
Abstract
Portal hypertension is the main non-neoplastic complication of chronic liver disease, being the cause of important life-threatening events including the development of ascites or variceal bleeding. The primary factor in the development of portal hypertension is a pathological increase in the intrahepatic vascular resistance, due to liver microcirculatory dysfunction, which is subsequently aggravated by extra-hepatic vascular disturbances including elevation of portal blood inflow. Evidence from pre-clinical models of cirrhosis has demonstrated that portal hypertension and chronic liver disease can be reversible if the injurious etiological agent is removed and can be further promoted using pharmacological therapy. These important observations have been partially demonstrated in clinical studies. This paper aims at providing an updated review of the currently available data regarding spontaneous and drug-promoted regression of portal hypertension, paying special attention to the clinical evidence. It also considers pathophysiological caveats that highlight the need for caution in establishing a new dogma that human chronic liver disease and portal hypertension is reversible.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Approximately 2 million people die each year from complications of chronic liver disease (CLD) in spite of recent major progresses in this field [1]. CLD originates due to chronic injury, which induces excessive extracellular matrix (ECM) deposition and microvascular dysfunction that, over time, hinder intrahepatic circulation and induce portal hypertension (PH) [2].
PH is a clinical syndrome defined as an increased blood pressure in the portal venous system, being the primary cause of clinically relevant complications such as ascites, jaundice, variceal bleeding and an increased risk of spontaneous bacterial peritonitis or other bacterial infections, hepatic encephalopathy, hepatorenal syndrome and liver failure [3] (Fig. 1). The current gold standard for diagnosing and staging cirrhotic (sinusoidal) portal hypertension is HVPG measurement, which allows estimation of the portal pressure by calculating the difference between the wedged hepatic venous pressure (WHVP) and the free hepatic venous pressure (FHVP).

Schematic representation of complications of portal hypertension. Clinically significant portal hypertension may lead to hepatic encephalopathy, gastroesophageal varices prone to hemorrhage, liver failure, hepatorenal syndrome and ascites and it is also associated with an increased risk for hepatocellular carcinoma development. GEV gastroesophageal varices, SBP spontaneous bacterial peritonitis
In normal conditions, HVPG ranges from 1 to < 5 mmHg. Values greater than 5 mmHg indicate portal hypertension; while, a HVPG greater than 10 mmHg indicates clinically significant portal hypertension (CSPH), which may result in the above-described life-threatening clinical complications [3].
Initially, PH develops due to increased intrahepatic vascular resistance (HVR) to blood flow. This increased HVR is most commonly caused by chronic liver disease as a result of multiple pathological events in the sinusoidal circulation [4]. Indeed, during the process of liver injury, and regardless of the etiology, liver sinusoidal endothelial cells (LSECs) are rapidly de-regulated and begin to de-differentiate, acquiring a “capillarized” phenotype. They become proinflammatory and produce soluble factors that reach neighboring cells and determine their phenotype [5]. At the same time, exogenous liver injury induces a transcriptional change in hepatocytes, promoting their proliferation and death. This, in turn, leads to the release of apoptotic bodies that further contribute to the activation of other hepatic cells [6, 7]. Due to these injuring stimuli, hepatic stellate cells (HSCs) leave their quiescent state, becoming proliferative, pro-contractile and start synthesizing ECM components, becoming the most direct contributors to hepatic fibrosis. Persistent fibrosis, then, leads to distortion of the liver parenchyma and vascular structures, contributing to the stiffening of the organ and perturbing many cellular functions [8], ultimately leading to increased HVR and PH (Fig. 2). In advanced stages of the disease, the splanchnic tissue senses the reduced blood flow and increased pressure upstream of the liver, and produces an extrahepatic vasodilatory response as a compensatory mechanism. However, this leads to increased blood flow to the portal vein (hyperdynamic circulation), further aggravating PH [9].

Main cellular mechanisms contributing to the progression of chronic liver disease in response to liver damage. Chronic liver injury induces the expression of cell adhesion molecules by LSEC, leading to a recruitment of macrophages to the tissue. These, together with liver damage, lead to necroinflammation, capillarization of LSEC, polarization of macrophages, HSC activation and liver fibrosis. aHSC activated hepatic stellate cell, cLSEC capillarized liver sinusoidal endothelial cell, dxHep dysfunctional hepatocyte
Currently, therapeutics for PH consist of drugs targeting the extrahepatic vascular bed (mostly non-selective beta-blockers) that ameliorate systemic circulation and the hyperdynamic syndrome, leading to a reduction in portal blood flow. Despite the fact that these approaches may accomplish a reduction in PH in some cases, they do not have an effect on increased HVR (the primary cause of PH). Until recently, advanced fibrosis was thought to be irreversible, liver transplantation being the only option to cure CLD in most cases [10]. However, recent data from both human and animal models have challenged this view and suggest that after removal of the etiologic factor liver fibrosis and even cirrhosis may regress [11]. This is the case of a fraction of the patients cured from hepatitis B, hepatitis C, hepatitis Delta or metabolic hepatitis [12], that achieved normal or near-normal liver histology and function after the etiological agent was removed. Therefore, such insights may provide valuable data in determining the underlying mechanisms of PH and potential future therapeutic strategies.
Regression of portal hypertension: spontaneous mechanisms
Pre-clinical evidence for PH regression upon etiologic treatment
Spontaneous resolution of fibrosis has been observed in the gold-standard models of cirrhosis, including the carbon tetrachloride (CCl4), thioacetamide (TAA) and bile duct ligation models [13, 14]. Furthermore, regression of fibrosis and PH was also observed in animals with NASH after replacement of the high-fat diet for standard diet [15], altogether suggesting that reversion of fibrosis and PH may be achieved in different etiological backgrounds.
Indeed, Abdel-Aziz and colleagues studied the reversibility of fibrosis in experimentally induced cholestasis in rats. Three weeks after the ligation of bile ducts, they observed bile duct proliferation and periportal fibrosis. Three weeks after relief of the bile duct ligation, there was resorption normalization of periportal fibrosis, except for the persistence of collagen IV in the sinusoids [13]. Iredale and colleagues examined spontaneous recovery from liver fibrosis in CCl4-treated rats. After four weeks of induction and four additional weeks of recovery, they observed dissolution of the collagen fibrous matrix and a return to essentially normal liver structure [16]. Additional studies confirmed these observations [17, 18].
Clinical evidence for PH regression upon etiologic treatment
As exposed above, one of the first and most important steps towards achieving regression of CLD is the removal of the injurious agent. The first observations regarding the issue date back in the early 1990s, when the resolution of esophageal varices was demonstrated in a small group of alcoholic cirrhotics who managed to abstain from alcohol for a long period of time [19]. Also, one-year alcohol abstinence determined a 46% reduction in hepatic vein wedge pressure (as a surrogate of portal pressure) [20]. The importance of achieving this goal has been demonstrated in CLD of several etiologies (Table 1), most convincingly in viral CLD.
