
Abstract
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection is at present an emerging global public health crisis. Angiotensin converting enzyme 2 (ACE2) and trans-membrane protease serine 2 (TMPRSS2) are the two major host factors that contribute to the virulence of SARS-CoV-2 and pathogenesis of coronavirus disease-19 (COVID-19). Transmission of SARS-CoV-2 from animal to human is considered a rare event that necessarily requires strong evolutionary adaptations. Till date no other human cellular receptors are identified beside ACE2 for SARS-CoV-2 entry inside the human cell. Proteolytic cleavage of viral spike (S)-protein and ACE2 by TMPRSS2 began the entire host–pathogen interaction initiated with the physical binding of ACE2 to S-protein. SARS-CoV-2 S-protein binds to ACE2 with much higher affinity and stability than that of SARS-CoVs. Molecular interactions between ACE2-S and TMPRSS2-S are crucial and preciously mediated by specific residues. Structural stability, binding affinity and level of expression of these three interacting proteins are key susceptibility factors for COVID-19. Specific protein–protein interactions (PPI) are being identified that explains uniqueness of SARS-CoV-2 infection. Amino acid substitutions due to naturally occurring genetic polymorphisms potentially alter these PPIs and poses further clinical heterogeneity of COVID-19. Repurposing of several phytochemicals and approved drugs against ACE2, TMPRSS2 and S-protein have been proposed that could inhibit PPI between them. We have also identified some novel lead phytochemicals present in Azadirachta indica and Aloe barbadensis which could be utilized for further in vitro and in vivo anti-COVID-19 drug discovery. Uncovering details of ACE2-S and TMPRSS2-S interactions would further contribute to future research on COVID-19.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The novel coronavirus (nCoV-19) later named as severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) by International Committee on Taxonomy of Viruses (ICTV) is the causative agent of COVID-19 pandemic. SARS-CoV-2 originated in the Wuhan province of China and spread over the world mostly due to human-to-human contacts during migration (Chinazzi et al. 2020; Zhou et al. 2020). While the origin, natural reservoir and intermediate host of the virus are still debatable, it had crossed territories and created havoc to the human race. Until 7th December, 2020, SARS-CoV-2 had infected 66,243,918 individuals and caused 1,528,984 deaths across the globe, with an overall case fatality rate 2.31%. While death rate among aged individuals (age>60 years) are found to be significantly higher, type-2 diabetes (T2D), cardiovascular diseases (CVD), hypertension (HT) and compromise immunity (CI) are identified as major comorbidities of COVID-19 (Chen et al. 2020; Gupta et al. 2020b; Santesmasses et al. 2020; Wang et al. 2020b).
Human receptor angiotensin converting enzyme 2 (ACE2), trans-membrane protease serine 2 (TMPRSS2), and SARS-CoV-2 surface spike (S)-protein are the major host–pathogen determinants known to influence the infection (Hoffmann et al. 2020). Other membrane proteases such as CD26/DPP4, trypsin, cathepsin L, TMPRSS11d, which play prominent role in Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV infections were also thought to play important role in COVID-19 (Millet and Whittaker 2015). In SARS-CoV-2 infection pathogenesis, major implications of ACE2 along with TMPRSS2 were highlighted in recent studies (Hoffmann et al. 2020; Yan et al. 2020). Viral replication and life cycle events are independent of these proteins, and mostly dependent on other viral nonstructural proteins (main protease: Mpro; RNA dependent RNA polymerase: RdRp, Nsp4, Nsp6 etc.) and human cellular system (Prajapat et al. 2020). Considerable efforts are thus being made to understand and predict the structural and functional characteristics of S-protein of SARS-CoV-2. Spike protein of SARS-CoV-2 has substantial sequence dissimilarity (80% homology) with SARS-CoV, which resulted into very limited monoclonal antibody cross-reactivity (Wrapp et al. 2020; Zhou et al. 2020). Recently, cryogenic electron microscopy (cryo-EM) and crystal structure of the functionally essential receptor-binding domain (RBD) of SARS-CoV-2 S-protein are determined (Lan et al. 2020; Wrapp et al. 2020).
Viral S-protein physically interacts and utilizes membrane bound TMPRSS2 and ACE2 to get primed and enter into the human cell. Amino acid substitutions at key positions on RBD have significantly increased the affinity of SARS-CoV-2 RBD to ACE2 by the factor of 10 to 15 while compared to SARS-CoV (Lan et al. 2020; Wang et al. 2020a, 2020b). These viral and human proteins are therefore believed to influence the differential incidence and case fatality rate (CFR) of COVID-19 in different populations (Khafaie and Rahim 2020). Recent studies on S-protein, TMPRSS2 and ACE2 identified interface of their interactions and highlighted different amino acid substitutions which alter their structural stability and potentially affect protein–protein interactions (PPIs) (Hussain et al. 2020; MacGowan and Barton 2020; Yan et al. 2020). Several regulatory genetic variants were also identified to influence the expression of ACE2 and TMPRSS2 in human, which potentially alter the COVID-19 susceptibility (Bhattacharyya et al. 2020; Gupta et al. 2020b; Senapati et al. 2020; Sharma et al. 2020).
We reviewed recent reports on predicted or confirmed host–pathogen interactions (PPI) between SARS-CoV-2 S-protein, human ACE2 and TMPRSS2. We also reviewed up-to-date information on critical amino acid residues and type of interactions involved in PPI interfaces between SARS-CoV-2 S-protein, human ACE2 and TMPRSS2 protein. Further, recent reports on substitution polymorphisms at the key amino acids and their effect on structure–function relationship of these proteins were also discussed in detail. This review is expected to help in studying the genetic susceptibility of COVID-19 and novel/repurposed drug designing against SARS-CoV-2.
Genomic organization of SARS-CoV-2
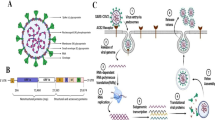
SARS-CoV-2 is an enveloped positive strand RNA (+ssRNA) virus which has a ~30 kb (29.9 kb) RNA genome and encodes ~9860 amino acid long polypeptide, translated in 14 open reading frames (ORFs) and 27 proteins (figure 1) (Potdar et al. 2020). The ORF1a and ORF1ab/b at the 5´-terminus of the genome encode the pp1a and pp1ab proteins, respectively. This poly-protein segregated into 16 nonstructural proteins (Nsps), Nsp1-Nsp11 and Nsp12-Nsp16 following proteolytic cleavage by the viral main protease (Mpro). The 3´-terminus of the genome encodes four structural proteins, the spike surface glycoprotein (S) having two domains, peripheral (S1) and transmembrane (S2), the small envelope protein (E), membrane protein (M) and nucleocapsid protein (N) (Prajapat et al. 2020). There are six accessory proteins currently denoted as ORF3a, ORF6, ORF7a, ORF7b, ORF8, and ORF10 according to reference sequence (ref seq of SARS-CoV-2) GenBank: MN908947.

Schematic diagram of SARS-CoV-2 viral particle and its genomic organization.
Genetic variations determined the phylogenetic position of SARS-CoV-2
SARS-CoV-2 are novel coronavirus (nCoV), named because of the crown-like spikes (spike protein) on their outer surface with diameter range of 80–120 nm. SARS-CoV-2 belongs to the order Nidovirales and family Coronaviridae and subfamily Orthocoronavirinae (Zhu et al. 2020). Based on the serology, coronaviruses are classified into four genera, namely α-coronavirus (α-COV), β-coronavirus (β-COV), γ-coronavirus (γ-COV), and δ-coronavirus (δ-COV) (Chan et al. 2013). Coronaviruses broadly infect vertebrates including humans, birds, bats, snakes, mice and other wild animals (Weiss and Leibowitz 2011; Balasuriya 2017). Six coronaviruses, namely HCoV-229E, HCoV-NL63, HCoV-OC43, HCoV-HKU1, SARS-CoV and MERS-CoV are previously known to cause diseases in human. SARS-CoV-2 is the seventh member of the Coronaviridae family that infects human. SARS-CoV-2, like SARS-CoV and MERS-CoV, belongs to β-coronavirus (Wang et al. 2020a, b). Genomic sequence of SARS-CoV-2 shows close resemblance with SARS-CoV, however positioned distinctly in phylogenetic tree when compared with six other human coronaviruses and a bat coronavirus (Bat-CoV-RaTG13) (figure 2a). Characteristic Spike protein of coronaviruses, which play central role in determining virulence, however shows dissimilar phylogenetic relationship compared to the whole genome. Spike protein seemingly has a close sequence homology with SARS-CoV and Bat-CoV compared to other human coronaviruses (figure 2b). Full-length genome of SARS-CoV-2 isolates obtained from patients had a 79.5% sequence homology with SARS-CoV and a homology of 96% with that of Bat-CoV-RaTG13 (Zhou et al. 2020). Protein coding segments, namely ORF6, ORF7b and ORF10 have been found to be most conserved sequences across 3950 clinical isolates of SARS-CoV-2 (Singh et al. 2020). Recent evidence suggested the presence of 11 common phylogenetic types of SARS-CoV-2, namely O, B, B1, B2, B4, A3, A6, A7, A1a, A2 and A2a across the globe. ‘O’ type is considered the ancestral type that was originated in the Wuhan province of China. Other 10 types are derived due to random nonsynonymous mutations accumulated over the time and spread. Gradual shifting of dominance occurred since December 2019 until April 2020 and A2a type had emerged as most dominant type in different countries (Biswas and Majumder 2020).

