Ras antibody
Because the localization of H-Ras-GFP was lost in caveolin-1-null MEF, we considered the possibility that palmitoylation of this exogenous protein was altered but not the palmitoylation of endogenous H-Ras. We therefore transiently transfected caveolin-1-expressing and -deficient cells with H-Ras-GFP. After recovery, the cells were incubated with [3H]palmitate and cell lysates were immunoprecipitated with rabbit anti-H-Ras antibody for 18 h followed by protein A Sepharose for 60 min. This antibody is able to immunoprecipitate the fusion protein, but not endogenous H-Ras and thus is not used up (titrated away) by the endogenous H-Ras. In control samples, the H-Ras antibody was replaced by rabbit non immune IgG. The immunoprecipitates were resolved by 12% SDS-PAGE and analyzed by fluorography (Fig. 5b). The palmitoylation of H-Ras-GFP was not decreased in the absence of caveolin-1. Taken together, our results clearly show that the absence of caveolin-1 expression does not impair palmitate incorporation into H-Ras in cultured cells.
Discussion
Caveolin-1 is known to regulate cellular signaling by multiple mechanisms, including (1) the compartmentalization of signal transduction molecules in caveolae, (2) the regulation of the activity of signaling proteins by interaction with the scaffolding domain of caveolin-1, and (3) the internalization of caveolae (del Pozo et al. 2005; White and Anderson 2005). Our results point to a new role for caveolin-1 in cellular signaling, namely the regulation of protein palmitoylation. This post translational modification modulates the function of target proteins in multiple ways (Resh 2006): palmitoylation controls the conformation, protein-protein and protein–lipid interactions, trafficking and targeting of the modified proteins as well as their association with the plasma membrane and/or membrane subdomains. All of these are important determinants of the activity of signaling proteins.
Several mechanisms by which caveolin-1 expression may influence protein palmitoylation can be proposed. First, the trafficking of a protein may be altered in the absence of caveolin-1 and/or caveolae, preventing it from reaching its palmitoylation location or on the contrary, retaining it at its palmitoylation location. Another possibility is that in the absence of caveolin-1, the palmitoylation/depalmitoylation cycle of a protein might be disrupted, for example due to the loss of proximity with an upstream signaling protein. Finally, the possibility that the function of palmitoylating enzymes, such as the recently identified family of DHHC protein acyl transferases, is altered in the absence of caveolin-1 or of caveolae can be suggested. This could occur through mislocalization, change in conformation or altered autoacylation of these enzymes in caveolin-1-deficient cells.
It is interesting to note that caveolin-1 is itself palmitoylated on three cysteine residues (Cys-133, Cys-143 and Cys-156) located near the C-terminus of the protein (Dietzen et al. 1995). Caveolin-1 palmitoylation is unusual in two aspects: it does not influence caveolin-1 targeting to caveolae (Dietzen et al. 1995), and it is essentially irreversible (Parat and Fox 2001). The function of caveolin-1 palmitoylation is not well understood. It may increase the stability of the oligomers and therefore the scaffold structure of caveolae (Monier et al. 1996). In addition, caveolin-1 palmitoylation may regulate its interaction with other proteins. A palmitoylation-deficient mutant of caveolin-1 exhibits reduced interaction with Gi1α (Galbiati et al. 1999). Moreover, palmitoylation of caveolin-1 on Cys-156 is required for coupling to the c-Src tyrosine kinase (Lee et al. 2001), and palmitoylation of at least two sites on caveolin-1 is required for binding and transport of cholesterol (Uittenbogaard and Smart 2000) and for transport of GPI-anchored proteins to the cell surface (Sotgia et al. 2002).
Our results show that the plasma membrane localization of H-Ras is lost in caveolin-1-null cells. Palmitoylated proteins such as Fyn or SNAP23 showed unaltered localization in the absence of caveolin-1 (unshown data). Other lipid-modified proteins, such as glycosylphosphatidyl inositol-linked protein or c-Src have been previously shown to exhibit altered subcellular localization in the absence of caveolin-1 or caveolae (Sotgia et al. 2002). In the case of H-Ras, intracellular retention in a perinuclear compartment is of particular interest because recent studies have shown that Ras is actively signaling not only from the plasma membrane but also from intracellular membranes, with different signaling outputs depending on the compartment from which Ras signals: artificially ER-tethering of H-Ras was shown to favor activation of the Jnk pathway, while artificially Golgi-tethering was shown to favor the Erk and Akt pathways and more recently the Ral pathway (Chiu et al. 2002; Matallanas et al. 2006). The kinetics of activation also differ between plasma membrane and endomembrane compartments, with delayed and sustained activation of Ras on the Golgi (Quatela and Philips 2006). Altering the subcellular localization of Ras may therefore be a new mechanism by which expression of caveolin-1 modulates cellular signaling.