-
HBV: Long-term studies (5–7 years) showed a histologically proven progressive reduction in necroinflammation and fibrosis scores in a vast majority of antiviral treatment (AVT)-responsive patients with HBV advanced fibrosis or cirrhosis [21, 22]. In addition, several studies demonstrated the beneficial effects of viral suppression on non-malignant decompensating events [23, 24], low grade esophageal varices (EV) progression [25, 26], EV development rate [26], and clinical scores and transplant-free survival [27]. Although the evaluation of EV dynamics is based on subjective judgement, the evidence provided by these trials suggest an underlying decrease in PH. One study including 19 patients with HBV-related CSPH showed a median reduction of 18.7% in HVPG during the 12 months of follow-up, with no significant systemic hemodynamic changes [28], suggesting a reduction in HVR, possibly through decreased hepatic necroinflammation.
-
HCV, pre-DAA: While sustained virological response (SVR) after HCV therapy is significantly higher since the introduction of direct-acting antivirals (DAA), several pre-DAA studies already held promise of PH regression induced by etiological intervention. As is the case with HBV AVT, treatment of HCV can also prevent the development of EV or slow down progression towards decompensation [29]. However, it seems that this effect is less consistent once EV is already present before treatment initiation, suggesting a reduced effect on already established PH [29, 30]. Moreover, in spite of PH decrease, decompensation is not always prevented and regression below the CSPH threshold is not always achievable, even with long-term follow-up (5.2 years after end of treatment) [31]. Short-term hemodynamic and/or histology studies in compensated patients demonstrated HVPG reduction paralleled by reduction in necroinflammatory scores, but either no or very weak reduction in fibrosis scores, possibly due to the short follow-up time [32, 33]. Around 60% of treated HCV patients with advanced fibrosis or cirrhosis followed up over a long time period (2–5 years) showed a progressive reduction in fibrosis score on repeated biopsy and significant reduction in scar collagen content, even in those without obvious fibrosis regression [34,35,36]. However, no changes in sinusoidal capillarization, as assessed by CD34 positivity or α-SMA staining, were observed, pointing towards lack of intrahepatic vascular remodeling [34].
-
HCV, DAA: With the emergence of DAAs, even patients in advanced stages of CLD can achieve viral eradication, which leaves open the question whether and to what degree their disease can be reversed. It is now clear that SVR after DAA can result in clinically significant (≥ 10–20%) decrease in HVPG [37, 38], even in those patients with baseline CSPH and in difficult to treat populations, such as HCV-HIV co-infected patients [38]. However, this effect seems less pronounced and more heterogeneous in those patients with advanced-stage CLD (high Child–Pugh score, HVPG > 16 mmHg) [37, 39]. A study on post-transplant graft HCV reinfection showed fibrosis regression and HVPG decrease in 67% and 66% of patients which achieved SVR, however, among F4 biopsies, none of the patients displaying thick fibrous septa (Laennec C cirrhosis) had cirrhosis regression [40]]. Histological data show that HVPG reduction is lower in those patients who still have necroinflammatory activity after SVR [38], which points towards an initial phase of reduction in HVPG through decreased intrahepatic inflammation [41]. However, it seems that HVPG decreases progressively over a longer time period, suggesting a long-term possibility of further decrease in HVPG based on mechanisms other than reduced inflammation. Still, in a relatively high proportion of patients, CSPH did not resolve, leaving them a risk for future decompensation [42,43,44].
A significant decrease in HVPG in patients without baseline CSPH is particularly important given its potential to prevent progression of disease (Fig. 3). Indeed, achieving SVR seems to decrease disease severity (Child–Pugh and MELD scores), decompensation risk and EV grade [39, 45, 46]. These important improvements in HCV-induced CLD management are also mirrored in the shift in transplant indication from the pre-DAA era and even within the last few years [47]. The effect of DAA on systemic hemodynamics is still not well established, with some studies showing mild but significant increase in MAP and systemic vascular resistance [37]; whereas, others described no influence [38].
-
NAFLD: The vast majority of patients benefit from different weight loss surgery approaches, with normalization or reduction in tissue fibrosis [48, 49], although this is more evident in early fibrosis [50]. Lifestyle interventions have an important role in obese CLD patients (any etiology), with 42% of patients showing a significant decrease (≥ 10%) in HVPG from baseline after 16 weeks, paralleled by a decrease in insulin resistance and plasma leptin levels, giving an insight into possible mechanisms of regression in this patient population [51]. Moreover, this last study showed no change in portal blood flow, thus reinforcing the probable effect on HVR of etiological treatment in CLD. A subsequent study investigating the effects of physical activity in cirrhotic patients with CSPH (compensated or decompensated) also demonstrated a reduction in HPVG [52].

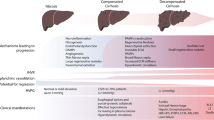
Stage-specific features determining the probability of regression of portal hypertension and chronic liver disease. It is accepted that the likeliness of regression is inversely correlated to the severity of the disease, usually determined by factors such as HVPG, thickness of fibrous septa or acellularization. HVPG hepatic venous pressure gradient, HE hepatic encephalopathy
Cellular and molecular events limiting spontaneous regression of PH
It is now evident that cirrhosis is at least partly reversible. However, the extent to which resolution can occur seems to be highly dependent on disease stage. Some of the most important elements that render PH resistant to regression are fibrotic tissue composition and stiffness, presence or absence of specific cell populations, and the profound (micro) vascular changes (Fig. 4), all elements which are significantly different in early versus advanced CLD.
-
Fibrotic tissue properties: In a murine CCl4 model of CLD, followed up for 1 year after cessation of toxicant administration, regression was only partial, with transformation from micronodular to macronodular pattern and incomplete resolution of broad mature septa [14]. This mirrors changes described during human cirrhosis regression [53]. In humans, small nodule size and increased septal thickness have both been correlated with higher HVPG and seem to be predictive of decompensation [54] and patients with Laennec C cirrhosis are unlikely to have a significant HVPG decrease even after removal of etiological agent [40]. In murine models, enzymes such as tissue-transglutaminase (tTG) and lysil oxidases (LOX) induce the cross-linking of collagens and elastins, creating acellular areas that are resistant to degradation [14, 55, 56]. A study using tTG2 KO mice showed that this molecule is not indispensable during fibrogenesis and mice lacking it do not have a resolution advantage compared to wild-type mice [57]. This could suggest alternative collagen cross-linking pathways. In this regard, elastin also plays an important role in the irreversibility of advanced fibrosis. Elastin to collagen ratio increases with disease progression and, despite an early increase in expression, its marked accumulation only occurs in advanced stages. This points towards an imbalance between synthesis and removal, also suggested by the increase in matrix metalloproteinase 12 (MMP12) bound to tissue inhibitor of matrix metalloproteinase-1 (TIMP-1), which renders it inactive. Moreover, MMP12 KO mice display significantly higher level of bridging fibrosis, further suggesting a defect in elastin removal [58]. Depletion of macrophages results in additional accumulation of elastin compared to wild-type mice and with failure of tissue remodeling. Indeed, pro-resolution macrophages are an important source of MMPs [58, 59].
-
Role of HSCs: Activated HSCs (aHSC) play a key role in fibrosis, and an important event necessary for its resolution is their disappearance through either senescence, apoptosis or inactivation [60]. HSCs situated within mature insoluble septa seem to be less prone to undergo apoptosis [14]. Indeed, it seems that persistence of scar tissue is associated with the maintenance of aHSC [61], which importantly also highlights the role of the underlying matrix in influencing cell phenotype [62]. aHSC are a major source of TIMPs and TIMP-1 overexpressing murine models fail to show resolution of fibrosis [63]. Moreover, it has been shown that TIMP-1 itself promotes survival of aHSC [64]. Although HSC inactivation may occur during fibrosis regression, these cells seem to remain more sensitive to renewed exposure to fibrogenic stimuli compared to their normal counterparts [65].
