
Abstract
Real-time PCR is a widely used technique for quantification of gene expression. However, commercially available kits for real-time PCR are very expensive. The ongoing coronavirus pandemic has severely hampered the economy in a number of developing countries, resulting in a reduction in available research funding. The fallout of this will result in limiting educational institutes and small enterprises from using cutting edge biological techniques such as real-time PCR. Here, we report a cost-effective approach for preparing and assembling cDNA synthesis and real-time PCR mastermixes with similar efficiencies as commercially available kits. Our results thus demonstrate an alternative to commercially available kits.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Real-time polymerase chain reaction (real-time PCR) is a powerful technique to measure the level of gene expression. It is a quantitative PCR technique where data is collected simultaneously as the PCR amplification proceeds. It is an extremely sensitive technique with a large dynamic range and high sequence specificity (Wong and Medrano 2005), with ability to even detect a single copy of a specific transcript (Palmer et al. 2003). Real-time PCR is characterized by a Ct (Cycle threshold) value which indicates the cycle where the fluorescence intensity of the PCR product is greater than the background fluorescence (Heid et al. 1996). Detection of the amplicon in real-time PCR can be done in multiple ways. The amplicon can be detected by using hybridization probes such as Taqman probes (Holland et al. 1991), molecular beacons (Tyagi and Kramer 1996), Eclipse Probes (Lukhtanov et al. 2007), LUX PCR Primers (Vilcek et al. 2010) and Scorpions (Whitcombe et al. 1999). While these probes are target sequence-specific, they are not commonly used due to their high cost. For the detection of a large number of genes, fluorescent DNA binding dyes, which are not sequence-specific, and intercalate between double-stranded DNA can be employed. Various DNA binding dyes such as SYBR Green I (Green and Sambrook 2018) and SYTO dyes (Gudnason et al. 2007; Eischeid 2011) are commercially used to detect the amplified PCR product, of which SYBR Green I is the most widely used. However, the commercially available SYBR Green I mastermixes are expensive. Hence, we set out with a goal to design a low-cost, in-house, real-time PCR mastermix using reagents easily available in labs, and which are as efficient as commercially available mastermixes.
SYBR Green I is the most widely used dye in real-time PCR mastermixes. Recently, EvaGreen dye has also been used for quantification in real-time PCR (Dhami and Kumarasinghe 2014). Reaction efficiency for EvaGreen has been shown to outperform SYBR Green I (Eischeid 2011). EvaGreen dye is spectrally similar to other dyes such as SYBR Green I and FAM (Mao et al. 2007). Hence, no changes are required in the optical settings of the instrument while using EvaGreen dye. Here, we demonstrate a protocol to purify in-house Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT) (Graham et al. 2021) for cDNA synthesis and designed an in-house real-time PCR mastermix using EvaGreen or SYBR Green I dye and in-house Hot start Taq DNA polymerase. The Ct values, amplification plots, dissociation curves were comparable between commercially available SYBR Green I PCR mastermix, and our in-house real-time PCR mastermix with either SYBR Green I or Evagreen dye. The in-house PCR mastermix is both sensitive, cost-effective and can be easily assembled. Our results therefore provide an effective solution towards developing an in-house real-time PCR mastermix which can be used as a cost-effective and efficient alternative towards commercially available real-time PCR mastermixes.
2 Materials and methods
2.1 RNA isolation
Total RNA was isolated from mESCs using TRIzol (Invitrogen Cat no. 15596018). Culture media was removed and cells were washed once with 1XDPBS. 500µl TRIzol was added to lyse the cells and plates were kept on the rocker for 5 minutes. Cells were scraped and the lysate was collected in an eppendorf. 100 µl of chloroform was added to the TRIzol lysate and mixed thoroughly by shaking. Samples were kept at room temperature for 5 minutes and then centrifuged for 15 min at 12000g at 4°C. The upper aqueous phase containing RNA was collected in a fresh eppendorf tube and 250 µl isopropanol was added to the sample. Sample was mixed and kept at room temperature for 10 min and later centrifuged for 10 min at 12000g at 4°C. Supernatant was discarded and the pellet was washed with 70% ethanol. Sample was centrifuged for 5 min at 7500g at 4°C. The pellet was air dried and RNase free water or DEPC-treated water was added to the sample. RNA was quantified using Nanodrop spectrophotometer.