The mechanism by which H-Ras localization is altered in the absence of caveolin-1 expression is still unresolved. We have shown that it is not due to altered H-Ras palmitoylation. It is probably not due to plasma membrane cholesterol depletion, because expression of a dominant negative caveolin mutant which disrupted cholesterol-enriched domains, reduced the abundance of caveolae and altered H-Ras-mediated Raf activation, did not alter H-Ras plasma membrane localization (Roy et al. 1999). It is also unlikely that the plasma membrane localization of H-Ras in wild type cells is due to its direct interaction with caveolin-1, because we showed no co-localization of the two proteins at the plasma membrane. We can therefore only speculate on possible mechanisms underlying the lack of plasma membrane localization of H-Ras in caveolin-1-deficient cells. Recent evidence points to an alternative itinerary from the endoplasmic reticulum to the plasma membrane for newly synthesized H-Ras that circumvents conventional vesicular transport (Zheng et al. 2007). This pathway, which was reported to convey a significant amount of H-Ras to the plasma membrane, might be impaired in the absence of caveolin-1. Alternatively, the expression of a protein essential for stabilization of H-Ras interaction with the plasma membrane may be decreased in caveolin-1-null cells. Down-regulation of galectin-1 was shown to alter plasma membrane targeting of GTP-loaded, but not GDP-bound H-Ras (Paz et al. 2001).
In summary, our results show two important new mechanisms by which loss of caveolin-1 expression may perturb intracellular signaling, namely the mislocalization of signaling proteins and alterations in protein palmitoylation. In the case of H-Ras however, these two observations were unrelated as the altered plasma membrane localization of H-Ras in caveolin-1-deficient cells was palmitoylation-independent.
References
Anderson RG, Kamen BA, Rothberg KG, Lacey SW (1992) Potocytosis: sequestration and transport of small molecules by caveolae. Science 255:410–411
Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF (2000) H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol 20:2475–2487
Bivona TG, Philips MR (2003) Ras pathway signaling on endomembranes. Curr Opin Cell Biol 15:136–142
Camp LA, Hofmann SL (1993) Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J Biol Chem 268:22566–22574
Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL, Cox AD, Philips MR (2002) Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol 4:343–350
Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR (1999) Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98:69–80
Das AK, Dasgupta B, Bhattacharya R, Basu J (1997) Purification and biochemical characterization of a protein-palmitoyl acyltransferase from human erythrocytes. J Biol Chem 272:11021–11025
del Pozo MA, Balasubramanian N, Alderson NB, Kiosses WB, Grande-Garcia A, Anderson RG, Schwartz MA (2005) Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat Cell Biol 7:901–908
Dietzen DJ, Hastings WR, Lublin DM (1995) Caveolin is palmitoylated on multiple cysteine residues. Palmitoylation is not necessary for localization of caveolin to caveolae. J Biol Chem 270:6838–6842
Drab M et al (2001) Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293:2449–2452
Duncan JA, Gilman AG (1998) A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21 (RAS). J Biol Chem 273:15830–15837
Dunphy JT, Greentree WK, Manahan CL, Linder ME (1996) G-protein palmitoyltransferase activity is enriched in plasma membranes. J Biol Chem 271:7154–7159
Engelman JA, Wykoff CC, Yasuhara S, Song KS, Okamoto T, Lisanti MP (1997) Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J Biol Chem 272:16374–16381
Engelman JA, Zhang XL, Razani B, Pestell RG, Lisanti MP (1999) p42/44 MAP Kinase-dependent and -independent Signaling Pathways Regulate Caveolin-1 Gene Expression. Activation of ras-map kinase and protein kinase a signaling cascades transcriptionally down-regulates caveolin-1 promoter activity. J Biol Chem 274:32333–32341
Fra AM, Williamson E, Simons K, Parton RG (1995) De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc Natl Acad Sci U S A 92:8655–8659
Galbiati F, Volonte D, Meani D, Milligan G, Lublin DM, Lisanti MP, Parenti M (1999) The dually acylated NH2-terminal domain of Gi1alpha is sufficient to target a green fluorescent protein reporter to caveolin-enriched plasma membrane domains. Palmitoylation of caveolin-1 is required for the recognition of dually acylated g-protein alpha subunits in vivo. J Biol Chem 274:5843–5850
Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC (1997) Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem 272:25437–25440