-
Vascular phenotype: In addition to fibrosis, the other crucial component and the major causative factor of pathophysiological consequences of cirrhosis are the vascular changes, both intra- and extrahepatic. Hepatic endothelial de-differentiation and neo-angiogenesis depend on the initial injury pattern and it may well be that the endothelial and vascular changes are in fact the most important determinants of regression capacity [5, 66, 67]. As recently proposed by Wanless, advanced stage CLD can progress independent of the initial etiological agent, due to a vicious circle in which vascular injury promotes vascular obstruction which leads to renewed vascular injury and hepatocellular damage; the so-called ‘congestive escalator’ [68]. In line with this, a recent study in murine cirrhosis shows persistent liver hyperarterialization, in spite of cirrhosis regression [69].
-
Aging and other factors: Advanced age has been shown to be an important determinant of CLD severity in murine models, results that were corroborated by HVPG and gene expression differences in human CLD patients with more advanced age [70]. Age seems to also be involved in CLD regression capacity, as shown in a murine CCl4 model, in which old mice were significantly less prone towards CLD reversal, as assessed by liver histology and ECM remodeling pathways (including macrophage populations) compared to their young counterparts [71]. Moreover, genetic and epigenetic factors likely play a role in the capacity and speed of CLD regression [72]. Last but not least, it is important to consider additional pro-fibrogenic factors which could influence the rate of regression in patients even after causative treatment, such as metabolic risk factors or excessive alcohol intake.

Molecular mechanisms modulating regression of chronic liver disease. Regression of cirrhosis and portal hypertension is usually impaired by liver architecture (altered extracellular matrix and acellularization) and microcirculatory dysfunction. All these may induce an activation response to hepatic cells, promoting progression rather than regression of chronic liver disease. On the other hand, modulation of the phenotype of hepatic cells (green box) may inhibit said alterations and represent potential targets for regression of chronic liver disease. ECM extracellular matrix, HSC hepatic stellate cell, LSEC liver sinusoidal endothelial cell
Regression of portal hypertension: therapy-driven strategies
Despite the evidence of spontaneous regression after removal of the etiologic cause, there is still a great percentage of patients whose disease does not regress or even progresses. Therefore, in the recent years, there has been an increasing effort to develop new therapies that could have an impact in regression of cirrhosis and PH [73]. Indeed, regression of PH has been assessed by targeting the different intrahepatic alterations associated with the disease, which are the primary cause of increased HVR (Table 2). These approaches mainly achieve vasodilation or reduced inflammation, leading to amelioration of the dynamic and structural components of HVR.
Vasomodulators
Statins: the first studies testing statins in humans evaluated acute hemodynamic responses to simvastatin and demonstrated a decrease in estimated HVR accompanied by increased hepatic NO production and a 50% reduction in post-prandial splanchnic hyperemia [74]. Longer periods of simvastatin treatment contributed to a significant decrease in PP as measured by HVPG [75, 76], to an improvement in liver function measured as IGC [75] and to a significant survival benefit related to decreased mortality due to EV bleeding and infection [77]. Importantly, the decrease in HVPG was further augmented when statins were used in combination with propranolol. Similarly, a study investigating simvastatin addition to carvedilol non-responders had positive results, increasing the number of patients with hemodynamic response [78]. In contrast, a recent study in the same patient category, using carvedilol in combination with simvastatin over a period of 3 months failed to show any additional benefit of combination therapy in reducing HVPG [79]. Ongoing trials are further testing statins in the context of CLD [80, 81].
Renin–angiotensin–aldosterone system (RAAS) inhibitors: angiotensin converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs) and aldosterone antagonists (AAs) have shown beneficial effects on fibrosis in other organs. In CLD, they could potentially target the excess sodium and water retention while acting as antifibrotics. NAFLD patients under treatment with ACEIs or ARBs had slower fibrosis progression rate, more pronounced in a subgroup with concomitant type 2 diabetes mellitus [82]. Conversely, in the HALT-C cohort, a post hoc analysis did not show any differences in fibrosis progression compared to untreated controls [83]. Regarding their effect on HVPG, it seems that ARBs and/or ACEIs (possibly with the addition of mineralocorticoid antagonists) could be useful in Child–Pugh A patients, but probably not in more advanced, where the risk of adverse events (renal, electrolytic disturbances, hypotension) is much greater [84].
PDE5 inhibitors: Clinical results using several PDE5 inhibitors are contradictory, with some studies showing an acute HVPG decrease [85, 86], and other observing no change in HVPG probably due to simultaneous increase in PBF counteracting the decrease in HVR [87]. Considering certain systemic effects of PDE5 inhibitors [88], and that even a relatively minor decrease in MAP can be deleterious in patients with advanced CLD, the combination of these agents with NSBB may be unsafe in advanced CLD. This approach might be promising in patients in early disease stages, given the importance of HVR as pathogenetic mechanism and the still relatively normal systemic hemodynamic.
Rho-kinase inhibitors: Fasudil produced a statistically significant acute hemodynamic response in HVPG and reduction in portal vascular resistance in a small RCT, but with negative systemic hemodynamic effects evidenced by significantly decreasing MAP and SVR [89].
Endothelin antagonism has shown promising results in treating PH complications, such as hepatorenal syndrome and portopulmonary hypertension [90]. Regarding PP, continuous infusion of tezosentan (dual endothelin receptor antagonist) did not cause relevant changes in HVPG, hepatic blood flow or IGC in a cohort of cirrhotic patients with CSPH [91]. ETA or ETB receptor antagonists in Child–Pugh A cirrhotic patients showed opposing actions on systemic hemodynamics: while blocking of ETA decreased MAP, MPAP and systemic vascular resistance, inhibition of ETB increased MAP and SVR, with no effect on pulmonary hemodynamics [92], but no net effect on HVPG was observed. Since there might be a shift in the ETA to ETB receptor ratio in the liver during the development of cirrhosis [93], and the responsiveness to ET1 may be altered during the course of CLD, a more selective targeting of this pathway is required. Indeed, a recent study has shown a beneficial effect of intrahepatic administration of ETA antagonist, highlighting the important local effects. Additionally, the same study has demonstrated the efficacy of ambrisentan on lowering PP, without clinically significant systemic changes, even in advance CLD [94]. Currently, a clinical trial investigating the effect of the ETA antagonist ambrisentan on PH is recruiting patients [95].
Sapropterin, an oral synthetic analog of tetrahydrobiopterin (BH4), which is an essential co-factor for NO synthesis and is reduced in cirrhotic livers, has not shown an effect on HVPG, IGC or markers of endothelial dysfunction and oxidative stress during a 2-week period of treatment in cirrhotic patients with CSPH [96].
Serelaxin, a recombinant human relaxin-2 analog has demonstrated no effect in HVPG acute response; however, the trial cohort was small due to permanent drug discontinuation [97]. It is conceivable that, in an accurately powered trial, the drug might show an effect on PP.