The phylogenetic analysis of the (a) whole genome of human coronaviruses (HCoV) with bat coronavirus (Bat-CoV-RaTG13); (b) S (spike) protein of HCoV with bat coronavirus (Bat-CoV-RaTG13). Virus species and sequence ids: HCoV-229E (NP_073551.1); HCoV-OC43 (YP_009555241.1); HCoV-NL63 (YP_003767.1); HCoV-HKU1 (YP_173238.1); SARS-CoV (NP_828851.1); MERS-CoV (YP_007188579.1); SARS-CoV-2 (YP_009724390.1); Bat-CoV-RaTG13 (EPI_ISL_402131).
SARS-CoV-2 viral entry mechanism into human cells
The spike protein expresses on the surface of coronavirus particle as trimers. Each of the S-protein monomer has two domains, namely S1 and S2. S1 domain binds to ACE2 receptors and S2 domain mediates membrane fusion that facilitates cellular entry of the viral particle. SARS-CoV-2 uses ACE2 receptors as the entry point to the human cell. RBD of the S1 domain mediates the interaction with ACE2 receptors. In vitro study has shown inability of SARS-CoV-2 to infect ACE2 null Vero E6 and Hela cells, suggesting pivotal role of this receptor in cellular viral entry (Zhou et al. 2020). Membrane bound serine protease TMPRSS2 is essential for cleaving ACE2 and S-protein for viral entry through membrane fusion, and hence, plays significant role in COVID-19 infection (figure 3). In addition to ACE2 and TMPRSS2, coronaviruses use several other cell surface components to facilitate cellular entry; these include DPP4 and furin for MERS-CoV, ANPEP for HCoV-299E, TMPRSS11D for SARS-CoV, and ST6GAL1 and ST3GAL4 for HCoV-OC43 and HCoV-HKU1 (Bertram et al. 2011, 2012; Zumla et al. 2016). S-protein of SARS-CoV-2 has ~80% sequence identity with that of SARS-CoV and most of the remaining positions are conserved. In RBD, residue between Leu335 and Phe515 of SARS-CoV-2 is homologous to Leu322 to Phe501 of SARS-CoV, except for the insertion of Val483 in SARS-CoV-2. Throughout the article, amino acid residues of SARS-CoV have been written in italics for distinct representation. Within this region, a receptor binding motif (RBM) of SARS-CoV-2 with ~50% sequence homology with SARS-CoV has three ACE2 interactive regions, namely CR1, CR2 and CR3 (Wang et al. 2020b).

Schematic diagram depicting the cellular entry and replication mechanism of SARS-CoV-2 in a human cell. SARS-CoV-2 enters cell through aerosol transmission and binds to ACE2 receptor (also present in bat and other species) which is widely present in alveolar cells of human lungs and fuses with membrane, this requires the two domains S1 and S2 of spike (S) protein to be cleaved using TMPRSS2 (serine protease). The positive sense single-stranded RNA (+ssRNA) genome translates two ORFs (1a and 1b) which can further transcribe and replicate into structural (S, M, E, N) and nonstructural proteins (NSPs). The virion proteins are translated and processed through rough endoplasmic reticulum (RER), Golgi and endoplasmic reticulum Golgi intermediate compartment (ERGIC) and inside endosomal vesicles they assemble and then through exocytosis the virocells are released. The viruses are ingested by antigen presenting cell (APCs) which presents viral S peptides to T helper cells which activates B cell and cytotoxic T cells.
PPIs between S-protein and ACE2 receptor
ACE2 located on chromosome Xp22.2 encode protein belongs to the angiotensin-converting enzyme family and is a functional receptor for the Spike (S) glycoprotein of SARS-CoVs. ACE2 is member of the renin angiotensin aldosterone system (RAAS) and convert angiotensin I and angiotensin II into angiotensin 1-9 and angiotensin 1-7, respectively. ACE2 receptor frequently presents itself as dimeric form (form A and B) and are stabilized by amino acid transporter B0AT1 (also known as SLC6A19) (Kowalczuk et al. 2008). However, B0AT1 does not help in ACE2 dimerization, and ACE2 can be dimerized even in the absence of B0AT1. It is noteworthy that two S-protein trimers simultaneously bind to an ACE2 homodimer, each through a monomer of ACE2 (Yan et al. 2020).
Full-length ACE2 receptor has N-terminal peptidase domain (PD) and C-terminal collectrin-like domain (CLD). PD and CLD consist of 597 amino acids (residues 19 to 615) and 153 amino acids (residues 616–768) respectively. CLD has a small extracellular domain, a long linker and ends with a 40 amino acid long single trans-membrane (TM) helix (Donoghue et al. 2000; Zhang et al. 2001). CLD helps in ACE2 dimerization, where residues Arg652, Glu653, Ser709, Arg710 and Asp713 of ACE2-A interacts with Tyr641, Tyr633, Asn638, Glu639, Asn636 and Arg716 of ACE2-B in a dimeric ACE2.
Within the PD region, the interactive interface between ACE2 and S-protein can be divided into hydrophobic and H-bonding halves. PD of ACE2 facilitates binding of highly conserved RBD present at the distal part of S1 domain of virals S-protein (Hoffmann et al. 2020; Lung et al. 2020). Striking sequence homology (~74%) has been observed in RBD of S protein, between SARS-CoV and SARS-CoV-2. A region of 72 amino acid residues between Asn437 and Tyr508 of S-protein RBD termed as RBM harbouring CR1, CR2 and CR3 regions which physically interact with ACE2 receptor. Compared to SARS-CoV, most of the sequence variations were aggregated in CR1 (Thr470-Phr486) and CR3 (Gln498-Val503) regions. While CR3 accumulated charge preserving substitutions, sequence alteration in CR1 and CR2 affects the surface electrostatics (Wang et al. 2020b).
Binding free energy between the SARS-CoV-2 and human ACE2 is significantly higher (–50.6 kcal mol–1) than that of SARS-CoV (–78.6 kcal mol–1), which is attributed by the loss of H-bond due to replacement of Arg426 with Asn439 in the SARS-CoV-2 S-protein (Xu et al. 2020). Six residues of human ACE2, namely Lys31, Glu35, Asp38, Tyr41, Met82 and Lys352 and five residues of RDB of SARS-CoVs, namely Tyr442, Leu472, Asn479, Asp480 and Thr487 are key interactive amino acids that determines the host specificity of coronaviruses (Li 2013). Between SARS-CoV and SARS-CoV-2, a number of RDB domain amino acid sequence variations, namely Val404>Lys417, Tyr442>Leu455, Leu443>Phe456, Phe460>Tyr473, Leu472>Phe486, Asn479>Gln493, Tyr484>Gln498 and Thr487>Asn501 were observed to potentially alter the binding affinity with ACE2. Replacement of Arg426>Asn439 however in contrary weaken the interaction by disrupting an important salt bridge with Asp329 on ACE2 (Yan et al. 2020). Val404>Lys417 along with Arg403 and Arg408 creates a positive electrostatic patch on RBD-CR1, which facilitate interaction with Asp30 on the surface of ACE2. Additional sequence variations at the RBD-CR1, i.e. Val458>Glu471, Lys465>Thr478, Pro470>Glu484 enhances the negative potential on RBD, which facilitate greater binding with ACE2 receptor (Wang et al. 2020b).