2.2 DNase treatment
1 µg of total RNA was used for DNAseI treatment (Invitrogen cat no. 18068-015). Details of the reaction are given below:
-
RNA sample – 1µg
-
10x DNase I buffer – 1 µl
-
DNase I – 1 µl
-
DEPC-treated water – upto 10 µl
Samples were incubated at room temperature for 15 min. 1 µl of 25mM EDTA solution was added to the reaction to deactivate DNase I. Samples were kept at 65°C for 10 min followed by cDNA synthesis.
2.3 Complementary DNA (cDNA) synthesis
DNAseI treated total RNA from mESCs was used to synthesize complementary DNA (cDNA) using random primers (Promega Cat no. C1181), dNTP mix (LAROVA Cat no. DMIX10_100ML) and in-house generated Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT). Details of the reaction assembly are given below.
2.4 cDNA synthesis reaction
-
5x BEAR Buffer – 4µl
-
10 mM dNTP – 2µl
-
Random Hexamers (100 ng/µl) – 1 µl
-
M-MLV Enzyme – 1 µl
-
Dnase treated RNA reaction volume – 11 µl
-
Total reaction volume – 20 µl
The above reaction was assembled in PCR tubes and was placed in Eppendorf mastercycler PCR machine using the following program: 52°C – 1 h; 95°C – 2 min
2.5 Real-time PCR
Complementary DNA (cDNA) was diluted (1:10) times and used as a template for Real-time PCR. ABI Power SYBR Green PCR master mix was used as commercial reagent. 2x in-house SYBR Green I and 2x in-house EvaGreen mix was prepared as described below. SYBR Green I (Lonza Cat no. 50513) or EvaGreen dye (Biotium Cat no. 31000), dNTP mix (LAROVA Cat no. DMIX10_100ML) were used to prepare mastermix. ABI 384 well plate (cat no. AB 1384) was used to set up real time PCR. 5µl of mastermix was added to each well. 1µl of (1:10) diluted cDNA was added as template. Total reaction volume was 6µl. ABI 7900 HT machine was used to perform Real-time PCR. Detailed composition of SYBR Green I and EvaGreen PCR mastermix is given below:
2X In-house SYBR Green I mix composition | 2X In-house EvaGreen mix composition |
|---|---|
2X In-house buffer – 1000 µl 10 mM dNTP – 25 µl 10000X SYBR Green I – 0.1 µl Hotstart Taq – 10 µl | 2X In-house buffer – 1000 µl 10 mM dNTP – 25 µl 20X Evagreen – 100 µl Hotstart Taq – 10 µl |
Both, 2X In-house SYBR Green I mix and 2X In-house EvaGreen mix can be stored at 4°C for up to 2 weeks. For long term storage, mastermix was stored at −20°C.
2.6 Real-time PCR reaction
-
2X In-house SYBR Green I mix or 2X In-house EvaGreen mix – 3 µl
-
100 µM Forward primer – 0.6 µl
-
100 µM Reverse primer – 0.6 µl
-
Milli Q water – 1.88 µl
-
Total reaction volume – 6 µl
The above reaction was set up with the given concentration of primers in 384-well plate.