Therapies leading to reduced inflammation/fibrosis
Belapectin: this galectin 3 inhibitor did not show significant changes in HVPG, liver histology or development of complications in a phase 2 NASH trial, with the exception of a subgroup of patients without band-ligated varices, where there was a significant decrease in HVPG and in the development of de novo EV [98]. There is an ongoing trial in this category of patients [99].
The FXR agonist obeticholic acid (OCA) is currently under evaluation in several trials. In a small trial presented in abstract form, OCA treatment for 7 days significantly reduced HVPG in more than half of the cohort of alcoholic cirrhosis patients investigated, opening an avenue for future investigations in this direction [100].
Sorafenib, a multikinase inhibitor used as HCC therapy, decreased baseline HVPG by ≥ 20% in 36% of cirrhotic patients with HCC after 2 weeks of treatment. Moreover, it significantly decreased liver tissue mRNA levels of VEGF, PDGF, PlGF, RhoA kinase and TNFα [101].
Taurine: In a trial of 28-day taurine treatment in cirrhotic patients with CSPH, 58% of treated patients had a drop of ≥ 10% in HVPG, without any systemic hemodynamic effects [102].
Probiotics: While some authors described an important decrease in HVPG after VSL#3 probiotic treatment, with a higher proportion of patients achieving hemodynamic response in case of concomitant NSBB treatment [103], other patients seem to not benefit from this [104]. In addition, other beneficial effects of VSL#3 have been described, such as improvement in systemic hemodynamics, decrease in plasma TNFα levels [105] and improvement in serum Na+ concentration [103].
Antioxidants: acute administration of dark chocolate decreased the magnitude of postprandial increase in HVPG when compared to the control group, without significantly affecting PBF, suggesting a possible intrahepatic mechanism of action. Moreover, patients receiving dark chocolate had a mild increase in mean arterial pressure [106]. Similarly, a clinical trial using ascorbic acid attenuated post-prandial increase in HVPG without changing HBF [107].
Simtuzumab, a monoclonal antibody against lysis oxidase-like 2 (LOXL2), has shown no effect on any of the study endpoints in NASH fibrosis or cirrhosis, HCV/HIV or primary sclerosing cholangitis [108,109,110]. However, targeting LOX family members could still be a promising approach, as highlighted in a recent review [111], with new molecules engaging intracellular LOXL2 or targeting several other LOX family members.
Emricasan, a pan-caspase inhibitor, significantly decreased HVPG and aminotransferases in a subgroup of cirrhotic patients (any etiology) with CSPH ≥ 12 mmHg in an exploratory study [112]. However, in a follow-up RCT, emricasan did not achieve a significant reduction in HVPG in NASH cirrhosis patients with severe PH [113].
Antibiotics: 4 weeks of norfloxacin treatment was either non-superior to placebo or showed a non-significant trend towards HVPG reduction [114,115,116]. However, norfloxacin showed systemic hemodynamic effects in these studies, by increasing SVR and MAP. It is possible that, due to decreased NO caused by attenuated bacterial translocation, norfloxacin causes a decrease in portal blood flow and an increase in HVR, which counteract each other and could explain the lack of HVPG effect [114, 116]. Rifaximin has shown a significant effect on short-term HVPG decrease [117] and also reduced the risk of developing complications and improved survival when administered for up to 5 years [118]. However, this last study included only the HVPG responders from the previous short-term study, which might bias results. Conversely, a more recent RCT failed to demonstrate any effect of short-term rifaximin treatment on hepatic or systemic hemodynamics [119]. It seems that addition of rifaximin to NSBB has a further favorable effect [120].
Studies not evaluating HVPG
Several studies, although not directly assessing HVPG reduction, have investigated the potential of different drugs to modify either components of CLD, such as fibrosis, or the natural history of the disease (Table 3).
Biomarkers of PH regression
While HVPG measurement remains the gold-standard approach for monitoring the dynamics of PP, a multitude of non-invasive tests have been designed and evaluated for diagnosis, stratification of disease severity and progression monitoring [132]. However, these are likely inaccurate for evaluating regression of fibrosis and PH [72, 133]. Furthermore, treatment or removal of etiological agents can modify individual score components independently of the evolution of the underlying CLD [134, 135]. A summary of studies investigating noninvasive markers can be found in Table 4.
Regarding the monitoring of CLD complications during PH regression, recently, the Baveno VI criteria for EV surveillance have been validated in HBV and/or HCV compensated patients, post-SVR [139].
Serum biomarkers of fibrogenesis and fibrolysis are another area of research. Collagen fragments can serve as such biomarkers: PRO-C3 and C6M have been shown to identify progressors, while PRO-C5 identified fibrosis regressors [140]. Moreover, combinations of these markers have been shown to correlate with the degree of portal hypertension [141, 142]. However, if and to what extent these markers correlate with HVPG reduction and clinical outcomes during cirrhosis regression, especially in patients with advanced CLD, remains a subject of future investigations.
Conclusions and future perspective
Alcohol, NASH and viral hepatitis are the most common etiologies of CLD. Even if these are usually treatable (even more so after the recent development of direct antiviral strategies), removal of the etiologic agent may stop progression of the disease and lead to regression of fibrosis only in some of the cases.
The mechanisms of cirrhosis regression are still widely unknown, in part due to the limitations of preclinical models, which develop and partially revert cirrhosis in a short time as opposed to the slow clinical progression and regression of the disease. This is also true for the study of pharmacologic therapies, which are usually prophylactic during the pre-clinical induction of CLD or administered for a short time after mild disease is established, while in the case of clinical trials require consistent study design and usually repeated assessment with invasive techniques like liver biopsy. Other factors including the age and gender of the animals used in pre-clinical studies may also play a key role for future successful developments [143].
Despite these limitations, new advances in the study of PH regression point towards a clear role of the sinusoidal biomechanical properties in CLD, which could determine the cellular phenotype, vascular function, proliferation and overall drive the disease towards progression or regression.
