
Abstract
The Tree of Life is the result of the interplay of changes in information and speciation. Almost 100 years after publication of Darwin’s Origin, the inception of Phylogenetic Systematics has resulted in a revolution in data inference. I briefly trace the development of this revolution and show examples of how data are interpreted relative to phylogenetic trees. I then provide brief discussions of how to read tree diagrams and the need to access the quality of phylogenetic inference.
Similar content being viewed by others


As a first principle, we adopt the Darwinian idea that all life is related. Life is diverse, being composed of many species, not one. So while there may have been only one line of descent initially, there are now many lines of descent, many “families” reproducing through time. This means that evolution is not simply “change through time,” although it certainly is that; it means, minimally, that speciation is also occurring such that lines of descent are divided by various processes into two or more lines of descent which can then follow their own, independent, evolutionary pathways. I say minimally because speciation mechanisms are diverse, sometimes two lineages found a third through other mechanisms, or one lineage spins off a new lineage through other processes. So we can conceive of the Tree of Life in nature as a diverging hierarchy of lineages composed of one or more populations with a few too many individual organisms, with most of the divergence being caused by the establishment of new lineages through speciation. Thus there are two general processes at work in evolutionary descent. One is change in information; ultimately change in the genetic code and how genes interact during development. When played out over time, this general process is termed “anagenesis” and the mechanisms include natural selection, sexual selection, and genetic drift operating on single evolving lineages. The other general process is speciation, the origin of new species. Although speciation can take many forms (various modes of speciation), these forms involve the establishment of two or more lineages where only a single lineage existed before: an ancestral species gives rise to daughter species through lineage splitting. This lineage splitting has been called “cladogenesis” and this is the origin of the term “cladist.”
Over the past 40 or so years, a revolution has occurred in the way that many biologists look at data. The revolution is fairly simple but profound. Data are interpreted relative to trees of descent which are the inferred genealogical relationships of entities linked by history. From this perspective, the data are dynamic; information changes through time, and these changes can be studied by following lines of genealogical descent. Trees can depict our hypotheses of the histories of individual organisms, populations, genes, proteins, morphological characters, developmental patterns, species, groups of species, and even areas of the Earth. In short, trees can convey our ideas of the historical relationships that exist among entities that share a common history and serve to organize and summarize where and how information has changed during historical descent.
In evolutionary biology, the more common kind of tree portrays the inferred evolutionary histories of species. They represent attempts to estimate the macroscopic properties of the Tree of Life, the genealogical nexus that ties together all of the living organisms on Earth. Such trees have been around for some time; Darwin (1859) included one as the only illustration in the Origin. However, it took more than 100 years for biologists to put together a coherent and logical methodology that allows them both to consistently estimate the Tree of Life and to estimate it in a manner that can be tested by new data in a rigorous manner. This is the “long march” from Darwin to Hennig, who proposed an integrated framework for the research program.
The Phylogenetics Revolution
From 1859, when Darwin first published a hypothetical genealogy of species, until just after World War II, there was no unified method for reconstructing the genealogical relationship among species. This is not to say there were no attempts to do so or that there were no trees; rather, that there was a lack of empirical rigor in the way those trees were formulated. For the most part, trees represented scientists’ opinions, based on their experience. Experience is frequently a good guide, but it lacks a mechanism for independent confirmation using new data from other sources or a consistent way to resolve conflicting ideas. Building on the work of such biologists as Karl Zimmerman (1943), the German entomologist Willi Hennig began synthesizing a method of reconstructing phylogenetic relationships before World War II and published his first synthesis, in German, in 1950. This received a bit of attention (e.g., Simpson 1961 mentions the work) but was overshadowed by the “phenetics revolution” (e.g., Sokal and Michener 1958) in the U.S. until the publication of his second synthetic work, Phylogenetic Systematics, in English, in 1966. In the U.S., this book caught the attention of a core of future phylogeneticists lead by Gareth Nelson of the American Museum of Natural History. Hennig seems to have thought himself a Darwinian, and his method as firming up basic Darwinian principles, forging a method of reconstructing phylogenies, and bringing Darwinian principles to the classification of organisms. Hennig’s basic ideas are fairly simple.
-
1.
“Relationship” in the Darwinian sense means genealogical relationship. It does not mean anything like the pre-Darwinian ideas of “similarity” or conformation to an ideal type.