2.7 Real-time PCR program
The conditions used for real-time PCR were:
Stage 1 | 50°C – 2 min |
Stage 2 | 95°C – 18 min |
Stage 3 | 95°C – 15 s |
60°C – 1 min | |
Repeat stage 3 (40 cycles) | |
Stage 4 (Dissociation stage) | 95°C – 15 s |
60°C – 15 s |
2.8 Buffer compositions
The buffer composition for 2X in-house buffer and 5X BEAR buffer was as follows:
2x In-house buffer composition – 14 ml | 5x BEAR (Basic Economical Amplification Reaction) buffer composition |
|---|---|
90 µl 2M Tris (pH 8.1) 250 µl 3M KCl 75 µl 1M MgCl2 850 mg Trehalose 30 µl 100% Tween-20 150 µl 20 mg/ml BSA | 250 mM Tris-HCL (pH 8.4) 375 mM KCl 15 mM MgCl2 10% Trehalose 50 mM Dithiothreitol (DTT) 0.5mM EDTA |
2.9 Buffers for M-MLV reverse transcriptase purification
The buffer composition for M-MLV purification was:
Lysis buffer | SP buffer A | Elution buffer | M-MLV storage buffer |
|---|---|---|---|
50 mM Tris-Hcl, pH 8 100 mM NaCl 10 mM Imidazole 1 mM DTT 0.1% Triton X-100 | 50 mM Tris-HCl, pH 8 100 mM NaCl 0.1 mM EDTA 5 mM DTT 0.1% Triton X-100 | 50 mM Tris-HCl, pH 8 100 mM NaCl 250 mM Imidazole 1 mM DTT 0.1% Triton X-100 | 50 mM Tris-HCl, pH 8 100 mM NaCl 0.1 mM EDTA 5 mM DTT 0.1% Triton X-100 50% glycerol |
2.10 Buffers for hot-start Taq polymerase preparation
The buffer composition for hot-start Taq polymerase purification was:
Lysis buffer | Heparin dialysis buffer | Taq storage buffer |
|---|---|---|
50 mM Tris-HCl, pH 8 500 mM NaCl 0.1% NP-40 0.1% Triton X-100 | 50 mM Tris-HCl, pH 8 100 mM NaCl 0.05% NP-40 10% glycerol 5 mM BME 1 mM benzamidine | 50 mM Tris-HCl, pH 8 100 mM NaCl 0.1 mM EDTA 50% glycerol 3mM DTT |
3 Results
3.1 Optimization of in-house SYBR Green I and EvaGreen PCR mastermix
Real-time PCR technique is an integration of 3 processes. RNA isolation, cDNA synthesis and real-time PCR (figure 1A). One of the essential steps after RNA isolation is cDNA synthesis. There are a large number of commercially available kits for cDNA synthesis which are very expensive. Here, we have used a protocol for synthesis and purification of Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT) (Graham et al. 2021) which was used for the synthesis of cDNA. Briefly, expression plasmid pET-28a_6H-MMLV_RT_D524N-6H (Addgene plasmid # 166945) was transformed into BL21 competent cells. Cultures were grown overnight at 37°C, induced with 1 mM IPTG, pelleted down and flash-frozen in liquid nitrogen and stored at −80°C until further use. Pellets were resuspended in lysis buffer and subjected for Ni-NTA purification followed by HiTrap SP sepharose purification (figure 1B and C). Proteins fractions were eluted in storage buffer and stored at −80°C until further use. (For buffer compositions refer materials and methods, and for detailed purification protocol refer (Graham et al. 2021)). cDNA synthesis was carried using 1µg of RNA from mouse ESCs as described in materials and methods. The efficiency of cDNA synthesis was analysed by GAPDH PCR (supplementary figure 1A). Next, cDNA was diluted to 1:10 v/v and used as template for real-time PCR. Hot-start Taq polymerase was purified as per the previously published protocol (Graham et al. 2021). Briefly, expression plasmid pET-28a_6H-TAQ_E602D (Addgene #166944) was transformed into BL21 competent cells, cultures were grown overnight at 37°C, induced with 1 mM IPTG, pelleted down and flash-frozen in liquid nitrogen and stored at −80°C until further use. Pellets were resuspended in lysis buffer and subjected to Ni-NTA based purification followed by HiTrap heparin purification. Proteins fractions were eluted in storage buffer and stored at −80°C until further use (for buffer composition refer materials and methods and for detailed purification protocol refer (Graham et al. 2021)). This hot-start Taq polymerase was used in the preparation of the in-house PCR mastermix (figure 1D and E). The dNTP concentration was optimized using a final concentration of 400 µM, 300µM and 250µM with EvaGreen I in-house mastermix (supplementary table 1). Amplification was observed with all the concentrations, however there was a lot of background noise with 400 µM dNTP which was absent with 250µM dNTP (data not shown). We therefore recommend using a final concentration of 250µM dNTP for all experiments. Ct values for Oct4, Sox2, Nestin, Brachyury, Gapdh and Rpl7 were consistent at all the above dNTP concentrations. Also, Ct values for duplicates of each sample were very close to each other implying the efficiency of in-house mastermixes (supplementary table 1). For optimized brew buffer composition refer materials and methods.