References
Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol 2019;70(1):151–71
Bosch J. Vascular deterioration in cirrhosis: the big picture. J Clin Gastroenterol 2007;41(SUPPL 3):247–53
Garcia-Tsao G, Abraldes JG, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: risk stratification, diagnosis, and management: 2016 practice guidance by the American Association for the study of liver diseases. Hepatology 2017;65(1):310–35
Gracia-Sancho J, Marrone G, Fernández-Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol 2019;16(4):221–34
Gracia-Sancho J, Caparros E, Fernández-Iglesias A, Francés R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol 2021. https://doi.org/10.1038/s41575-020-00411-3
Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003;38(5):1188–98
Jiang JX, Mikami K, Venugopal S, Li Y, Török NJ. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-κB-dependent pathways. J Hepatol 2009;51(1):139–48
Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol 2010;53:976–80
Møller S, Bendtsen F. The pathophysiology of arterial vasodilatation and hyperdynamic circulation in cirrhosis Liver Int. 2018;38:570–80
Sauerbruch T, Schierwagen R, Trebicka J. Managing portal hypertension in patients with liver cirrhosis F1000Res. 2018;7:1–17
Brenner DA. Reversibility of liver fibrosis. Gastroenterol Hepatol (N Y) 2013;9(11):737–9
Lee YA, Friedman SL. Reversal, maintenance or progression: what happens to the liver after a virologic cure of hepatitis C? Antiviral Res 2014;107:23–30
Abdel-Aziz G, Lebeau G, Rescan PY, Clement B, Rissel M, Deugnier Y, et al. Reversibility of hepatic fibrosis in experimentally induced cholestasis in rat. Am J Pathol 1990;137(6):1333–42
Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MDA, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology 2004;126(7):1795–808
Ding Z-M, Xiao Y, Wu X, Zou H, Yang S, Shen Y, et al. Progression and regression of hepatic lesions in a mouse model of NASH induced by dietary intervention and its implications in pharmacotherapy. Front Pharmacol 2018;9(MAY):410
Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, et al. Mechanisms of spontaneous resolution of rat liver fibrosis: hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 1998;102(3):538–49
Kisseleva T, Cong M, Paik YH, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A 2012;109(24):9448–53
Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012;143(4):1073-1083.e22
Müting D, Kalk JF, Fischer R, Wiewel D. Spontaneous regression of oesophageal varices after long-term conservative treatment. Retrospective study in 20 patients with alcoholic liver cirrhosis, posthepatitic cirrhosis and haemochromatosis with cirrhosis. J Hepatol 1990;10(2):158–62
Klein CP, Kalk JF, Muting D, Klein CG. Influence of alcohol on portal-vein haemodynamics in nutritional-toxic cirrhosis of the liver. Dtsch Med Wochenschr 1993;118(4):89–93
Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 2013;381(9865):468–75
Chang T, Liaw Y, Wu S, Schiff E, Han K, Lai C, et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology 2010;52:886–93
Liaw Y-F, Sung JJY, Chow WC, Farrell G, Lee C-Z, Yuen H, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med 2004;351(15):1521–31
Su TH, Hu TH, Chen CY, Huang YH, Chuang WL, Lin CC, et al. Four-year entecavir therapy reduces hepatocellular carcinoma, cirrhotic events and mortality in chronic hepatitis B patients. Liver Int 2016;36(12):1755–64
Koga H, Ide T, Oho K, Kuwahara R, Hino T, Ogata K, et al. Lamivudine treatment-related morphological changes of esophageal varices in patients with liver cirrhosis. Hepatol Res 2007;37(7):503–9
Lampertico P, Invernizzi F, Viganò M, Loglio A, Mangia G, Facchetti F, et al. The long-term benefits of nucleos(t)ide analogs in compensated HBV cirrhotic patients with no or small esophageal varices: a 12-year prospective cohort study. J Hepatol 2015;63(5):1118–25
Jang JW, Choi JY, Kim YS, Woo HY, Choi SK, Lee CH, et al. Long-term effect of antiviral therapy on disease course after decompensation in patients with hepatitis B virus-related cirrhosis. Hepatology 2015;61(6):1809–20
Manolakopoulos S, Triantos C, Theodoropoulos J, Vlachogiannakos J, Kougioumtzan A, Papatheodoridis G, et al. Antiviral therapy reduces portal pressure in patients with cirrhosis due to HBeAg-negative chronic hepatitis B and significant portal hypertension. J Hepatol 2009;51(3):468–74
Bruno S, Crosignani A, Facciotto C, Rossi S, Roffi L, Redaelli A, et al. Sustained virologic response prevents the development of esophageal varices in compensated, child-pugh class A hepatitis C virus-induced cirrhosis. A 12-year prospective follow-up study. Hepatology 2010;51(6):2069–76
Di Marco V, Calvaruso V, Ferraro D, Bavetta MG, Cabibbo G, Conte E, et al. Effects of eradicating hepatitis C virus infection in patients with cirrhosis differ with stage of portal hypertension. Gastroenterology 2016;151(1):130-139.e2
Lens S, Rincón D, Albillos A, Calleja L, Bañares R, González-abraldes J, et al. Association between severe portal hypertension and risk of liver decompensation in patients with hepatitis C, regardless of response to antiviral therapy. Clin Gastroenterol Hepatol 2015;13:1046–53
Rincon D, Ripoll C, Iacono OL, Salcedo M, Catalina MV, Alvarez E, et al. Antiviral therapy decreases hepatic venous pressure gradient in patients with chronic hepatitis C and advanced fibrosis. Am J Gastroenterol 2006;18:2269–74
Roberts S, Gordon A, Mclean C, Pedersen J, Bowden S, Thomson K, et al. Effect of sustained viral response on hepatic venous pressure gradient in hepatitis C—related cirrhosis. Clin Gastroenterol Hepatol 2007;5:932–7
D’Ambrosio R, Aghemo A, Rumi MG, Ronchi G, Donato MF, Paradis V, et al. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology 2012;56(2):532–43
Casado JL, Quereda C, Moreno A, Pérez-Elías MJ, Martí-Belda P, Moreno S. Regression of liver fibrosis is progressive after sustained virological response to HCV therapy in patients with hepatitis C and HIV coinfection. J Viral Hepat 2013;20(12):829–37
Pan JJ, Bao F, Du E, Skillin C, Frenette CT, Waalen J, et al. Morphometry confirms fibrosis regression from sustained virologic response to direct-acting antivirals for hepatitis C. Hepatol Commun 2018;2(11):1320–30
Lens S, Alvarado-Tapias E, Mariño Z, Londoño MC, LLop E, Martinez J, et al. Effects of all-oral anti-viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus-associated cirrhosis. Gastroenterology 2017;153(5):1273–83
Schwabl P, Mandorfer M, Steiner S, Scheiner B, Chromy D, Herac M, et al. Interferon-free regimens improve portal hypertension and histological necroinflammation in HIV/HCV patients with advanced liver disease. Aliment Pharmacol Ther 2017;45(1):139–49
Mandorfer M, Kozbial K, Schwabl P, Freissmuth C, Schwarzer R, Stern R, et al. Sustained virologic response to interferon-free therapies ameliorates HCV-induced portal hypertension. J Hepatol 2016;65(4):692–9