-
2.
Darwinian classifications are purely genealogical. Historians and biologists who think they can interpret history argue over whether Darwin advocated purely genealogical classifications. It appears he did (Ghiselin 2004), but how to translate that thought into a functioning system has taken over 150 years, and we are still working on it. Here are the problems. First, one had to develop a methodology to consistently reconstruct phylogeny in a way that we could argue about different hypotheses in a rigorous manner, without appeal to “authority.” This did not happen in a consistent manner until the rise of phylogenetic thinking brought on by Hennig and his advocates some 100+ years after the publication of the Darwin/Wallace thesis. We should not forget that Hennig built on the work of others, in particular Othenio Abel, Adolf Naef, and Walter Zimmermann (Willmann 2003). But it was left to Hennig to forge the now accepted principles of classification used by phylogeneticists today. Second, there was the pervasive idea, a holdover of pre-Darwininan thinking, that classifications could be based on similarity even at the expense of what we think we know about phylogenetic relationships. Third, a particular term, “monophyly,” was as confused as the term “homology.” Interestingly, the second problem is wrapped up in the third problem, discussed below.
-
3.
“Similarity” is a complex concept requiring parsing. There is nothing wrong with similarity per se, but we must parse out similarity that denotes unique, immediate, common ancestry from similarity that denotes ancient common ancestry from similarity unrelated to common ancestry (i.e., similarity due to convergence). Homology is basically similarity due to descent of information from a common ancestor to its descendants, and sharing homologous similarities may signal unique ancestry or it may signal more ancient ancestry. For example, hair is homologous in horses and humans, and toes are homologous in horses, humans, and lizards. When we ask if horses are more closely related to humans than to lizards, we would answer “yes” because hair originated in the common ancestor of horses and humans but not in the common ancestor of all three species. When we ask if humans are more closely related to lizards than to horses because humans and lizards have multiple toes while (living) horses have only one toe, we would answer “no,” because having multiple toes is found in the common ancestor of humans, horses, and lizards, not simply in the common ancestor of humans and lizards. There is nothing wrong with the homology of human and lizard toes; it is just that this particular homology originated in an earlier ancestor, an ancestor that was common to lizards, horses, and humans. It signals a more ancient ancestor, one common to salamanders as well as lizards, horses, and humans. Since we think that the homologous similarity of having multiple digits arose once in evolutionary descent, we use it only once, at the level signaling the common ancestry of all tetrapods.
Hennig (1966) used a particular set of terms to describe homologous characters. Characters that demonstrated a unique common ancestry relative to other organisms in analysis (humans+horses versus lizards+humans) were termed apomorphic characters or apomorphic homologies. Organisms that had such characters were said to share a synapomorphy. Homologous characters that denoted a deeper relationship (humans+horses+lizards) were termed plesiomorphic characters at that level of inquiry. Lizards and humans share a symplesiomorphy, multiple digits, when we also consider horses in the mix. It is important to understand that these are relative terms. The common ancestor of all tetrapods, which includes horses, humans, lizards, salamanders, turtles, dinosaurs, etc. as well as some advanced lobe-finned fishes, is hypothesized to have multiple digits attached to legs. A more ancient common ancestor, the ancestor of tetrapods and bony fishes, had only fin rays and fins. An even more ancient ancestor, the ancestor of sharks, bony fishes, and tetrapods, also had fins with rays. Unless we are quite wrong about the relationships, we can conclude that sharks and bony fishes share the homology of having fins. Relative to having legs, the presence of fins is a symplesiomorphy of sharks and bony fishes, a “shared primitive character.” Relative to having fins, having legs is a synapomorphy of lizards, horses, and humans, a “shared advanced character.” Deeper in the phylogeny, having fins is a synapomorphy of jawed vertebrates. The ancestor of sharks, bony fishes, tetrapods, etc. is thought to have had fins. Having a relatively unmodified body wall is a plesiomorphy of lampreys and a symplesiomorphy of lampreys and hagfishes. So, apomorophy and plesiomorphy are relative terms; they describe the dynamics of character change of homologous features over the phylogeny. The unmodified body wall of lampreys was transformed by changes in information (probably using the same genes in different ways during development) to fins in some (unknown at this point) ancestor that gave rise to jawed vertebrates. Fins were transformed to limbs with multiple digits in the ancestor of tetrapods and their closest lobefin relatives, the multiple digits of mammals and early horses were transformed into the single digit we see today in the ancestor of our living species of horses.