Purification of Moloney murine leukemia virus reverse transcriptase (M-MLV RT) and Hot-start Taq polymerase: (A) Flow chart showing various steps from RNA isolation, cDNA synthesis and real-time PCR set up under different conditions. (B) Representative image of SDS-PAGE stained with Coomassie brilliant blue showing purified fractions F1-F6 (Fraction 1–6) after Ni-NTA purification of M-MLV RT. (C) Representative image of SDS-PAGE stained with Coomassie brilliant blue showing purified fractions 1–4 of M-MLV RT after HiTrap SP sepharose purification images for purification. (D) Representative image of SDS-PAGE stained with Coomassie brilliant blue showing purified fractions F1-F6 (Fraction 1–6) after Ni-NTA purification of Hot start Taq DNA polymerase. (E) Representative image of SDS-PAGE stained with Coomassie brilliant blue showing purified fractions F1-F6 (Fraction 1–6) after HiTrap heparin purification of Hot start Taq DNA polymerase.
3.2 Efficiency of in-house SYBR Green I and EvaGreen PCR mastermix is comparable to commercial SYBR Green I mastermix
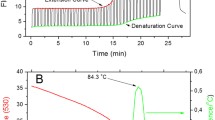
Oct4, Sox2 and Nanog transcription factors are highly expressed in embryonic stem cells (ESCs) (Boyer et al. 2005). Hence, we selected transcription factors Oct4, Sox2, Nanog genes for real-time PCR optimization in mouse ESCs (mESCs) with Gapdh and Rpl7 as housekeeping controls. Real-time PCR was set up using commercially available SYBR Green I mastermix PowerUp SYBR Green Master Mix (Applied Biosystems Cat no. A25742) and in-house SYBR Green I or EvaGreen mastermix. Ct values for Oct4, Sox2, Nanog, Gapdh and Rpl7 were comparable in commercial SYBR Green I mastermix and in-house SYBR Green I or EvaGreen mastermix. A comparative table with the Ct values of different genes using all the mastermixes is given in supplementary table 2. Amplification plots represent the accumulation of the PCR product over successive PCR cycles. Amplification plots were comparable between the commercial SYBR Green I mastermix (figure 2A), and in-house SYBR Green I (figure 3A), or EvaGreen mastermix (figure 4A). Melting curve is used to analyze if there is nonspecific amplification in the PCR along with the specific band (Ririe et al. 1997). The single amplified product is detected by the presence of single peak whereas non-specific amplification results in appearance of multiple peaks. Melting curve for all the samples amplified with commercial SYBR Green I mastermix (figure 2B), and in-house SYBR Green I (figure 3B) or EvaGreen mastermix (figure 4B) had a single peak implying the presence of a single and specific amplicon in the PCR reaction. Real-time PCR products for Oct4, Sox2 and Gapdh amplified using commercial SYBR Green I mastermix and in-house EvaGreen PCR mastermix were run on 1.5% agarose gel. A specific and unique single band was observed for Oct4, Sox2 and Gapdh at 119bp, 99bp and 107bp corresponding to the specificity of the in-house EvaGreen PCR mastermix (supplementary figure 1B and supplementary table 4). To demonstrate the sensitivity of the in-house PCR mastermix we set up real-time PCR for Oct4, Sox2 and Gapdh with either commercial SYBR Green I mastermix or in-house EvaGreen PCR mastermix using mESC cDNA at different concentrations. 1µl of (1:2.5), (1:10), (1:20) and (1:40) diluted cDNA was used as template. A corresponding and proportionate increase in Ct value was observed for Oct4, Sox2 and Gapdh with increase in cDNA dilution indicating good sensitivity of the commercial SYBR Green I mastermix as well as of in-house Evagreen PCR mastermix (supplementary figure 2A).