Mauro E, Crespo G, Montironi C, Londoño MC, Hernández-Gea V, Ruiz P, et al. Portal pressure and liver stiffness measurements in the prediction of fibrosis regression after sustained virological response in recurrent hepatitis C. Hepatology 2018;67(5):1683–94
Winters A, Luedtke S, Moreland A, Jangouk P, Weng G, Silveira M, et al. In hepatitis C virus-related advanced fibrosis and cirrhosis, early decline of liver stiffness following antiviral therapy with DAAs is related to decline in liver inflammation. J Hepatol 2017;66(1):S280
Afdhal N, Everson GT, Calleja JL, McCaughan GW, Bosch J, Brainard DM, et al. Effect of viral suppression on hepatic venous pressure gradient in hepatitis C with cirrhosis and portal hypertension. J Viral Hepat 2017;24(10):823–31
Mandorfer M, Kozbial K, Schwabl P, Chromy D, Semmler G, Stättermayer AF, et al. Changes in hepatic venous pressure gradient predict hepatic decompensation in patients who achieved sustained virologic response to interferon-free therapy. Hepatology 2020;71(3):1023–36
Lens S, Baiges A, Alvarado E, LLop E, Martinez J, Fortea JI, et al. Clinical outcome and hemodynamic changes following HCV eradication with oral antiviral therapy in patients with clinically significant portal hypertension. J Hepatol 2020;73:1415–24
Garg G, Dixit VK, Shukla SK, Singh SK, Sachan S, Tiwari A, et al. Impact of direct acting antiviral drugs in treatment Naïve HCV cirrhosis on fibrosis and severity of liver disease: a real life experience from a tertiary care center of North India. J Clin Exp Hepatol 2018;8(3):241–9
Carrat F, Fontaine H, Dorival C, Simony M, Diallo A, Hezode C, et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: a prospective cohort study. Lancet 2019;393(10179):1453–64
Belli LS, Perricone G, Adam R, Cortesi PA, Strazzabosco M, Facchetti R, et al. Impact of DAAs on liver transplantation: Major effects on the evolution of indications and results. An ELITA study based on the ELTR registry. J Hepatol 2018;69(4):810–7
Manco M, Mosca A, De Peppo F, Caccamo R, Cutrera R, Giordano U, et al. The benefit of sleeve gastrectomy in obese adolescents on nonalcoholic steatohepatitis and hepatic fibrosis. J Pediatr 2017;180(31–37):e2
Parker BM, Wu J, You J, Barnes DS, Yerian L, Kirwan JP, et al. Reversal of fibrosis in patients with nonalcoholic steatohepatosis after gastric bypass surgery. BMC Obes 2017;4(1):1–9
Lassailly G, Caiazzo R, Buob D, Pigeyre M, Verkindt H, Labreuche J, et al. Bariatric surgery reduces features of non-alcoholic steatohepatitis in morbidly obese patients. Gastroenterology 2015;149:379–88
Berzigotti A, Albillos A, Villanueva C, Genescá J, Ardevol A, Augustín S, et al. Effects of an intensive lifestyle intervention program on portal hypertension in patients with cirrhosis and obesity: the SportDiet study. Hepatology 2017;65(4):1293–305
Macías-rodríguez RU, Ilarraza-lomelí H, Ruiz-margáin A. Changes in hepatic venous pressure gradient induced by physical exercise in cirrhosis: results of a pilot randomized open clinical trial. Clin Transl Gastroenterol 2016;7:e180
Wanless I, Stueck A. The mechanism of irreversibility of late stage cirrhosis. J Hepatol 2018;2018(68):S692
Garcia-Tsao G, Friedman S, Iredale J, Pinzani M. Now there are many (stages) where before there was one: in search of a pathophysiological classification of cirrhosis. Hepatology 2010;51:1445–9
Fallowfield J, Hayes P. Pathogenesis and treatment of hepatic fibrosis: is cirrhosis reversible? Clin Med (Lond) 2011;11(2):179–83
Puente A, Fortea JI, Cabezas J, Arias Loste MT, Iruzubieta P, Llerena S, et al. LOXL2-A new target in antifibrogenic therapy? Int J Mol Sci 2019;20(7):1634
Popov Y, Sverdlov DY, Sharma AK, Bhaskar KR, Li S, Freitag TL, et al. Tissue transglutaminase does not affect fibrotic matrix stability or regression of liver fibrosis in mice. Gastroenterology 2011;140(5):1642–52
Pellicoro A, Aucott RL, Ramachandran P, Robson AJ, Fallowfield JA, Snowdon VK, et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology 2012;55(6):1965–75
Iredale JP, Thompson A, Henderson NC. Extracellular matrix degradation in liver fibrosis: biochemistry and regulation. Biochim Biophys Acta 2013;1832(7):876–83
Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol Nat Research 2020
Issa R, Zhou X, Trim N, Millward-Sadler H, Krane S, Benyon C, et al. Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. FASEB J 2003;17(1):47–9
Guixé-Muntet S, Ortega-Ribera M, Wang C, Selicean S, Andreu I, Kechagia JZ, et al. Nuclear deformation mediates liver cell mechanosensing in cirrhosis. JHEP Rep 2020;2(5):100145
Yoshiji H. Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology 2002;36(4):850–60
Murphy FR, Issa R, Zhou X, Ratnarajah S, Nagase H, Arthur MJP, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition. Implications for reversibility of liver fibrosis. J Biol Chem 2002;277(13):11069–76
Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64(5):830–41
Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol 2009;50:604–20
Saffioti F, Pinzani M. Development and regression of cirrhosis. Dig Dis 2016;34:374–81
Wanless IR. The role of vascular injury and congestion in the pathogenesis of cirrhosis: the congestive escalator and the parenchymal extinction sequence. Curr Hepatol Rep 2020;19(1):40–53
Hsu SJ, Tsai MH, Chang CC, Hsieh YH, Huang HC, Lee FY, et al. Extrahepatic angiogenesis hinders recovery of portal hypertension and collaterals in rats with cirrhosis resolution. Clin Sci 2018;132(6):669–83
Maeso-Díaz R, Ortega-Ribera M, Lafoz E, JoséLozano J, Baiges A, Francés R, et al. Aging influences hepatic microvascular biology and liver fibrosis in advanced chronic liver disease. Aging Dis 2019;10(4):684–98
Delire B, Lebrun V, Selvais C, Henriet P, Bertrand A, Horsmans Y, et al. Aging enhances liver fibrotic response in mice through hampering extracellular matrix remodeling. Aging (Albany NY) 2017;9(1):98–113
Pinzani M. Liver fibrosis in the post-HCV era. Semin Liver Dis 2015;35(2):157–65
Guixé-Muntet S, Zhu C-P, Xie W-F, Gracia-Sancho J. Novel therapeutics for portal hypertension and fibrosis in chronic liver disease. Pharmacol Ther 2020;215:107626
Zafra C, Abraldes JG, Turnes J, Berzigotti A, Fernández M, García-Pagán JC, et al. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology 2004;126(3):749–55
Abraldes JG, Albillos A, Bañares R, Turnes J, González R, García-Pagán JC, et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology 2009;136(5):1651–8
Pollo-Flores P, Soldan M, Santos UC, Kunz DG, Mattos DE, da Silva AC, et al. Three months of simvastatin therapy vs. placebo for severe portal hypertension in cirrhosis: a randomized controlled trial. Dig Liver Dis 2015;47(11):957–63
Abraldes JG, Villanueva C, Aracil C, Turnes J, Hernandez-Guerra M, Genesca J, et al. Addition of simvastatin to standard therapy for the prevention of variceal rebleeding does not reduce rebleeding but increases survival in patients with cirrhosis. Gastroenterology 2016;150(5):1160-1170.e3