It is also important to note that Hennig was not the first to understand this distinction. The importance of parsing homologous characters into those that denoted unique common ancestry and those that denoted more ancient common ancestry was recognized by several workers in the early half of the twentieth century. Willmann (2003) provides a detailed account of the early development of phylogenetics and points out many of the contributions of Hennig’s predecessors such as Sinai Tschulok, who provided criteria for parsing primitive and derived characters and the idea that it was the characters that are primitive and derived and not the whole organism (see Willmann 2003 and Rieppel 2010, for discussions of Tschulok’s contributions). But it was Hennig who melded these concepts and brought them to a wider audience.
-
4.
Monophyly is strict. Before Hennig, “monophyly” was applied inexactly. We had two commonly used terms, “monophyly” and “polyphyly” just as we had two terms “homology” and “convergence.” Everyone agreed that polyphyly was bad because the characters that support a polyphyletic group are known to be convergent. Mammals and birds are both warm-blooded, but they gained this character independently. Homeothermia is a class based on convergence. However, few took note of the fact that “monophyletic groups” could be based on either plesiomorphies (Pisces, with fins) or apomorphies (Tetrapods, with legs). The distinction between these two kinds of homologous characters was largely unrecognized. This created a tension: how do you justify calling a group “monophyletic?” There were no less than three reactions.
Pheneticists advocated abandoning the pursuit of phylogeny reconstruction and monophyly entirely (Sneath and Sokal 1973). Simply group by some measure of overall similarity and be done with it. This didn’t work for two reasons. First, pheneticists could not agree among themselves as to exactly what constituted a measure of overall similarity; there were simply too many measures from which to choose. Second, there is no standard by which one could judge the resulting classifications. Is a 70% difference the mark of one genus from another, or is it 85% dissimilarity? And, of course, there was a third reason. Who would be interested in phenograms (trees of overall similarity) when one could work with phylogenetic trees (trees of genealogy)? If we can reconstruct phylogeny, such trees are much more useful as prediction machines (see examples below) because they parse homology and convergence, which phenograms cannot accomplish.
The “old guard,” evolutionary biologists such George Simpson (1961) and Ernst Mayr advocated a hybrid system (Mayr and Bock 2002). Some groups are groups of unique common ancestry, but other groups can exclude some descendants of a common ancestor if they are really different. The usual criterion for “really different” was the occupation of a unique adaptive zone. For example, birds have descended from the common ancestor of reptiles and birds. But birds are really distinctive; they fill an adaptive zone much different than the adaptive zone of, say, crocodiles. So they will be placed in their own class Aves, while reptiles will be placed in the class Reptilia. Humans have their own family, Hominidae, while their relatives, the great apes, are classified in a different family, Pongidae. But, there are problems with such “half-measures.” Without even being aware of Hennig’s work, David Hull (1964) pointed out that this approach resulted in classifications that were logically inconsistent (read illogical) with the phylogenies they were supposed to summarize. Inexplicably, Hull’s conclusions were largely ignored (but see Wiley 1981); yet, they form the necessary and sufficient conditions for rejecting the entire “school” of evolutionary taxonomy.
Hennig’s choice, made independently of Hull’s observations, was genealogy. The problem was that the commonly used term “monophyly” was a complex term. In some cases, a monophyletic group included an (inferred) ancestral species and all of its descendants; in other cases, a monophyletic group included an (inferred) ancestral species and only some of its descendants. Groups that include an ancestral species and only some of its descendants were, to Hennig, incomplete groups. The analogy is including your cousins but not your sister in your family. Hennig called such groups as Reptilia (excludes birds) and Pongidae (excludes humans) “paraphyletic,” while he called complete groups “monophyletic.” Using Hull’s choice (1964; also see Wiley, 1981), only classifications containing monophyletic groups and only monophyletic groups were logical classifications relative to the phylogenies they represent. Only these kinds of classifications were truly “Darwinian.” This attitude was expressed as early as 1919 by Naef who advocated dissolving “stem groups” into their component branches if one wished a strictly evolutionary classification (a step Naef did not take, fearing disruption of existing classifications; see Willmann 2003). Classifications, it would seem, can express some ideas, but not every idea you choose: do you wish to express similarity or genealogy? Take your choice. One route leads to phenetics, the other to phylogenetics. To put it bluntly, no one would argue with a pheneticist who claimed that his similarity tree was consistent with his phenetic classification, given the pheneticist’s own criteria of grouping by a particular measure of similarity. No one would argue with a phylogeneticist whose classification contained only monophyletic groups found in his phylogenetic tree. But when you mix the two, the result is an illogical system that does not fully cover either phenomenon (see Wiley 1981 for additional discussion).