Three-dimensional EM reconstruction of RDB-ACE2-B0AT1 complex helped in identifying details of molecular interactions. Gln498, Thr500 and Asn501 of RDB interact with Tyr41, Gln42 and Arg357 of ACE2 receptor through H-bonds. Other key interactions include Lys417, Tyr453, Gln474 and Phe486 residues of RDB with Gln24, Asp30 and His34 residues of ACE2, respectively (Yan et al. 2020). Molecular docking models showed that Glu54 and Lys314 of ACE2 receptor interacts with S1 domain, via electrostatic interactions, H-bonding, and hydrophobic interactions (Zhang et al. 2005). Among many others, a major residue of ACE2 that interacted with S1 domain of SARS-CoV-2 was identified to be the amino acid Met82, strongly interacting with amino acid Phe486 on the viral protein (Luan et al. 2020). The involvement of amino acids Gln492 and Asn501 are the key residues of SARS-CoV-2 RDB for energetically favourable interaction with ACE2 as compared to SARS-CoV by the production of electrostatically stabilizing interactions (Ortega et al. 2020) (table 1).
Among several other residues Lys26, Thr27, Glu37, Lys68, Asp206, Gly211, Arg219, Gly326, Lys341, Gly352, Val447, Ile468, Phe486, Arg559 of ACE2, and Ser438, Gly476, Asn479, Val483 of S-protein were polymorphic sites which were also critical in determining the strength of S-ACE2 interaction (table 1). Several missense mutations/variations are known to potentially influence the virulence of SARS-CoV-2. Asp614Gly substitution known to influence the S1–S2 cleavage reportedly facilitating the viral entry into the lung cells (Follis et al. 2006). This mutation is characteristic of SARS-CoV-2 type A2a, which is presently most common and dominant type (Biswas and Majumder 2020).
TMPRSS2 promote priming of S-protein
TMPRSS2 interact with both human ACE2 and viral S-protein separately. Immunofluorescence experiments identified that TMPRSS2 colocalized with ACE2 on the surface of type II pneumocytes (Shulla et al. 2011). TMPRSS2 protein belongs to the serine protease family. The encoded protein contains a type II transmembrane domain (N-termini), a receptor class-A domain, a scavenger receptor cysteine-rich domain and a protease domain (C-terminal) (Yan et al. 1999; Heald-Sargent and Gallagher 2012). TMPRSS2 is a zymogen which becomes active by its autocatalytic activity. The catalytically active form of TMPRSS2 interacts with ACE2 receptor and proteolytically cleaves it to generate a secreted form (Ko et al. 2015; Zmora et al. 2015; Hoffmann et al. 2018; Qing et al. 2020). Other serine proteases, such as TMPRSS11D and HPN/TMPRSS1 and ADAM17 were also shown to be able to cleave ACE2 receptors (Fan et al. 2019; Cava et al. 2020; Wang et al. 2020a, b; Xu et al. 2020; Zhou et al. 2020).
TMPRSS2 cleaves the trans-membrane C-terminal domain (residue 697 to 716) of ACE2, which resulted into conformational changes to S1–ACE2 complex and facilitate S-protein driven viral entry (Shulla et al. 2011). Above mentioned ACE2 residues overlap with the ACE2 symmetric dimerization interface, which is also physically associated and guarded by B0AT1 from the outside and form ACE2-B0AT1 hetero-tetramer. Hence this hetero-tetramer potentially may block the access of TMPRSS2 to the cleave site on ACE2 and thus suppress SARS-CoV-2 infection (Yan et al. 2020). However, it is noteworthy that B0AT1 coexpressed with ACE2 only in kidney and intestine (Kleta et al. 2004). TMPRSS2 cleaves S-protein at multiple arginine rich positions (Arg667 and Arg797) at the S1–S2 fusion site / fusion peptide, which were found to be crucial for SARS-CoV-2 entry into the human cell (Matsuyama et al. 2005; Reinke et al. 2017; Iwata-Yoshikawa et al. 2019; Hoffmann et al. 2020). Such S1–S2 fusion protein cleavage is known as S proteolytic priming. This ‘priming’ of the S2 domain generates a mature peptide (S2’) for membrane fusion, while S1 domain is released extracellular (Yan et al. 2020; Heurich et al. 2014; Hoffmann et al. 2018) (figure 3). S2 domain helps in membrane fusion, which is a pH dependent event, where low pH was shown to inhibit membrane fusion event by S2 domain (Simmons et al. 2004). In contrary, attachment of SARS-CoVs spike (S1) protein to ACE2, in the absence of membrane proteases (like TMPRSS2), an endosomal entry of viral particle gets initiated, where cysteine protease cathepsin L (CatL) cleaves the S-protein within the endosomal vesicle (Hoffmann et al. 2018). By cleaving S1 from infected-cell surfaces, TMPRSS2 releases S1 fragments that bind and incapacitate neutralizing antibodies (Glowacka et al. 2011). In silico molecular docking studies identified several key amino acid residues of TMPRSS2 and S-protein that interacts. Along several other residues Trp64, Thr95, Val213, Ala262, Lys187, Arg214, Gly261, Asp215, His66, Leu242, Asp80, Phe79, His245, Arg246, Asp614, Arg667 and Asp614 of S-protein were involved in interaction with TMPRSS2 and exert critical role in stabilizing the binding (Bhattacharyya et al. 2020; Hoffmann et al. 2020; Nelson-Sathi et al. 2020; Senapati et al. 2020) (table 2).
Virus replication and release
The virion releases RNA genome into the cell and translation of structural and nonstructural proteins (NSPs) follows. ORF1a and ORF1ab are translated to produce pp1a and pp1ab polyproteins, which are cleaved by the main proteases (Mpro) to yield 16 different Nsp proteins. To replicate its genome, a positive single-strand RNA serves as a template for an RNA-dependent RNA polymerase (RdRp), yielding a complementary RNA strand, which forms a dimer with the template strand. The dsRNA formed subsequently serves as the template for a new positive sense genome which further forms an independent infectious (+) ssRNA genome (Berman 2019). This is followed by assembly and budding into the lumen of the endoplasmic reticulum Golgi intermediate compartment (ERGIC). Virions are then released from the infected cell through exocytosis to further activate host immune system (figure 3) (Adnan Shereen et al. 2020).
Coexpression of ACE2 and TMPRSS2
While ACE2, even without its intrinsic ectopeptidase activity, has been an efficient viral entry factor, the colocalization of TMPRSS2 along with ACE2 was found to be enhancing the viral entry into the host cells (Shulla et al. 2011; Lu et al. 2015). It is worth noting that TMPRSS2 was only expressed in a subset of ACE2+ cells, suggesting that the virus might use alternative pathways (Hoffmann et al. 2020). Indeed, other proteases were more promiscuously expressed than TMPRSS2, such as cathepsin B/L, which is expressed in more than 70–90% of ACE2+ cells (Zhou et al. 2015; Iwata-Yoshikawa et al. 2019). However, while TMPRSS2 activity is documented to be important for viral transmission, cathepsin B/L activity may be able to substitute for TMPRSS2 (Hoffmann et al. 2020; Sungnak et al. 2020). Tissue specific expression of ACE2 overlaps with the primarily affected organs following SARS-CoV-2 infection, these include lungs (type II alveolar cells, AT2), heart (myocardial cells), oesophagus (oesophagus epithelial cells), kidney (proximal tubule cells of the kidney), bladder (oesophagus epithelial cells), and small intestine (ileum) (Zhou et al. 2020). Interestingly, this receptor is highly expressed in epithelial cells of tongue and mucosa of oral cavity (Xu et al. 2020). ACE2 and TMPRSS2 are coexpressed in lung AT2 cells, oesophageal upper epithelial and gland cells, and surface of type II pneumocytes cells lining, the alveoli of the lungs in the human airway (Bertram et al. 2010; Shulla et al. 2011; Glowacka et al. 2011).