Wani ZA, Mohapatra S, Khan AA, Mohapatra A, Yatoo GN. Addition of simvastatin to carvedilol non responders: a new pharmacological therapy for treatment of portal hypertension. World J Hepatol 2017;9(5):270–7
Vijayaraghavan R, Jindal A, Arora V, Choudhary A, Kumar G, Sarin SK. Hemodynamic effects of adding simvastatin to carvedilol for primary prophylaxis of variceal bleeding. Am J Gastroenterol 2020;115:729–37
U.S. National Library of Medicine; NCT03654053
U.S. National Library of Medicine; NCT03150459
Pelusi S, Petta S, Rosso C, Borroni V, Fracanzani L, Dongiovanni P, et al. Renin-angiotensin system inhibitors, type 2 diabetes and fibrosis progression: an observational study in patients with nonalcoholic fatty liver disease. PLoS ONE 2016;11:e0163069
Abu Dayyeh BK, Yang M, Dienstag JL, Chung RT. The effects of angiotensin blocking agents on the progression of liver fibrosis in the HALT-C Trial cohort. Dig Dis Sci 2011;56(2):564–8
Tandon P, Abraldes JG, Berzigotti A, Garcia-pagan JC, Bosch J. Renin–angiotensin–aldosterone inhibitors in the reduction of portal pressure: a systematic review and meta-analysis research article angiotensinogen renin ACE inhibitors portal hypertension angiotensin aldosterone antagonists aldosterone. J Hepatol 2010;53(2):273–82
Deibert P, Schumacher YO, Ruecker G, Opitz OG, Blum HE, Rössle M, et al. Effect of vardenafil, an inhibitor of phosphodiesterase-5, on portal haemodynamics in normal and cirrhotic liver—Results of a pilot study. Aliment Pharmacol Ther 2006;23(1):121–8
Kreisel W, Deibert P, Kupcinskas L, Sumskiene J, Appenrodt B, Roth S, et al. The phosphodiesterase-5-inhibitor udenafil lowers portal pressure in compensated preascitic liver cirrhosis. A dose-finding phase-II-study. Dig Liver Dis 2015;47(2):144–50
Lee KC, Yang YY, Wang YW, Hou MC, Lee FY, Lin HC, et al. Acute administration of sildenafil enhances hepatic cyclic guanosine monophosphate production and reduces hepatic sinusoid resistance in cirrhotic patients. Hepatol Res 2008;38(12):1186–93
Tandon P, Inayat I, Tal M, Spector M, Shea M, Groszmann RJ, et al. Sildenafil has no effect on portal pressure but lowers arterial pressure in patients with compensated cirrhosis. Clin Gastroenterol Hepatol 2010;8(6):546–9
Fukuda T, Narahara Y, Kanazawa H, Matsushita Y, Kidokoro H, Itokawa N, et al. Effects of fasudil on the portal and systemic hemodynamics of patients with cirrhosis. J Gastroenterol Hepatol 2014;29(2):325–9
Sitbon O, Bosch J, Cottreel E, Csonka D, De GP, Hoeper MM, et al. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir 2019;7(7):594–604
Lebrec D, Bosch J, Jalan R, Dudley FJ, Jessic R, Moreau R, et al. Hemodynamics and pharmacokinetics of tezosentan, a dual endothelin receptor antagonist, in patients with cirrhosis. Eur J Clin Pharmacol 2012;68:533–41
Disease L. Endothelin-1 contributes to maintenance of systemic but not portal haemodynamics in patients with early cirrhosis: a randomised controlled trial. Gut 2006;55:1290–5
Ling L, Kuc RE, Maguire JJ, Davie NJ, Webb DJ, Gibbs P, et al. Comparison of endothelin receptors in normal versus cirrhotic human liver and in the liver from endothelial cell-specific ETB knockout mice. In: Life Sciences. Pergamon; 2012. p. 716–22
Zipprich A, Gittinger F, Winkler M, Dollinger MM, Ripoll C. Effect of ET-A blockade on portal pressure and hepatic arterial perfusion in patients with cirrhosis: a proof of concept study. Liver Int 2020. https://doi.org/10.1111/liv.14757
U.S. National Library of Medicine; NCT03827200
Reverter E, Mesonero F, Seijo S, Martínez J, Abraldes JG, Peñas B, et al. Effects of sapropterin on portal and systemic hemodynamics in patients with cirrhosis and portal hypertension : a bicentric double-blind placebo-controlled study. Am J Gastroenterl. 2015;110:98592
Gifford FJ, Dunne PDJ, Weir G, Ireland H, Graham C, Tuck S, et al. A phase 2 randomised controlled trial of serelaxin to lower portal pressure in cirrhosis (STOPP). Trials 2020;21(1):1–10
Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology 2020;158(5):1334-1345.e5
U.S. National Library of Medicine; NCT04365868
Mookerjee R, Rosselli M, Pieri G, Beecher-Jones T, Hooshmand-Rad R, Chouhan M, et al. Effects of the FXR agonist obeticholic acid on hepatic venous pressure gradient (HVPG) in alcoholic cirrhosis: a proof of concept phase 2a study. J Hepatol 2014;60(1):S7–8
Pinter M, Sieghart W, Reiberger T, Ferlitsch A. Alimentary pharmacology and therapeutics the effects of sorafenib on the portal hypertensive syndrome in patients with liver cirrhosis and hepatocellular carcinoma—a pilot study. Aliment Pharmacol Ther 2011;35:83–91
Schwarzer R, Kivaranovic D, Mandorfer M, Paternostro R, Heinisch B, Wolrab D, Reiberger T, Ferlitsch M, et al. Randomised clinical study: the effects of oral taurine 6/day g vs placebo daily on portal hypertension. Aliment Pharmacol Ther 2018;47:86–94
Rincón D, Vaquero J, Hernando A, Galindo E, Ripoll C, Puerto M, et al. Oral probiotic VSL#3 attenuates the circulatory disturbances of patients with cirrhosis and ascites. Liver Int 2014;34(10):1504–12
Jayakumar S, Carbonneau M, Hotte N, Befus AD, St. Laurent C, Owen R, et al. VSL#3® probiotic therapy does not reduce portal pressures in patients with decompensated cirrhosis. Liver Int 2013;33(10):1470–7
Gupta N, Kumar A, Sharma P, Garg V, Sharma BC, Sarin SK. Effects of the adjunctive probiotic VSL#3 on portal haemodynamics in patients with cirrhosis and large varices: a randomized trial. Liver Int 2013;33(8):1148–57
De Gottardi A, Berzigotti A, Seijo S, D’Amico M, Thormann W, Abraldes JG, et al. Postprandial effects of dark chocolate on portal hypertension in patients with cirrhosis: results of a phase 2, double-blind, randomized controlled trial. Am J Clin Nutr 2012;96(3):584–90
Hernández-Guerra M, García-Pagán JC, Turnes J, Bellot P, Deulofeu R, Abraldes JG, et al. Ascorbic acid improves the intrahepatic endothelial dysfunction of patients with cirrhosis and portal hypertension. Hepatology 2006;43(3):485–91
Meissner EG, McLaughlin M, Matthews L, Gharib AM, Wood BJ, Levy E, et al. Simtuzumab treatment of advanced liver fibrosis in HIV and HCV-infected adults: results of a 6-month open-label safety trial. Liver Int 2016;36(12):1783–92
Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology 2018;155(4):1140–53
Muir AJ, Levy C, Janssen HLA, Montano-Loza AJ, Shiffman ML, Caldwell S, et al. Simtuzumab for primary sclerosing cholangitis: phase 2 study results with insights on the natural history of the disease. Hepatology 2019;69(2):684–98
Chen W, Yang A, Jia J, Popov YV, Schuppan D, You H. Lysyl oxidase (LOX) family members: rationale and their potential as therapeutic targets for liver fibrosis. Hepatology 2020;72:729–41
Garcia-Tsao G, Fuchs M, Shiffman M, Borg BB, Pyrsopoulos N, Shetty K, et al. Emricasan (IDN-6556) lowers portal pressure in patients with compensated cirrhosis and severe portal hypertension. Hepatology 2019;69(2):717–28