Of What Use Are Phylogenetic Trees?
As dynamic hypotheses of genealogy and character change, phylogenetic trees can be used both to describe and understand character evolution and, as devices, to predict what we do not yet know. If Theodosius Dobzhansky (1973) was correct in stating that “nothing in biology makes sense except in the light of evolution,” and if all similarities and differences among organisms are the result of the evolutionary processes of cladogenesis (lineage splitting) and anagenesis (character change), then trees should be very useful to a wider audience. Indeed, “tree thinking” is beginning to be felt in many disciplines (see Baum and Offner 2008, for a perspective on tree thinking and the classroom). I illustrate some examples of the use of trees below. Two of these come directly from a review paper by Bull and Wichman (2001), a paper I highly recommend to educators and one that is required reading in my systematics course.
-
Case 1:
The origin of HIV in humans (from Bull and Wichman 2001). Retroviruses evolve and HIV is a notoriously fast evolving virus. There are actually two different forms, HIV-1 and HIV-2. By performing phylogenetic analysis on human HIV strains as well as HIV strains from a number of primate species, Gao et al. (1999) were able to demonstrate that HIV-1 was more closely related to the HIV strains in chimpanzees while Hahn et al. (2000) traced HIV-2 to the sooty mangabey monkey. Interestingly, HIV-2 is both less prevalent and less often fatal than HIV-1 in humans.
-
Case 2:
Diagnosing cancer. Abu-Asab et al. (2006) have proposed a novel way of diagnosing cancer through a combination of proteomics and phylogenetic analysis (“phyloproteomics”). The resulting phylogenetic analyses of three types of cancer (ovarian, prostate, and pancreatic) that included samples from non-cancerous individuals grouped all cancerous samples into one group, at the bottom was a healthy group or groups and in between are what Abu-Asb and colleagues call a transitional zone. This raises the exciting possibility of relatively simple diagnoses of cancers in very early stages of development since the cancers have a predictable phylogenetic position relative to healthy and cancerous samples. Note the power of using phylogenetics. Such analyses do not depend on a “magic bullet” approach to diagnosing a complex disease but rather using the history and evolution of the development of the serum proteins in cancer cells to provide a broad spectrum diagnostic tool.
-
Case 3:
Phylogenetics and the law (from Bull and Wichman 2001). In December 1994, the former mistress of a Louisiana physician was diagnosed with HIV and hepatitis C. She had tested negative only a few months before the diagnosis. She suspected that the physician was the source. Since he was HIV negative, the HIV had to come from another source, which turned out to be one of the physician’s patients, while another patient had hepatitis C. A phylogenetic analysis of the woman’s HIV DNA sequence clustered with another patients HIV sequence: the physician had used the tainted blood in a vitamin injection given to the mistress in August 1994. The physician is now serving a 50-year sentence for attempted murder. Just like the cancer example, the ultimate origin of the unfortunate women’s HIV viruses is not dependent on some sort of exact match with the original sample. HIV evolves rapidly enough that an exact match may or may not obtain. Rather, the outcome hinged on placing the woman’s HIV strain within the historical context of the evolution of HIV and showing the historical origin of her strain, which lay with the sample of another patient of the felonious physician.