Angiotensin-converting enzyme inhibitor (ACEI) or angiotensin II receptor blocker (ARB) therapy given to individuals suffering from CVD, and HT was found to significantly increase the expression of ACE2 receptors in cardiac tissue (Ferrario et al. 2005), kidney (Ferrario and Varagic 2010) and duodenum (Vuille-dit-Bille et al. 2015). Higher expression of ACE2 was also observed among subjects with male sex, older age, HT and T2D (Walters et al. 2017; Uri et al. 2014; Úri et al. 2016). Therefore, subjects under treatment with ACEI and ARBs could be at higher risk to get infected with SARS-CoV-2. On the other hand post infection SARS-CoV-2 reportedly downregulate the ACE2 expression which is otherwise needed to prevent lung injury (Kuba et al. 2005). Therefore, post infection administration of ARBs potentially prevents and treats lung injury induced by COVID-19 (Vaduganathan et al. 2020).
Viral S-protein binding potential to ACE 2 receptors in different species
The amino acid sequence identities between human ACE2, and ACE2 of Chinese horseshoe bats, civet, swine and mouse are 80.75%, 83.48%, 81.37%, 82.11%, respectively. Interestingly, mouse ACE2 which cannot be used by SARS-CoV-2 show higher similarity to human ACE2 than those of Chinese horseshoe bats and swine which can be used by SARS-CoV-2, indicating that whole amino acid sequence identities is not a reliable indicator for predicting binding efficiency by SARS-CoV-2 (Qiu et al. 2020). A study reported that pangolin, cat, cow, buffalo, goat, sheep and pigeon ACE2 receptors might be utilized by SARS-CoV-2, indicating potential interspecies transmission of the virus from bat to these animals and among these animals (Fan et al. 2019; Li et al. 2020). In a breakthrough study, Menachert et al. constructed two chimeric SARS coronaviruses, namely SHC014-MA15 and SHC014-CoV, where they have used pathogenic spike protein of RsSHC014-CoV in the background of mouse-adopted SARS-CoV and SARS-CoV, respectively. Both, SHC014-MA15 and SHC014-CoV could infect the cell but could not lead to severe disease or cause lethality. This indicated that beside, sequence adaptation on S-protein and ACE2, other genomic (structural and/or nonstructural) variations are also instrumental for successful infection by SARS-CoVs (Menachery et al. 2015). Bats are considered the natural host of SARS-CoV-2, which is genetically 96% identical to two SARS-like Bat-CoVs, namely bat-SL-CoVZX45 and bat-SL-CoVZX21 (Dong et al. 2020; Lu et al. 2020; Zhou et al. 2020), although the transmission route is yet to be elucidated. Ji et al. (2020) proposed snakes as a carrier of the virus from bats to humans (Ji et al. 2020). Pangolins are also identified as the potential intermediate host of SARS-CoV-2, as CoVs that are natural to them has 99% genetic homology with SARS-CoV-2 (Xiao et al. 2020).
Impact of genetic polymorphisms in ACE2 and TMPRSS2 on COVID-19
The role of structural and regulatory variants of ACE2 and TMPRSS2 in susceptibility to COVID-19 is a major point of investigation. Initial observations reported that African populations show genetic predisposition for expressing significantly lower levels of ACE2 and TMPRSS2, explaining probability of the lower incidence of COVID-19 in Africans, while allelic frequencies contributing to higher levels of ACE2 and TMPRSS2 expressions are observed in south Asian, Southeast Asian, and east Asian populations in accordance with the higher infection rate observed (Ortiz-Fernandez and Sawalha 2020; Cao et al. 2020). Results of a ‘Firedock modelling’ identifies ACE2 variant p.Ser19Pro/c.55T>C (rs73635825) to decrease ACE2-S-protein interactions and was found to be common among an African population thereby conferring a protective effect, while p.Lys26Arg/c.77A>G (rs4646116) was found to increase its affinity for S-protein and common among European population (Calcagnile et al. 2020). p.Ile468Val/c.1402A>G (rs191860450), another ACE2 variant, was found to be mutated more frequently among the Asians (Li et al. 2020). p.Ser19Pro/c.55T>C (rs73635825) and p.Glu329Gly/c.986A>G (rs143936283) of ACE2 were also among the majorly noticed functional variations which potentially alters major PPIs with viral S-protein (table 3). Low or reduced binding affinity and/or stability induced by the key residues of ACE2 receptor in the formation of complex with SARS-CoV-2 S-protein might be suggestive of a level of resistance in a way (Hussain et al. 2020). Mapping of ACE2 amino acid residues between 697 and 716 upon ACE2 structure (61M7-Protein Data Bank) reveals its significance in symmetric homo-dimerization of ACE2 in the hetero-tetrameric complex of B0AT1, which disabled TMPRSS2 dependent cleavage. When the tetrameric complex was allowed to interact with RBD of SARS-CoV-2 S protein, a reduced affinity for ACE2 dimer was observed, as compared to its interactions with monomeric ACE2 (Sharma et al. 2020). Two ACE2 intronic variants, rs1978124 (g.7130A>T) and rs2106809 (g.7132T>C) were observed to have different distribution of allelic frequencies in different populations (Sharma et al. 2020).
Analysis of TMPRSS2 functional variants among 17 populations revealed that African population had higher percentage of loss of function mutation alleles globally, mirroring that of ACE2, with East Asians are predisposed to higher expression levels while African populations showed the lowest. The nonsynonymous pathogenic variants were found to have highest allelic frequencies in Ashkenazi Jews (Russo et al. 2020; Ortiz-Fernandez and Sawalha 2020). Two missense variations, p.Gly8Val/c.23G>T (rs75603675) and p.Val197Met/c.589G>A (rs12329760) were identified that might have a role in influencing its interaction with ACE2, and S-protein. While p.Gly8Val was identified to disrupting the local protein structure but increase the overall stability TMPRSS2, p.Val197Met leads to decrease in stability of TMPRSS2 protein structure and inhibits its binding to ACE2 in silico. Contrastingly, it also shows increased affinity towards S-protein domains (table 4) (Sharma et al. 2020). Common TMPRSS2 missense variation rs12329760 was found not involving in direct interaction with S-protein, but potentially influence the structural confirmation (Senapati et al. 2020). Four common regulatory SNPs present at the 3’UTR (rs112657409, rs11910678, rs77675406) and 5’ flanking (rs713400) of TMPRSS2 were found to significantly influence the expression (eQTL P ≤ 2.00E-05) of MX1 and TMPRSS2. MX1 encodes a GTP metabolizing protein and participate in cellular anti-viral response (PMID: 24296571). These coding and regulatory SNPs were found to have considerably variable allelic frequencies in different global populations, which might determine COVID-19 incidence (Senapati et al. 2020). Another regulatory SNP rs35074065 was found to influence the expression (eQTL) of TMPRSS2 and its upstream MX1 gene (Russo et al. 2020). It is noteworthy that besides TMPRSS2, MX1 levels are major determinants of cellular antiviral response.
Viral–human protein interaction interface in drug discovery
Till date vaccines are not available for any of the coronavirus infections. Presently, several potential vaccines against SARS-CoV-2 spike protein, using platforms such as nonreplicating viral vector, replicating viral vector, protein subunit, virus like particle, DNA based, inactivated virus, live attenuated virus against SARS-CoV-2 are either being tested or under clinical study (https://www.who.int/blueprint/priority-diseases/key-action/novel-coronavirus-landscape-ncov.pdf). In view of their crucial role in COVID-19, these interface of interactions are being utilized for designing novel or repurposed therapeutic inhibitors or blockers (Cao et al. 2020; Wu et al. 2020). Conceiving COVID-19 massive health emergency approaches to design novel drugs or repurposing specific drugs are warranted. Computer aided drug designing identified several natural compounds such as isothymol, captopril, chitosan, chitosan-spike trimer, anabsinthin, absinthin, 3,4,5-tricaffeoylquinic acid, quercetin compounds, geniposide, excavatolide M having potential to disrupt ACE2-S and TMPRSS2-S PPIs (Kalathiya et al. 2020; Joshi et al. 2020; Rahman et al. 2020; Abdelli et al. 2020; Qing et al. 2020). Approved drugs against these proteins, such as B38 (IgG), hydroxychloroquine, chloroquine, arbidol, azithromycin, cepharanthine, selamectin, mefloquine, DX600, CP-1 (peptide from HR2), camostat, nafamostat, gabexate mesylate, rivaroxaban, letaxaban among others were also reported to have potential repurposed value (Abdel-Mottaleb and Abdel-Mottaleb 2020; Bittmann et al. 2020; Liu et al. 2020; Mckee et al. 2020; Meyer et al. 2013; Rensi et al. 2020; Sandeep and McGregor 2020; Wu et al. 2020; Yamamoto et al. 2016).