Garcia-Tsao G, Bosch J, Kayali Z, Harrison SA, Abdelmalek MF, Lawitz E, et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J Hepatol 2020;72(5):885–95
Albillos A. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology 2003;37(1):208–17
Rasaratnam B, Kaye D, Jennings G, Dudley F, Chin-Dusting J. The effect of selective intestinal decontamination on the hyperdynamic circulatory state in cirrhosis. Ann Intern Med 2003;139(3):186
Kemp W, Colman J, Thompson K, Madan A, Vincent M, Chin-Dusting J, et al. Norfloxacin treatment for clinically significant portal hypertension: results of a randomised double-blind placebo-controlled crossover trial. Liver Int 2009;29(3):427–33
Vlachogiannakos J, Saveriadis AS, Viazis N, Theodoropoulos I, Foudoulis K, Manolakopoulos S, et al. Intestinal decontamination improves liver haemodynamics in patients with alcohol-related decompensated cirrhosis. Aliment Pharmacol Ther 2009;29(9):992–9
Vlachogiannakos J, Viazis N, Vasianopoulou P, Vafiadis I, Karamanolis DG, Ladas SD. Long-term administration of rifaximin improves the prognosis of patients with decompensated alcoholic cirrhosis. J Gastroenterol Hepatol 2013;28(3):450–5
Kimer N, Pedersen JS, Busk TM, Gluud LL, Hobolth L, Krag A, et al. Rifaximin has no effect on hemodynamics in decompensated cirrhosis: a randomized, double-blind, placebo-controlled trial. Hepatology 2017;65(2):592–603
Lim YL, Kim MY, Jang YO, Baik SK, Kwon SO. Rifaximin and propranolol combination therapy is more effective than propranolol monotherapy for the reduction of portal pressure: an open randomized controlled pilot study. Gut Liver 2017;11(5):702–10
Moreau R, Elkrief L, Bureau C, Perarnau JM, Thévenot T, Saliba F, et al. Effects of long-term norfloxacin therapy in patients with advanced cirrhosis. Gastroenterology 2018;155(6):1816-1827.e9
Villa E, Cammà C, Marietta M, Luongo M, Critelli R, Colopi S, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology 2012;143(5):1253-1260.e4
U.S. National Library of Medicine; NCT02643212
Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016;387:679–90
Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized Ph III STELLAR trials. J Hepatol 2020;73:26–39
Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018;67(5):1754–67
U.S. National Library of Medicine; NCT03028740
U.S. National Library of Medicine; NCT03517540
Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362(18):1675–85
Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. The pan-PPAR agonist lanifibranor induces both resolution of NASH and regression of fibrosis after 24 weeks of treatment in non-cirrhotic NASH: results of the NATIVE phase 2b TRIAL Sven. Hepatology 2020;72(S1):1–1159
Newsome PN, Fox R, King AL, Barton D, Than NN, Moore J, et al. Granulocyte colony-stimulating factor and autologous CD133-positive stem-cell therapy in liver cirrhosis (REALISTIC): an open-label, randomised, controlled phase 2 trial. Lancet Gastroenterol Hepatol 2018;3(1):25–36
Qi X, Berzigotti A, Cardenas A, Sarin SK. Review emerging non-invasive approaches for diagnosis and monitoring of portal hypertension. Lancet Gastroenterol Hepatol 2018;3(10):708–19
Castera L. Non-invasive tests for liver fibrosis progression and regression. J Hepatol 2016;64(1):232–3
Sayyar M, Saidi M, Zapatka S, Deng Y, Ciarleglio M, Garcia-Tsao G. Platelet count increases after viral elimination in chronic HCV, independent of the presence or absence of cirrhosis. Liver Int 2019;39(11):2061–5
Ghosh Laskar M, Eriksson M, Rudling M, Angelin B. Treatment with the natural FXR agonist chenodeoxycholic acid reduces clearance of plasma LDL whilst decreasing circulating PCSK9, lipoprotein(a) and apolipoprotein C-III. J Intern Med 2017;281(6):575–85
Radu C, Stancu O, Sav R, Bugariu A, Suciu A, Grigoras C, et al. Liver stiffness better predicts portal hypertension after HCV eradication. J Gastrointestin Liver Dis 2018;27:201–5
Ravaioli F, Colecchia A, Dajti E, Marasco G, Alemanni LV, Tamè M, et al. Spleen stiffness mirrors changes in portal hypertension after successful interferon-free therapy in chronic-hepatitis C virus patients. World J Hepatol 2018;27(10):731–42
Kim HY, So YH, Kim W, Ahn DW, Jung YJ, Woo H, et al. Non-invasive response prediction in prophylactic carvedilol therapy for cirrhotic patients with esophageal varices. J Hepatol 2019;70(3):412–22. https://doi.org/10.1016/j.jhep.2018.10.018
Thabut D, Bureau C, Layese R, Bourcier V, Hammouche M, Cagnot C, et al. Validation of Baveno VI criteria for screening and surveillance of esophageal varices in patients with compensated cirrhosis and a sustained response to antiviral therapy. Gastroenterology 2019;156(4):997-1009.e5
Karsdal MA, Hjuler ST, Luo Y, Rasmussen DGK, Nielsen MJ, Nielsen SH, et al. Assessment of liver fibrosis progression and regression by a serological collagen turnover profile. Am J Physiol 2019;316(1):G25–31
Jansen C, Leeming DJ, Mandorfer M, Byrjalsen I, Schierwagen R, Schwabl P, et al. PRO-C3-levels in patients with HIV/HCV-Co-infection reflect fibrosis stage and degree of portal hypertension. PLoS ONE 2014;9(9):e108544
Leeming DJ, Karsdal MA, Byrjalsen I, Bendtsen F, Trebicka J, Nielsen MJ, et al. Novel serological neo-epitope markers of extracellular matrix proteins for the detection of portal hypertension. Aliment Pharmacol Ther 2013;38(9):1086–96
Nevzorova YA, Boyer-Diaz Z, Cubero FJ, Gracia-Sancho J. Animal models for liver disease – A practical approach for translational research. J Hepatol 2020;73(2):423–40
Acknowledgements
S.S. is recipient of a Juan Rodés fellowship from the European Association for the Study of the Liver (EASL). S.G.-M. has a Sara Borrell Contract from the Instituto de Salud Carlos III.
Funding
Open Access funding provided by Universität Bern. J.G.-S. acknowledges funding from the Swiss National Science Foundation (SNF 320030_189252/1), the Swiss Foundation against Liver Cancer, the Spanish Ministry of Science and Innovation—Instituto de Salud Carlos III (FIS PI20/0220), European Union FEDER Funds, “una manera de hacer Europa”, AGAUR 2017-SGR-517 and the CERCA Program from the Generalitat de Catalunya. CIBEREHD is funded by Instituto de Salud Carlos III.
Author information
Authors and Affiliations
Contributions
S.S., C.W., S.G.-M. bibliography search, draft writing and preparation of figures and tables; H.S. and N.K. critical revision of work; J.G.-S. conceived, wrote and critically revised the work.
Corresponding author
Ethics declarations
Conflict of interest
The author(s) declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Selicean, S., Wang, C., Guixé-Muntet, S. et al. Regression of portal hypertension: underlying mechanisms and therapeutic strategies. Hepatol Int 15, 36–50 (2021). https://doi.org/10.1007/s12072-021-10135-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-021-10135-4