-
Case 4:
Global climate change and the fate of species. Every species is associated with a complex set of environmental parameters that characterize its Grinnellian niche, which are essentially the general environmental parameters that allow the species to live and prosper. This niche is not some single set of parameters, such as a specific range of temperature and moisture, but a complex set of parameters than can vary geographically due to local adaptation. It can vary over time and space. Many of the broader parameters, such as maximum and minimum yearly temperature, total and seasonal rainfall, vegetation cover, and the like are those parameters subject to global climate change. Sets of these global environmental parameters can be successfully used to predict the potential niches of species and geographic information system technology can be used to project these predictions onto the surface of the Earth (for a good review, see Peterson 2003). This forms a prediction of where a species might potentially be found, its potential range. This is useful for all sorts of things, like prediction of the spread of invasive species. There are other uses when we consider the evolution of niches. Peterson et al. (1999) pointed out that the broader parameters of the Grinnellian niche are shared among closest taxonomic relatives. That is, these niches are conserved over speciation events and thus can be thousands, if not millions of years old and retained by the descendants of ancient ancestral species. McNyset (2009) modeled the dynamics of niche change over explicit phylogenies, demonstrating that this was not a taxonomic anomaly. The implication is clear: the broader aspects of species’ niches evolve slowly; the rate of change is slower than the speciation rate. This implies that the ability of species to adapt to phenomena such as global climate change may be very limited. We can feed the niche model of a species into a global climate change model and see where, in geographic space, the niche shifts in response to global climate change (Peterson et al. 2002).
Phylogenetic trees are so useful because they provide the historical narrative for explaining the similarities and differences among those entities placed on the tree. It is not so important that the DNA sequence of the HIV virus recovered from the victim exactly matches that of the former patient, what is important is that the two strains appear on the phylogenetic tree as more closely related that other HIV strains, indicating that they had a common origin. But, we must know exactly what information they convey.
The Tree of Life Versus Our Tree Hypotheses
When we draw a tree, we are attempting to capture a limited but accurate picture of the Tree of Life as it exists in nature. As such, trees are rather like highway maps that help us navigate along the path of evolutionary descent. All such trees have two things in common. First, they explicitly show ancestor and descendant relationships. Second, they all have a relative time axis. This makes them different from other kinds of graphs such as phenograms; there, the vertical axis is an axis of relative similarity, not time.
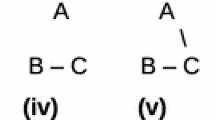
Figure 1 shows two basic kinds of tree diagrams. The one on the left (Fig. 1a) is what I term a “stem-based tree.” The ancestral species are symbolized by the lines (technically edges or internodes) and the branching points (technically nodes or vertices) are speciation events. This diagram shows that to account for the evolution of humans, chimps, and gorillas, we need a minimum of two speciation events and a minimum of two common ancestral species. Now, it is important to understand that two speciation events and two ancestors is the minimum number of speciation events and ancestors needed to account for these three species. It does not mean that these are all the ancestors in this part of the Tree of Life. In fact, as we add fossil chimps and fossil humans to our tree, we will add additional ancestors. It is also important to note that while the ancestors may be unsampled or unrecognized as ancestral species, they are not “hypothetical” in the sense that this term is commonly applied. To assert that ancestors are hypothetical is to assert that evolutionary descent itself is hypothetical. And, the monophyletic groups to which chimp and human are parts extend back to the split between the common ancestor of all chimps and humans which occurred after the split of the common ancestor of chimps, humans and gorillas. Finally, the common ancestor of chimps and humans is neither a chimp nor a human.

A hypothesis of relationships among gorillas, chimpanzees, and humans shown as two different, but complementary, tree graphs. a A phylogenetic tree. b A Hennig tree showing the identical genealogical relationships as (a) in alternative form. In (a), each lineage is traced back to a speciation event shown at each node. In (a), the ancestors (X and Y) are unsampled, encompass the entire lineage between speciation events, and represent only the minimum number of ancestors needed to account for descendant lineages. In (b), each ancestral lineage and descendant group is folded into a single node and the arrow lines represent statements of parent–child relationships, not lineages. In (b), speciation events are not shown but implied by the parent–child relationships. Two hypothetical synapomorphies uniting chimps and humans are placed on each tree graph. A similar mapping is shown in Hennig (1966)
The tree on the right shows exactly the same kind of relationship ancestors and descendants have, but it is organized differently. I term it a “node-based tree.” You could also term it a “Hennig tree” based on Hennig’s detailed description of the two kinds of tree he presented in Hennig (1966: see p. 59, Fig. 15 and p. 60, Fig. 15). It is much more like a human genealogy, turned upside down, with ancestors at nodes connected to descendants (children) at the tips. The edges are, symbolically, explicit statements of genealogical relationship, the equivalent of parent–child statements, just like a family tree of a human family except that there is usually only one parent. So, in Fig. 1b, we would read “X is the parent of gorillas and Y.” And we would read “gorillas are the children of X.”