Recently, Paniri et al. (2020) indicated that single-nucleotide polymorphisms (SNPs) in TMPRSS2 might influence SARS-CoV-2 entry into the host cell. The group also proposed potential therapeutic implication of TMPRSS2 in designing antiviral drugs. It has been reported that phytochemicals present in Withania somnifera (solonaceae) plant possess potential to inhibit TMPRSS2 protein and also TMPRSS2 mRNA level lowering efficacy in cells (Kumar et al. 2020). In a different study, Elmezayen et al. (2020) identified promising TMPRSS2 inhibitors (Rubitecan, Loprazolam, ZINC000015988935 and ZINC000103558522) in in silico study. Basit et al. (2020) reported that blocking the S glycoprotein through potential inhibitor may interfere its interaction with ACE2 and thus SARS-CoV-2 entry into the host cell. Recently scientists around the world identified phytochemicals (essential oil component of Ammoides verticillata, dithymoquinone from Nigella sativa, saikosaponins U and V etc.), probiotics (Plantaricin BN, Plantaricin JLA-9, Plantaricin W, Plantaricin D) which are able to bind at Spike-ACE2 interaction interface (Abdelli et al. 2020; Anwar et al. 2020; Ahmad et al. 2020; Sinha et al. 2020).
In this line of work we identified some phytochemicals present in Azadirachta indica (Meliaceae) and Aloe barbadensis (Asphodelaceae) have potential to bind to TMPRSS2 at their PPI interface using molecular docking approach. This piece of work was attended to report the natural lead compounds, which could be further validated using anti-COVID-19 in vitro and in vivo drug discovery approach. The natural compounds reported in A. indica (nimbochalcin, melianin B and vepaol) and A. barbadensis (LS-143447; (10R)‐2,8‐dihydroxy‐6‐(hydroxymethyl)‐1‐methoxy‐10‐ [(2R,3R,4R,5R,6R)‐3,4,5 ‐trihydroxy‐6‐(hydroxymethyl)oxan‐2‐yl]‐9,10‐dihydroanthracen‐9‐one; Barbaloin; 7-hydroxyaloin B; 10-hydroxyaloin A; CHEMBL518845) showed docking score more than respective standard compounds (VE607 and camostat mesylate, respectively). MM-GBSA analysis of the unbound TMPRSS2 protein and its complex with top lead A. barbadensis (10-hydroxyaloin A) and A. indica (Nimbochalcin) phytochemical was –13641.05, –13633.21, –13727.59 kcal/mole respectively which showed stable ligand binding. Docking pose of all the lead molecules against human TMPRSS2 protein are depicted in figure 4a. Surface structure of the top lead, each from A. indica and A. barbadensis, against TMPRSS2 protein is depicted in figure 4, b&c. Docking score, type of interaction and amino acids involved in the interaction are summarized in table 5. Structure of lead phytochemicals present in Azadirachta indica and Aloe barbadensis against human TMPRSS2 protein are depicted in figure 5. We hereby recommend further in silico and in vitro studies to validate the efficacy of these phytochemicals in the field of anti SARS-CoV-2 drug discovery.

Docking pose and surface structure of lead phytochemicals present in A. indica and A. barbadensis against Human TMPRSS2 proteins using molecular docking study. (a) Docking pose of lead phytochemicals at TMPRSS2 protein. (red, 10-hydroxyaloin A; cyan, CHEMBL518845; yellow, vepaol; green, nimbochalcin; megenta, melianin B; blue, camostat mesylate) (b) and (c) surface structure of A. barbadensis (10-hydroxyaloin A) and A. indica (nimbochalcin) phytochemical interacted with TMPRSS2 protein respectively. The lime green and orange colour in surface structure represents amino acids involved in hydrogen and hydrophobic bond formation, respectively. The selected phytochemical ligands for the study were downloaded from Indian Medicinal Plants, Phytochemistry and Therapeutics (IMPPAT) database (https://cb.imsc.res.in/imppat/basicsearch). The TMPRSS2 protein was modeled through Swiss-model server (https://swissmodel.expasy.org/). AutoDock and PyMol tools were used to perform the molecular docking study and visualization, respectively (Gupta et al. 2020a, b).

In silico identification of human TMPRSS2 protein natural lead inhibitors present in A. indica and A. barbadensis.
Conclusion and future aspects
SARS-CoV-2 is a highly infectious virus which has jolted the world at present. Recent genomic and proteomic studies revealed that SARS-CoV-2 has substantial genomic differences with SARS-CoV. Precise differences in molecular interaction between ACE2 and S-protein of SARS-CoV-2 compared to SARS-CoV have been well-explained (Wang et al. 2020b). From evolutionary aspect, several gain of function mutations in S-protein promoted supposedly cross-species transmission from bat to human, through an intermediate host. Recent reports identified several key residues of S-protein that physically interacts with human receptor ACE2 and membrane protease TMPRSS2 to initiate the infection (tables 1 & 2). Host–pathogen interactions through S-protein with ACE2 and TMPRSS2 determine the virulence, where ACE2 is the only known receptor for SARS-CoV-2, several other proteases are known to prime S-protein of related coronaviruses. Comprehensive studies on these interactive residues are expected to uncover the exact mechanism of S-protein priming and cellular internalization of SARS-CoV-2. This is noteworthy that ACE2 and TMPRSS2 are coexpressed in very limited number of tissues. Therefore, this is evident that SARS-CoV-2 also uses alternative mechanisms for cellular entry. Furin is another membrane bound protease expressed in multiple tissues, including neuroendocrine, gut, brain, lungs, liver and small intestine. Its expression is highest in the lungs (Belouzard et al. 2012). The S-protein of the SARS-CoV-2 possesses a polybasic cleavage site (PRRAR) at the junction of S1 and S2, the two domains of the spike, allows effective cleavage by furin (Coutard et al. 2020; Nao et al. 2017; Wrapp et al. 2020) where the proline inserted at this site creates a turn which is predicted to result in the addition of O-linked glycans to Ser673, Thr678 and Ser686, which flank the cleavage site and are unique to SARS-CoV-2. The furin activation site (FAS) makes the SARS-CoV-2 much different in cell entry than SARS-CoV, and affects the stability of the virus and, consequently, the transmission process (Yamada and Liu 2009; Millet and Whittaker 2014; Li et al. 2015). In comparison, SARS-CoV entry is activated by TMPRSS2 and cathepsins, but not furin. FAS has been observed in human β-coronaviruses including HKU1 (lineage A) which predicted O-linked glycans but have not been observed in related ‘lineage B’ β-coronaviruses (Belouzard et al. 2012; Andersen et al. 2020). Identification of the FAS allows to investigate how the virus spreads among humans, viruses including Influenza family viruses contain these sites that are easily spread among people, but the site of activation is found on a protein called Haemagglutinin, not on the S protein (Zmora et al. 2015; Hasan et al. 2020). Furin preactivation let the SARS-CoV-2 to be less dependent on target cells, particularly cells with relatively low expressions of TMPRSS2 and/or lysosomal cathepsins (Wrapp et al. 2020). The known level of genetic variations in S gene, deciphers that SARS-CoV-2-like viruses with partial or full polybasic cleavage sites will be discovered in other species too which is probably responsible for interspecies transmission and gain of infection (Andersen et al. 2020). Comprehensive studies on such alternative viral entry mechanisms are needed to obtain an overall understanding of COVID-19.