Amplification plots and dissociation curves for indicated genes with commercial SYBR Green I mastermix: (A) Amplification plots for pluripotency marker genes Oct4, Sox2, Nanog and housekeeping genes Gapdh and Rpl7. (B) Dissociation curves for pluripotency marker genes Oct4, Sox2, Nanog and housekeeping genes Gapdh and Rpl7.

Amplification plots and dissociation curves for different genes with in-house SYBR Green I mastermix: (A) Amplification plots for pluripotency marker genes Oct4, Sox2, Nanog and housekeeping genes Gapdh and Rpl7. (B) Dissociation curves for pluripotency marker genes Oct4, Sox2, Nanog and housekeeping genes Gapdh and Rpl7.

Amplification plots and dissociation curves for different genes with In-house EvaGreen PCR mastermix: (A) Amplification plots for pluripotency marker genes Oct4, Sox2, Nanog and housekeeping genes Gapdh and Rpl7. (B) Dissociation curves for pluripotency marker genes Oct4, Sox2, Nanog and housekeeping genes Gapdh and Rpl7.
3.3 Costing for commercial SYBR Green I mastermix and in-house SYBR Green I or EvaGreen PCR mastermix
Real-time PCR technique consists of 3 processes: RNA isolation, cDNA synthesis and real-time PCR. RNA isolation can be done with commercially available RNA isolation kits which are very expensive. The other traditional way is to use TRIzol for RNA isolation. Details of RNA isolation using TRIzol are given in materials and methods. The next step, cDNA synthesis is done with the help of commercially available kits. A wide range of cDNA synthesis kits are available commercially with price ranging from INR 25,000 - 50,000 per 50–100 reactions. Synthesis and purification of M-MLV RT for cDNA synthesis along with other components like MgCl2, dNTPs, oligo dT or random hexamers cost INR 21 per reaction. The final step is the real-time PCR set up. Commercially available SYBR Green I mastermix costs around INR 3000-4000 per ml. In-house SYBR Green I mastermix costs INR 50 per ml and EvaGreen PCR mastermix costs INR 510 per ml. For detailed costing in INR (Indian rupees) refer supplementary table 3. In summary, we demonstrate a cost-effective and efficient method to assemble mastermixes for cDNA synthesis and RT-PCR.
3.4 Discussion
Real-time PCR is the most favoured technique for measuring gene expression. Here, one can measure the expression of several genes with relatively low amounts of sample. However, commercially available RNA isolation kits, cDNA synthesis kits and SYBR Green I mastermix required to set up real-time PCR are very expensive and cannot be afforded by several educational institutions and small enterprises. The ongoing coronavirus pandemic has negatively impacted the research funding in a large number of developing countries, including India. This adds to the additional hurdle which limits research scholars from using expensive cutting edge techniques such as real-time PCR. Earlier, there have been attempts to prepare homemade SYBR Green I mastermix for real-time PCR. However, this mastermix contained commercial Taq polymerase and cDNA was also synthesised using commercially available kits (Karsai et al. 2002). This increases the expenses of conducting real-time PCR. While Graham et al demonstrate the assembly of home-made mastermixes for cDNA synthesis and real-time PCR, they do not provide a direct comparison with commercially available reagents for the same (Graham et al. 2021). Our study provides such a direct comparison, and also includes alternatives to SYBR Green I, such as EvaGreen. Through our manuscript, we have systematically integrated and validated protocols for RNA isolation, cDNA synthesis and real-time PCR using in-house reagents. We have compared the efficacy of these reagents with commercially available kits. Our results demonstrate that real-time PCR set up using in-house SYBR Green I or EvaGreen PCR mastermix generated Ct values, amplification plots and dissociation curves comparable to commercially available SYBR Green I PCR mastermix. These in-house reagents are not only cost-effective, but are also sensitive. Hence, these in-house reagents for real-time PCR are a promising alternative to commercially available kits.