I have garnished both trees with two hypothetical characters that are synapomorphies shared by humans and chimps, but not gorillas. In Fig. 1a, these are attached to the ancestral lineage, but do not be misled. Just because one is lower than two does not mean that we know that one arose before two. We do not even know if both characters arose in one ancestor or in two ancestors. The only sense of the plotting of these characters on Fig. 1a is that both were characters evolved or fixed sometime before the speciation event that established the human and chimp lineages. In Fig. 1b, we see that one and two are simply listed beside the ancestor that appears in Fig. 1a as an edge rather than as a node.
I use the terms node-based and stem-based trees as usefully neutral terms. But do not be misled; they are both phylogenetic trees, and one can be converted into the other. However, one can get in trouble if they are mixed. Nodes must either be taxa or speciation events and internodes must be either taxa or statement of relationships over the entire tree. There is another way of thinking of these trees. Stem-based trees (such as Fig. 1a) treat ancestral and descendant taxa as lineages. Node-based trees (Fig. 1b) treat taxa as objects. Figure 1a is probably the natural way that people think about phylogenetic trees but Fig. 1b is the way computers think about objects that are analyzed.
If there were only two kinds of trees in the world, then interpretation of trees would be easy and straightforward. Alas, graph theory is much richer. Stem-based and node-based trees of the sorts discussed by Hennig (1966) are simply two kinds of acyclic graphs and acyclic graphs are simply graphs with no loops. Gene trees are acyclic graphs and gene trees do not always portray the descent of the species of which the genes are a part. Phenetic trees (phenograms) are acyclic graphs. Cladograms are acyclic graphs usually thought of as common ancestry trees. Figures drawn by Louis Agassiz in the 1840s look very much like those drawn later by Romer. Yet, they are not meant to represent evolutionary descent (Agassiz rejected evolution). Imposing an evolutionary interpretation on an acyclic graph that is not meant to portray evolutionary descent is a category mistake; yet, the graphs may take exactly the same form. Thus, we must exercise caution: we must know the intention of the graph, what it is meant to portray; we cannot divine it purely from the form. There are other problems, relatively minor but vexing in our quest for full understanding of the diagrams we draw and the evolutionary biology they are meant to document. For example, when Baum et al. (2005) mark ancestral species at the nodes of their tree, do we assume that their tree is a node-based tree, as in Fig. 1b without the mark of the “circle” convention? Surely this must be so, for in a stem-based tree, a node (branch point) is an event (speciation) and not a thing (ancestral species). Fortunately, this should not cause major problems in interpretation of the relationships of descendants, but they are relevant to meaning. Ancestral species do not exist on a stem-based tree at nodes; they exist between nodes (Hennig 1966). Descent species may or may not exist only at tips on a phylogenetic tree, but the lineage to which they belong has existed since the speciation event that founded the edge that connects them to their closest analyzed relative. And, we have no idea how many other species join that edge until we have a full account of the diversity represented by that edge. In the chimp–human case, there are a number of other lineages that join along both edges. But, if we accept the hypothesis of chimp+human as opposed to chimp+gorilla, the tree is still accurate in giving an account of relative relationships among the organisms analyzed. Such graphs may be accurate in a relative sense without having to be accurate in an absolute sense. The analogy to a highway map is apt. Highway maps may not show all the intersections, but the intersection they do show must be accurately drawn.
Assessing Tree Quality
Robustness in phylogenetic inference refers to how well methods work in the face of violations of the assumption of the method or model used in an analysis. A robust tree would be one that is relatively immune to violations of the assumptions used to generate the tree hypothesis and might be expected to stand the test of new data, perhaps analyzed using different methods. Hopefully, a robust tree is an accurate tree. Phylogeneticists have put a great deal of effort in exploring how violations of assumption affect the results of an analysis (for example, Holder et al. 2008), and I will not review that extensive literature here. Suffice to say, how robust a phylogenetic tree needs to be depends on the use to which it is put. If the goal is to convict a physician of second-degree murder, then we require a very robust tree that is likely to closely estimate the actual descent of HIV strains. If the goal is to estimate rates of speciation, then not only must the tree be a robust estimate of the Tree of Life but it must also be populated by a significant number of species of the group. Every missing species represents an underestimation of speciation events. If the goal is to use the tree to forecast the potential distribution of an invasive species based on the ecological niche of it and its nearest relatives, then the result could influence policy decision on a national or international level. The major point is that before using a tree, one should access the relative strength of the hypothesis, and the greater the consequences, the more closely we should question the strength of the tree hypothesis. We must remind ourselves that tree hypotheses, like all scientific hypotheses, are conjectures, not facts.