Interactive residues of S-protein, ACE2 and TMPRSS2 that helps in PPI (host–pathogen interaction) could be considered for computer aided drug designing. Novel synthetic or natural products can be screened for their potential binding to these interactive residues to inhibit or block the host–pathogen interactions. Several such molecules have already been identified which have potential to bind to block or inhibit these proteins (Kalathiya et al. 2020; Joshi et al. 2020; Rahman et al. 2020; Wu et al. 2020; Abdel-Mottaleb and Abdel-Mottaleb 2020; Abdelli et al. 2020; Liu et al., 2020; Sandeep and McGregor 2020; Mckee et al. 2020; Li et al. 2019; Bittmann et al. 2020; Yamamoto et al. 2016; Rensi et al. 2020). Further studies are warranted to validate effectiveness of these repurposed and novel compounds and future applications for COVID-19 disease management.
References
Abdelli I., Hassani F., Bekkel Brikci S. and Ghalem S. 2020 In silico study the inhibition of Angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from western Algeria. J. Biomol. Struct. Dyn. 1-17. (https://doi.org/10.1080/07391102.2020.1763199).
Abdel-Mottaleb M. S. and Abdel-Mottaleb Y. 2020 In search for effective and safe drugs against SARS-CoV-2: Part I] simulated interactions between selected nutraceuticals ACE2 enzyme and S protein simple peptide sequences. ChemRxiv, https://doi.org/10.26434/chemrxiv.12155235.v1.
Ahmad S., Abbasi H. W., Shahid S., Gul S. and Abbasi S. W. 2020 Molecular docking simulation and MM-PBSA studies of Nigella Sativa compounds: a computational quest to identify potential natural antiviral for COVID-19 treatment. J. Biomol. Struct. Dyn. 1-16.
Andersen K. G., Rambaut A., Lipkin W. I., Holmes E. C. and Garry R. F. 2020 The proximal origin of SARA-CoV-2. Nat. Med. 26, 450-452.
Anwar F., Altayb H. N., Al-Abbasi F. A., Al-Malki A. L., Kamal M. A. and Kumar V. 2020 Antiviral effects of probiotic metabolites on COVID-19. J. Biomol. Struct. Dyn. 1-1.
Ali F., Elserafy M., Alkordi M. and Amin M. 2020 ACE2 coding variants in different populations and their potential impact on SARS-CoV-2 binding affinity. BioRxiv, https://doi.org/10.1101/2020.05.08.084384.
Balasuriya U. B. R. 2017 List of contributors. In Fenner’s veterinary virology (ed. N. J. MacLachlan and E. J. Dubovi), 5th edition, Academic Press, Boston, pp. xvii–xviii.
Basit A., Ali T. and Rehman S. U. 2020 Truncated human angiotensin converting enzyme 2; a potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. J. Biomol. Struct. Dyn. 1-7.
Belouzard S., Millet J. K., Licitra B. N. and Whittaker G. R. 2012 Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4, 1011–1033.
Berman J. J. 2019 Taxonomic guide to infectious diseases: understanding the biologic classes of pathogenic organisms. In Viruses, chapter 7, 2nd edition. Academic Press, London.
Bertram S., Glowacka I., Müller M. A., Lavender H., Gnirss K., Nehlmeier I. et al. 2011 Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 85, 13363–13372.
Bertram S., Glowacka I., Steffen I., Kühl A. and Pöhlmann S. 2010 Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 20, 298–310.
Bertram S., Heurich A., Lavender H., Gierer S., Danisch S., Perin P. et al. 2012 Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS One 74, e35876.
Bhattacharyya C., Das C., Ghosh A., Singh A. K., Mukherjee S., Majumder P. P. et al. 2020 Global spread of SARS-CoV-2 subtype with spike protein mutation D614G is shaped by human genomic variations that regulate expression of TMPRSS2 and MX1 genes. BioRxiv, https://doi.org/10.1101/2020.05.04.075911.
Biswas N. K. and Majumder P. P. 2020 Analysis of RNA sequences of 3636 SARS-CoV-2 collected from 55 countries reveals selective sweep of one virus type. Indian J. Med. Res., https://doi.org/10.4103/ijmr.IJMR_1125_20.
Bittmann S., Luchter E., Weissenstein A., Villalon G. and Moschuring-Alieva E. 2020 TMPRSS2-inhibitors play a role in cell entry mechanism of COVID-19: an insight into Camostat and Nafamostat. J. Regen. Biol. Med. 2, 1–3.
Calcagnile M., Forgez P., Iannelli A., Bucci C., Alifano M. and Alifano P. 2020 ACE2 polymorphisms and individual susceptibility to SARS-CoV-2 infection: insights from an in silico study. BioRxiv, https://doi.org/10.1101/2020.04.23.057042.
Cao Y., Li L., Feng Z., Wan S., Huang P., Sun X. et al. 2020 Comparative genetic analysis of the novel coronavirus 2019-nCoV/SARS-CoV-2 receptor ACE2 in different populations. Cell Discov. 6, 1–4.
Cava C., Bertoli G. and Castiglioni I. 2020 in silico discovery of candidate drugs against covid-19. Viruses 12, 404.
Chan J. F. W., To K. K. W., Tse H., Jin D. Y. and Yuen K. Y. 2013 Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol. 21, 544–555.
Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y. et al. 2020 Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan China: a descriptive study. Lancet 395, 507–513.
Chinazzi M., Davis J. T., Ajelli M., Gioannini C., Litvinova M., Merler S. et al. 2020 The effect of travel restrictions on the spread of the 2019 novel coronavirus COVID-19 outbreak. Science 368, 395–400.
Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N. G. and Decroly E. 2020 The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 176, 104742.
Dong N., Yang X., Ye L., Chen K., Chan E. W. C., Yang M. et al. 2020 Genomic and protein structure modelling analysis depicts the origin and infectivity of 2019-nCoV a new coronavirus which caused a pneumonia outbreak in Wuhan China. BioRxiv, https://doi.org/10.1101/2020.01.20.913368.
Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N. et al. 2000 A novel angiotensin-converting enzyme–related carboxypeptidase ACE2 converts angiotensin I to angiotensin 1–9. Circ. Res. 87, e1–e9.
Elmezayen A. D., Al-Obaidi A., Şahin A. T. and Yelekçi K. 2020 Drug repurposing for coronavirus COVID-19: in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 1-3. (https://doi.org/10.1080/07391102.2020.1758791).
Fan Y., Zhao K., Shi Z.-L. and Zhou P. 2019 Bat coronaviruses in China. Viruses 11, 210.
Ferrario C. M. and Varagic J. 2010 The ANG-1–7/ACE2/mas axis in the regulation of nephron function. Am. J. Physiol-Renal. 298, F1297–F1305.
Ferrario C. M., Jessup J., Chappell M. C., Averill D. B., Brosnihan K. B., Tallant E. A. et al. 2005 Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme. Circulation 111, 2605–2610.
Follis K. E., York J. and Nunberg J. H. 2006 Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 350, 358–369.
Glowacka I., Bertram S., Müller M. A., Allen P., Soilleux E., Pfefferle S. et al. 2011 Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 85, 4122–4134.
Gupta S., Singh A. K., Kushwaha P. P., Prajapati K. S., Shuaib M., Senapati S. and Kumar S. 2020a Identification of potential natural inhibitors of SARS-CoV-2 main protease by molecular docking and simulation studies. J. Biomol. Struct. Dyn. 1-19. (https://doi.org/10.1080/07391102.2020.1776157).
Gupta R., Ghosh A., Singh A. and K. and Misra A. 2020 Clinical considerations for patients with diabetes in times of COVID-19 epidemic. Diabetes. Metab. Syndr. 14, 211.
Hasan A., Paray B. A., Hussain A., Qadir F. A., Aziz F. M. et al. 2020 A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J. Biomol. Struct. Dyn. 1–13.
Heald-Sargent T. and Gallagher T. 2012 Ready set fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4, 557–580.
Heurich A., Hofmann-Winkler H., Gierer S., Liepold T., Jahn O. and Pöhlmann S. 2014 TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 88, 1293–1307.
Hoffmann M., Hofmann-Winkler H. and Pöhlmann S. 2018 Priming time: How cellular proteases arm coronavirus spike proteins. In Activation of Viruses by Host Proteases (ed. E. Böttcher-Friebertshäuser, W. Garten and H. Klenk), Springer, Cham, pp. 71–98.
Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S. et al. 2020 SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 1812, 271–280.
Hussain M., Jabeen N., Raza F., Shabbir S., Baig A. A., Amanullah A. and Aziz B. 2020 Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 92, 1580–1586.