Change history
12 January 2022
A Correction to this paper has been published: https://doi.org/10.1007/s12038-021-00242-7
References
Boyer LA, Lee TI, Cole MF, et al. 2005 Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122 947–956
Dhami MK and Kumarasinghe L 2014 A HRM Real-Time PCR assay for rapid and specific identification of the emerging pest spotted-wing Drosophila (Drosophila suzukii). PLOS ONE 9 e98934
Eischeid AC 2011 SYTO dyes and EvaGreen outperform SYBR Green in real-time PCR. BMC Res. Notes 4 263
Graham TGW, Dugast-Darzacq C, Dailey GM, Darzacq X and Tjian R 2021 Simple, inexpensive RNA isolation and one-step RT-qPCR methods for SARS-CoV-2 detection and general use. Curr. Protoc. 1 e130
Green MR and Sambrook J 2018 2018 Quantification of RNA by real-time reverse transcription-polymerase chain reaction (RT-PCR). Cold Spring Harb. Protoc.
Gudnason H, Dufva M, Bang DD and Wolff A 2007 Comparison of multiple DNA dyes for real-time PCR: effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res. 35 e127
Heid CA, Stevens J, Livak KJ and Williams PM 1996 Real time quantitative PCR. Genome Res. 6 986–994
Holland PM, Abramson RD, Watson R and Gelfand DH 1991 Detection of specific polymerase chain reaction product by utilizing the 5’–3’ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 88 7276–7280
Karsai A, Müller S, Platz S and Hauser M-T 2002 Evaluation of a Homemade SYBR® Green I reaction mixture for real-time pcr quantification of gene expression. BioTechniques 32 790–796
Lukhtanov EA, Lokhov SG, Gorn VV, Podyminogin MA and Mahoney W 2007 Novel DNA probes with low background and high hybridization-triggered fluorescence. Nucleic Acids Res. 35 e30
Mao F, Leung W-Y and Xin X 2007 Characterization of EvaGreen and the implication of its physicochemical properties for qPCR applications. BMC Biotechnol. 7 76
Palmer S, Wiegand AP, Maldarelli F, et al. 2003 New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J. Clin. Microbiol. 41 4531–4536
Ririe KM, Rasmussen RP and Wittwer CT 1997 Product differentiation by analysis of DNA Melting curves during the polymerase chain reaction. Anal. Biochem. 245 154–160
Tyagi S and Kramer FR 1996 Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 14 303–308
Vilcek S, Vlasakova M and Jackova A 2010 LUX real-time PCR assay for the detection of porcine circovirus type 2. J. Virol. Methods 165 216–221
Whitcombe D, Theaker J, Guy SP, Brown T and Little S 1999 Detection of PCR products using self-probing amplicons and fluorescence. Nat. Biotechnol. 17 804–807
Wong ML and Medrano JF 2005 Real-time PCR for mRNA quantitation. BioTechniques 39 75–85
Acknowledgements
This work was supported by funds to DS from the Department of Biotechnology (BT/PR25883/GET/119/105/2017), and ICMR (2020-3076/SCR/ADHOC-BMS). DS, AM, VS and VT acknowledge intramural funding from National Centre for Cell Science. SBS is a recipient of a Senior Research Fellowship from the Department of Biotechnology, India; MT and JS are recipients of a Senior Research Fellowship from University Grants Commission, India; HS is a recipient of a Senior Research Fellowship from CSIR, India. We thank members of the Subramanyam and Majumdar lab for constructive discussion.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by BJ Rao.
Corresponding editor: BJ Rao
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mote, R.D., Laxmikant, V.S., Singh, S.B. et al. A cost-effective and efficient approach for generating and assembling reagents for conducting real-time PCR. J Biosci 46, 109 (2021). https://doi.org/10.1007/s12038-021-00231-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-021-00231-w