Hancock JF (2003) Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 4:373–384
Hancock JF, Paterson H, Marshall CJ (1990) A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63:133–139
Koleske AJ, Baltimore D, Lisanti MP (1995) Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci U S A 92:1381–1385
Lee H, Woodman SE, Engelman JA, Volonte D, Galbiati F, Kaufman HL, Lublin DM, Lisanti MP (2001) Palmitoylation of caveolin-1 at a single site (Cys-156) controls its coupling to the c-Src tyrosine kinase. Targeting of dually acylated molecules (GPI-linked, transmembrane, or cytoplasmic) to caveolae effectively uncouples c-Src and caveolin-1 (Tyr-14). J Biol Chem 276:35150–35158
Li S, Okamoto T, Chun M, Sargiacomo M, Casanova JE, Hansen SH, Nishimoto I, Lisanti MP (1995) Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J Biol Chem 270:15693–15701
Li S, Couet J, Lisanti MP (1996) Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J Biol Chem 271:29182–29190
Lobo S, Greentree WK, Linder ME, Deschenes RJ (2002) Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J Biol Chem 277:41268–41273
Matallanas D, Sanz-Moreno V, Arozarena I, Calvo F, gudo-Ibanez L, Santos E, Berciano MT, Crespo P (2006) Distinct utilization of effectors and biological outcomes resulting from site-specific Ras activation: Ras functions in lipid rafts and Golgi complex are dispensable for proliferation and transformation. Mol Cell Biol 26:100–116
Michaelson D, Ahearn I, Bergo M, Young S, Philips M (2002) Membrane trafficking of heterotrimeric G proteins via the endoplasmic reticulum and Golgi. Mol Biol Cell 13:3294–3302
Mineo C, Anderson RG (2001) Potocytosis. Histochem Cell Biol 116:109–118
Mineo C, James GL, Smart EJ, Anderson RG (1996) Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J Biol Chem 271:11930–11935
Monier S, Dietzen DJ, Hastings WR, Lublin DM, Kurzchalia TV (1996) Oligomerization of VIP21-caveolin in vitro is stabilized by long chain fatty acylation or cholesterol. FEBS Lett 388:143–149
Parat MO, Fox PL (2001) Palmitoylation of caveolin-1 in endothelial cells is post-translational but irreversible. J Biol Chem 276:15776–15782
Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y (2001) Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 20:7486–7493
Prior IA, Harding A, Yan J, Sluimer J, Parton RG, Hancock JF (2001) GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol 3:368–375
Quatela SE, Philips MR (2006) Ras signaling on the Golgi. Curr Opin Cell Biol 18:162–167
Razani B et al (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276:38121–38138
Resh MD (2006) Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci STKE 2006: re14
Roy S, Luetterforst R, Harding A, Apolloni A, Etheridge M, Stang E, Rolls B, Hancock JF, Parton RG (1999) Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nat Cell Biol 1:98–105
Schlegel A, Pestell RG, Lisanti MP (2000) Caveolins in cholesterol trafficking and signal transduction: implications for human disease. Front Biosci 5:929–937
Smotrys JE, Linder ME (2004) Palmitoylation of intracellular signaling proteins: regulation and function. Annu Rev Biochem 73:559–587
Song SK, Li Shengwen, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP (1996) Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem 271:9690–9697
Sotgia F et al (2002) Intracellular retention of glycosylphosphatidyl inositol-linked proteins in caveolin-deficient cells. Mol Cell Biol 22:3905–3926
Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, Linder ME (2005) DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem 280:31141–31148
Uittenbogaard A, Smart EJ (2000) Palmitoylation of caveolin-1 is required for cholesterol binding, chaperone complex formation, and rapid transport of cholesterol to caveolae. J Biol Chem 275:25595–25599
White MA, Anderson RG (2005) Signaling networks in living cells. Annu Rev Pharmacol Toxicol 45:587–603
Yeh DC, Duncan JA, Yamashita S, Michel T (1999) Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 Is potentiated by Ca (2+)-calmodulin. J Biol Chem 274:33148–33154
Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC (2006) Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116:1284–1291
Zheng H, McKay J, Buss JE (2007) H-Ras does not need COP-I or COP-II-dependent vesicular transport to reach the plasma membrane. J Biol Chem 282:25760–25768
Acknowledgments
This work was supported by the Ohio Cancer Research Associates.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Baran, J., Mundy, D.I., Vasanji, A. et al. Altered localization of H-Ras in caveolin-1-null cells is palmitoylation-independent. J. Cell Commun. Signal. 1, 195–204 (2007). https://doi.org/10.1007/s12079-008-0017-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-008-0017-3