Iwata-Yoshikawa N., Okamura T., Shimizu Y., Hasegawa H., Takeda M. and Nagata N. 2019 TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J. Virol. 93, e01815–e01818.
Ji W., Wang W., Zhao X., Zai J. and Li X. 2020 Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 92, 433–440.
Joshi T., Sharma P., Mathpal S., Pundir H., Bhatt V. and Chandra S. 2020 In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur. Rev. Med. Pharmaco. 24, 4529–4536.
Kalathiya U., Padariya M., Mayordomo M., Lisowska M., Nicholson J., Singh A. et al. 2020 Highly conserved homotrimer cavity formed by the SARS-CoV-2 Spike glycoprotein: a novel binding site. J. Clin. Med. 9, 1473.
Khafaie M. A. and Rahim F. 2020 Cross-country comparison of case fatality rates of COVID-19/SARS-COV-2. Osong Public Health Res. Perspect. 11, 74.
Kleta R., Romeo E., Ristic Z., Ohura T., Stuart C., Arcos-Burgos M. et al. 2004 Mutations in SLC6A19 encoding B0 AT1 cause Hartnup disorder. Nat. Genet. 36, 999–1002.
Ko C. J., Huang C. C., Lin H. Y., Juan C. P., Lan S. W., Shyu H. Y. et al. 2015 Androgen-induced TMPRSS2 activates matriptase and promotes extracellular matrix degradation prostate cancer cell invasion tumor growth and metastasis. Cancer Res. 75, 2949–2960.
Kowalczuk S., Broer A., Tietze N., Vanslambrouck J. M., Rasko J. E. and Broer S. 2008 A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 22, 2880–2887.
Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B. et al. 2005 A crucial role of angiotensin converting enzyme 2 ACE2 in SARS coronavirus–induced lung injury. Nat. Med. 11, 875–879.
Kumar V., Dhanjal J. K., Bhargava P., Kaul A., Wang J., Zhang H. et al. 2020 Withanone and withaferin-A are predicted to interact with transmembrane protease serine 2 TMPRSS2 and block entry of SARS-CoV-2 into cells. J. Biomol. Struct. Dyn. 1-27.
Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S. et al. 2020 Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 1-6.
Li C. C., Wang X. J. and Wang H. C. R. 2019 Repurposing host-based therapeutics to control coronavirus and influenza virus. Drug Discov. Today 24, 726–736.
Li F. 2013 Receptor recognition and cross-species infections of SARS coronavirus. Antivir. Res. 100, 246–254.
Li J. Y., You Z., Wang Q., Zhou Z. J., Qiu Y., Luo R. and Ge X. Y. 2020 The epidemic of 2019-novel-coronavirus 2019-nCoV pneumonia and insights for emerging infectious diseases in the future. Microbes. Infect. 22, 80–85.
Li W., Wicht O., van Kuppeveld F. J., He Q., Rottier P. J. and Bosch B. J. 2015 A single point mutation creating a furin cleavage site in the spike protein renders porcine epidemic diarrhea coronavirus trypsin independent for cell entry and fusion. J. Virol. 89, 8077–8081.
Liu C., Zhou Q., Li Y., Garner L. V., Watkins S. P., Carter L. J. et al. 2020 Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS. Cent. Sci. 6, 315–331.
Lu R., Zhao X., Li J., Niu P., Yang B., Wu H. et al. 2020 Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574.
Lu G., Wang Q. and Gao G. F. 2015 Bat-to-human: spike features determining ‘host jump’of coronaviruses SARS-CoV MERS-CoV and beyond. Trends Microbiol. 23, 468–478.
Luan J., Lu Y., Jin X. and Zhang L. 2020 Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 5261, 165–169.
Lung J., Lin Y. S., Yang Y. H., Chou Y. L., Shu L. H., Cheng Y. C. et al. 2020 The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J Med. Virol. 92(6), 693–697.
MacGowan S. A. and Barton G. J. 2020 Missense variants in ACE2 are predicted to encourage and inhibit interaction with SARS-CoV-2 Spike and contribute to genetic risk in COVID-19. BioRxiv, https://doi.org/10.1101/2020.05.03.074781.
Matsuyama S., Ujike M., Morikawa S., Tashiro M. and Taguchi F. 2005 Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 102, 12543–12547.
McKee D. L., Sternberg A., Stange U., Laufer S. and Naujokat C. 2020 Candidate drugs against SARS-CoV-2 and COVID-19. Pharmaco. Res. 104859 (https://doi.org/10.1016/j.phrs.2020.104859).
Menachery V. D., Yount B. L. Jr., Debbink K., Agnihothram S., Gralinski L. E., Plante J. A. et al. 2015 A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 21, 1508.
Meyer D., Sielaff F., Hammami M., Böttcher-Friebertshäuser E., Garten W. and Steinmetzer T. 2013 Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 452, 331–343.
Millet J. K. and Whittaker G. R. 2014 Host cell entry of Middle East respiratory syndrome coronavirus after two-step furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 111, 15214–15219.
Millet J. K. and Whittaker G. R. 2015 Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 202, 120–134.
Nao N., Yamagishi J., Miyamoto H., Igarashi M., Manzoor R., Ohnuma A. et al. 2017 Genetic predisposition to acquire a polybasic cleavage site for highly pathogenic avian influenza virus hemagglutinin. Mol. Biol. 81, e02298–e02316.
Nelson-Sathi S., Umasankar P. K., Sreekumar E., Nair R. R., Joseph I., Nori S. R. C. et al. 2020 Structural and functional implications of non-synonymous mutations in the Spike protein of 2954 SARS-CoV-2 Genomes. BioRxiv, https://doi.org/10.1101/2020.05.02.071811.
Ortega J. T., Serrano M. L., Pujol F. H. and Rangel H. R. 2020 Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 19, 410–417.
Ortiz-Fernandez L. and Sawalha A. H. 2020 Genetic variability in the expression of the SARS-CoV-2 host cell entry factors across populations. BioRxiv, https://doi.org/10.1101/2020.04.06.027698.
Paniri A., Hosseini M. M. and Akhavan-Niaki H. 2020 First comprehensive computational analysis of functional consequences of TMPRSS2 SNPs in susceptibility to SARS-CoV-2 among different populations. J. Biomol. Struct. Dyn. 1-8. (https://doi.org/10.1080/07391102.2020.1767690).
Potdar V., Cherian S. S., Deshpande G. R., Ullas P. T., Yadav P. D., Choudhary M. L. et al. 2020 Genomic analysis of SARS-CoV-2 strains among Indians returning from Italy, Iran and China and Italian tourists in India. Indian J. Med. Res. 151(2), 255.
Prajapat M., Sarma P., Shekhar N., Avti P., Sinha S., Kaur H. et al. 2020 Drug targets for corona virus: A systematic review. Indian J. Pharmaco. 52, 56.
Qing E., Hantak M., Perlman S. and Gallagher T. 2020 Distinct Roles for Sialoside and Protein Receptors in Coronavirus Infection. Host-Microbe Biol. 11, e02764–e02819.
Qiu Y., Zhao Y. B., Wang Q., Li J. Y., Zhou Z. J., Liao C. H. et al. 2020 Predicting the angiotensin converting enzyme 2 ACE2 utilizing capability as the receptor of SARS-CoV-2. Microbes. Infect., https://doi.org/10.1016/j.micinf.2020.03.003.
Rahman N., Basharat Z., Yousuf M., Castaldo G., Rastrelli L. and Khan H. 2020 Virtual screening of natural products against type II transmembrane serine protease TMPRSS2 the priming agent of coronavirus 2 SARS-CoV-2. Molecules 25, 2271.
Reinke L. M., Spiegel M., Plegge T., Hartleib A., Nehlmeier I., Gierer S. et al. 2017 Different residues in the SARS-CoV spike protein determine cleavage and activation by the host cell protease TMPRSS2. PLoS One 12, e0179177.
Rensi S. B., Altman R., Liu T., Lo Y. C., McInnes G., Derry A. et al. 2020 Homology modeling of TMPRSS2 yields candidate drugs that may inhibit entry of sars-cov-2 into human cells. ChemRxiv, https://doi.org/10.26434/chemrxiv.12009582.v1.