References
Abu-Asab M, Chaouchi M, Amri H. Phyloproteomics: What phylogenetic analysis reveals about serum proteomics. J Proteome Res. 2006;5:2236–40.
Baum DA, Offner S. Phylogenies and tree thinking. Am Biol Teach. 2008;70:222–9.
Baum DA, DeWitt Smith S, Donovan SS. The tree thinking challenge. Science. 2005;310:979–80.
Bull JJ, Wichman HA. Applied evolution. Annu Rev Evol Syst. 2001;32:183–217.
Darwin C. On the origin of species by means of natural selection; or the preservation of favored races in the struggle for life (Reprinted 1st edition). Cambridge: Harvard University Press; 1859.
Dobzhansky T. Nothing in biology makes sense except in the light of evolution. Am Biol Teach. 1973;35:125–9.
Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397:436–41.
Ghiselin MT. Mayr and Bock versus Darwin on genealogical classification. J Zool Syst Evol Res. 2004;42:165–9.
Hahn BH, Shaw GM, De Cock KM, Sharp PM. AIDS as a zoonosis: scientific and public health implications. Science. 2000;287:607–14.
Hennig W. Grundzuge einer Theorie der phylogenetischen Systematik. Berlin: Deutscher Zentralverlag; 1950.
Hennig W. Phylogenetic systematics. Urbana: University of Illinois Press; 1966.
Holder MT, Zwickl DJ, Dessimoz C. Evaluating the robustness of phylogenetic methods to among-site variability in substitution processes. Philos Trans R Soc B Biol Sci. 2008;363:4013–2.
Hull DL. Consistency and Monophyly. Syst Zool. 1964;13:1–11.
Mayr E, Bock WJ. Classification and other ordering systems. J Zool Syst Evol Res. 2002;40:169–94.
McNyset KM. Ecological niche conservatism in North American freshwater fishes. Biol J Linn Soc. 2009;96:282–95.
Peterson AT. Predicting the geography of species’ invasions via ecological niche modeling. Quart Rev Biol. 2003;78:419–33.
Peterson A, Soberón TJ, Sanchez-Cordero V. Conservation of ecological niches in evolutionary time. Science. 1999;285:1265–7.
Peterson AT, Ortega-Huerta MA, Bartley J, Sanchez-Cordero V, Soberon J, Buddemeier RH, et al. Future projections for Mexican faunas under global climate change scenarios. Nature. 2002;416:626–9.
Rieppel O. Sinai Tschulok (1875-1945)—a pioneer of cladistics. Cladistics. 2010;26:103–11.
Simpson GG. The principles of animal taxonomy. New York: Columbia University Press; 1961.
Sneath PHA, Sokal RR. Numerical taxonomy. San Francisco: Freeman; 1973.
Sokal RR, Michener CD. A statistical method for evaluating systematic relationships. Univ Kans Sci Bull. 1958;38:1409–38.
Wiley EO. Convex groups and consistent classifications. Syst Bot. 1981;6:346–58.
Willmann R. From Haeckel to Hennig: the early development of phylogenetics in German-speaking Europe. Cladistics. 2003;19:449–79.
Zimmermann W. Die Methoden der Phylogenetik. In: Henberer G, editor. Dei Evolution der Organismsn 1, Aulf G. Jena: Justav Fisher; 1943. p. 20–56.
Acknowledgements
My thanks to Dr. Daniel R. Brooks (University of Toronto) for inviting me to participate in this special issue and to Teresa E. McDonald (University of Kansas) for her discussions of understanding of public awareness of the meaning of phylogenetic trees in educational and museum settings. This paper is supported by NSF DEB 0732819, the Euteleost Tree of Life project, which includes an educational component designed to increase understanding of phylogenetic trees.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wiley, E.O. Why Trees Are Important. Evo Edu Outreach 3, 499–505 (2010). https://doi.org/10.1007/s12052-010-0279-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12052-010-0279-0