Russo R., Andolfo I., Lasorsa V. A., Iolascon A. and Capasso M. 2020 Genetic analysis of the novel SARS-CoV-2 host receptor TMPRSS2 in different populations. BioRxiv, https://doi.org/10.1101/2020.04.23.057190.
Sandeep S. and McGregor K. 2020 Energetics based modeling of hydroxychloroquine and azithromycin binding to the SARS-CoV-2 Spike S protein-ACE2 complex. ChemRxiv, https://doi.org/10.26434/chemrxiv.12015792.v2.
Santesmasses D., Castro J. P., Zenin A. A., Shindyapina A. V., Gerashchenk M. V., Zhang B. et al. 2020 COVID-19 is emergent disease of aging. MedRxiv, https://doi.org/10.1101/2020.04.15.20060095.
Senapati S., Kumar S., Singh A. K., Banerjee P. and Bhagavatula S. 2020 Assessment of risk conferred by coding and regulatory variations from TMPRSS2 and CD26 in susceptibility of SARS-CoV-2 infection in human. J. Genet. 99, 53.
Sharma S., Singh I., Haider S., Malik M. Z., Ponnusamy K. and Rai E. 2020 ACE2 homo-dimerization human genomic variants and interaction of host proteins explain high population specific differences in outcomes of COVID19. BioRxiv, https://doi.org/10.1101/2020.04.24.050534.
Shereen M. A., Khan S., Kazmi A., Bashir N. and Siddique R. 2020 COVID-19 infection: origin transmission and characteristics of human coronaviruses. J. Adv. Res. 24, 91–98.
Sinha S. K., Shakya A., Prasad S. K., Singh S., Gurav N. S., Prasad R. S. and Gurav S. S. 2020 An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. J. Biomol. Struct. Dyn. 1-3. (https://doi.org/10.1080/07391102.2020.1762741).
Shulla A., Heald-Sargent T., Subramanya G., Zhao J., Perlman S. and Gallagher T. 2011 A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 85, 873–882.
Simmons G., Reeves J. D., Rennekamp A. J., Amberg S. M., Piefer A. J. and Bates P. 2004 Characterization of severe acute respiratory syndrome-associated coronavirus SARS-CoV spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA 101, 4240–4245.
Singh H., Singh J., Khubaib M., Jamal S., Sheikh J. A., Kohli S. et al. 2020 Mapping the genomic landscape and diversity of COVID-19 based on >3950 clinical isolates of SARS-CoV-2: Likely origin and transmission dynamics of isolates sequenced in India. Indian J Med. Res, https://doi.org/10.4103/ijmr.IJMR_1253_20.
Stawiski E. W., Diwanji D., Suryamohan K., Gupta R., Fellouse F. A., Sathirapongsasuti F. et al. 2020 Human ACE2 receptor polymorphisms predict SARS-CoV-2 susceptibility. BioRxiv, https://doi.org/10.1101/2020.04.07.024752.
Sungnak W., Huang N., Bécavin C., Berg M. and HCA Lung Biological Network 2020 SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26(5), 681–687.
Úri K., Fagyas M., Kertész A., Borbély A., Jenei C., Bene O. et al. 2016 Circulating ACE2 activity correlates with cardiovascular disease development. J. Renin Angiotensin Aldosterone Syst. 17, 1470320316668435.
Uri K., Fagyas M., Siket I. M., Kertesz A., Csanadi Z., Sandorfi G. et al. 2014 New perspectives in the renin-angiotensin-aldosterone system RAAS IV: circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS One 9, e87845.
Vaduganathan M., Vardeny O., Michel T., McMurray J. J., Pfeffer M. A. and Solomon S. D. 2020 Renin–angiotensin–aldosterone system inhibitors in patients with Covid-19. New England J. Med. 382, 1653–1659.
Veeramachaneni G. K., Thunuguntla V. B. S. C. and Bondili J. S. 2020 Structural and Simulation analysis of hot spot residues interactions of SARS-CoV-2 with Human ACE2 receptor. J. Biomol. Struct. Dyn. 1-16. (https://doi.org/10.1080/07391102.2020.1773318).
Vuille-dit-Bille R. N., Camargo S. M., Emmenegger L., Sasse T., Kummer E., Jando J. et al. 2015 Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids. 47, 693–705.
Walters T. E., Kalman J. M., Patel S. K., Mearns M., Velkoska E. and Burrell L. M. 2017 Angiotensin converting enzyme 2 activity and human atrial fibrillation: increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Ep Europace. 19, 1280–1287.
Wang Y., Liu M. and Gao J. 2020a Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 117, 13967–13974.
Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z. et al. 2020b Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181(4), 894–904.
Weiss S. R. and Leibowitz J. L. 2011 Coronavirus pathogenesis. In Advances in virus research. vol. 81. Academic Press, Cambridge, pp. 85–164.
Wrapp D., Wang N., Corbett K. S., Goldsmith J. A., Hsieh C. L., Abiona O. et al. 2020 Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263.
Wu Y., Wang F., Shen C., Peng W., Li D., Zhao C. et al. 2020 A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science. eabc2241.
Xiao K., Zhai J., Feng Y., Zhou N., Zhang X., Zou J. J. et al. 2020 Isolation and characterization of 2019-nCoV-like coronavirus from Malayan pangolins. BioRxiv, https://doi.org/10.1101/2020.02.17.951335.
Xu X., Chen P., Wang J., Feng J., Zhou H., Li X. et al. 2020 Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 63, 457–460.
Yamada Y. and Liu D. X. 2009 Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin-dependent entry syncytium formation and infectivity of coronavirus infectious bronchitis virus in cultured cells. J. Virol. 83, 8744–8758
Yamamoto M., Matsuyama S., Li X., Takeda M., Kawaguchi Y., Inoue J. I. et al. 2016 Identification of nafamostat as a potent inhibitor of Middle East respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob. Agents Chemother. 60, 6532–6539.
Yan R., Zhang Y., Li Y., Xia L., Guo Y. and Zhou Q. 2020 Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448.
Yan W., Sheng N., Seto M., Morser J. and Wu Q. 1999 Corin a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J. Biol. Chem. 274, 14926–14935.
Zhang H., Wada J., Hida K., Tsuchiyama Y., Hiragushi K., Shikata K. et al. 2001 Collectrin a collecting duct-specific transmembrane glycoprotein is a novel hololog of ACE2 and is developmentally regulated in embryonic kidneys. J. Biol. Chem. 276, 17132–17139.
Zhang Y., Zheng N., Hao P., Cao Y. and Zhong Y. 2005 A molecular docking model of SARS-CoV S1 protein in complex with its receptor human ACE2. Comput. Biol. Chem. 29, 254–257.
Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W. et al. 2020 A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273.
Zhou Y., Vedantham P., Lu K., Agudelo J., Carrion R. Jr., Nunneley J. et al. 2015 Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 116, 76–84.
Zhu N., Zhang D., Wang W., Li X., Yang B., Song J. et al. 2020 A novel coronavirus from patients with pneumonia in China 2019. New England J. Med. 382, 727–733.
Zmora P., Moldenhauer A. S., Hofmann-Winkler H. and Pöhlmann S. 2015 TMPRSS2 isoform 1 activates respiratory viruses and is expressed in viral target cells. PLoS One 10, e0138380.
Zumla A., Chan J. F., Azhar E. I., Hui D. S. and Yuen K. Y. 2016 Coronaviruses—drug discovery and therapeutic options. Nat. Rev. Drug Discov. 15, 327–347.
Acknowledgements
Authors acknowledge support from Central University of Punjab, Bathinda, India. SS is supported by DST-SERB, New Delhi, Government of India (ECR/2016/001660), and ICMR (33/6/2019TF/Rare/BMS). SK is supported by DST-SERB (EEQ/2016/000350), and UGC-BSR (F.30/372/2017-BSR). DST-FIST support (SR/FST/LS-I/2017/49-C) to Department of Human Genetics and Molecular Medicine is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: Shrish Tiwari
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
SENAPATI, S., BANERJEE, P., BHAGAVATULA, S. et al. Contributions of human ACE2 and TMPRSS2 in determining host–pathogen interaction of COVID-19. J Genet 100, 12 (2021). https://doi.org/10.1007/s12041-021-01262-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12041-021-01262-w