
Abstract
Several neurological disorders, neurodevelopmental disorders, and neurodegenerative disorders have a genetic element with various clinical presentations ranging from mild to severe presentation. Neurological disorders are rare multifactorial disorders characterized by dysfunction and degeneration of synapses, neurons, and glial cells which are essential for movement, coordination, muscle strength, sensation, and cognition. The cerebellum might be involved at any time, either during development and maturation or later in life. Herein, we describe a spectrum of NDDs and NDs in seven patients from six Egyptian families. The core clinical and radiological features of our patients included dysmorphic features, neurodevelopmental delay or regression, gait abnormalities, skeletal deformities, visual impairment, seizures, and cerebellar atrophy. Previously unreported clinical phenotypic findings were recorded. Whole-exome sequencing (WES) was performed followed by an in silico analysis of the detected genetic variants’ effect on the protein structure. Three novel variants were identified in three genes MFSD8, AGTPBP1, and APTX, and other previously reported three variants have been detected in “TPP1, AGTPBP1, and PCDHGC4” genes. In this cohort, we described the detailed unique phenotypic characteristics given the identified genetic profile in patients with neurological “neurodevelopmental disorders and neurodegenerative disorders” disorders associated with cerebellar atrophy, hence expanding the mutational spectrum of such disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inherited complex genetic disorders are common in Egypt and constitute a public health burden [1]. Neurodevelopmental disorders affect more than 3% of the pediatric population in many of their severe chronic and progressive forms that are expected to have an underlying genetic etiology that remains undiagnosed despite all available genetic tools [2].
Neurological disorders such as neurodevelopmental disorders and neurodegenerative disorders (NDs) are characterized by extreme genetic heterogeneity [3, 4] implying that whole-exome sequencing (WES) is the most appropriate first-tier test to cover more analyzed genes in neurogenetic disorders [5].
Neurodegenerative disorders are a heterogeneous group of mostly genetically determined diseases that result in progressive loss of neuronal structures or functions in different areas of the central and peripheral nervous system with a resultant loss of the previously acquired motor, sensory, and cognitive functions [4]. In the pediatric age group, the accretion of new developmental milestones does not exclude the existence of an ND disorder. On the clinical level, neurodegenerative disorders share similar manifestations including visual and hearing impairment, seizures, skeletal deformities, feeding, and intellectual difficulties [6]. Therefore, reaching a specific diagnosis could be quite challenging in the pediatric age group especially in resource-limited countries due to several reasons such as the ability of the clinicians to discriminate between the loss of a previously acquired and a delay in the achievement of specific developmental milestones, lack of expertise, and the long list of unaffordable potential investigations including molecular genetic analysis [7]. On the radiological level, the posterior fossa structures show variable degrees of involvement suggesting the period of affection such as growth cessation “prenatal,” growth cessation with concurrent atrophy “prenatal and postnatal,” or either stationary or progressive cerebellar atrophy “postnatal” [8]. On the molecular and biochemical level, NDs are characterized by depositions of misfolded, toxic conformations of various proteins, which generally accumulate to form insoluble deposits [9].
In this study, we will review the clinical features and radiological findings to explore the molecular and mutation spectrum in seven Egyptian patients with neurological “neurodevelopmental disorders and neurodegenerative disorders” disorders with an overlapping phenotype. We employed WES to screen the mutations and investigate the genotypic and phenotypic heterogeneities of molecularly characterized patients. With this, we aim to provide a better understanding of neurological disorders with an underlying genetic etiology among clinicians especially in resource-limited countries to help them offer appropriate management, prognosis expectations, and proper genetic counseling.
Material and Methods
Ethical Approval
The ethical approval was granted by the Medical Research Ethics Committee of the National Research Centre (NRC), Cairo, Egypt (Number: 932702021) according to the Declaration of Helsinki. Informed consent was obtained from the patient’s parents.
Patient Enrollment and Clinical Analysis
Seven patients (five males and two females) were recruited from the Multiple Congenital Anomalies Clinic (MCAs), Clinical Genetics Department, National Research Centre (NRC). Patients either presented primarily at the MCAs or have been referred by participating pediatric neurologists or ophthalmologists for further evaluation and workup completion.
Patients presented with developmental delay, neurodevelopmental regression (NDR), neurobehavioral disorders, visual or hearing impairment, short stature, abnormal gait, skeletal abnormalities, seizures, or family history of early unexplained death with uneventful perinatal or postnatal course were included in this study.
Once patients were identified as having possible neurological neurodevelopmental disorders or neurodegenerative disorders disorder, the caregivers were counseled about the possible genetic diagnosis and the required genetic test. The initial evaluation of patients comprised detailed history taking including “family history of a similar condition or other genetic disorders and perinatal history of possible prenatal insult or postnatal complications”, neurodevelopmental milestones assessment, and thorough physical and neurological examination. Patients with a perinatal history of maternal infection or postnatal complications such as kernicterus, meningitis, stroke, posterior fossa surgery, radiotherapy, head trauma, or suspected primary mitochondrial disorders were excluded from the study.
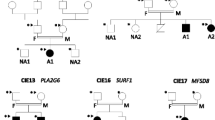
Detailed demographic data revealed positive consanguinity of all the examined patients and positive family history in four patients. The age range was from 1.5 to 18 years old at presentations. Anthropometric measurements including “head circumference, weight, and height/length” were plotted according to the recommendation of the International Biological Program (IBP) and showed underweight in two patients while height was not evaluated in one patient due to the associated scoliosis and joint contractures [10]. The detailed family pedigree is shown in Fig. 1. Detailed patients’ demographic data and anthropometric measurements are shown in Table 1. A special emphasis on the presence of dysmorphic features, skeletal deformities, and a review of other body systems was carried out to analyze the phenotypic presentation of the index patients. Physical and neurological examination was carried out for both parents and available siblings. The core clinical presentation of our patients included dysmorphic features (two patients), neurodevelopmental delay or regression (seven patients), gait abnormalities (three patients), skeletal deformities (five patients), visual impairment (seven patients), and seizures (five patients). Additional clinical features and diagnostic workup are described in Table 2.

Family pedigrees of the six studied unrelated families. A P1, B P2, C P3, D P4, E P5&P6, F P7
Other Ancillary Tests
A skeletal survey including plain X-rays of the “skull, spine, pelvis, short and long bones,” brain magnetic resonance imaging (MRI), electroencephalogram (EEG), electroretinogram (ERG), visual evoked potential (VEP), and echocardiography (Echo) was carried out whenever indicated.
Cytogenetic Analysis
Karyotype analysis was performed for all patients to exclude the presence of any chromosomal abnormalities. Chromosomal preparations were done from peripheral blood samples collected on lithium heparin vacutainers, following standard protocols [11].
Molecular Analyses
DNA Extraction and WES
A total of 3-ml venous blood was collected in EDTA tubes from all patients and their available family members. Genomic DNA was extracted from peripheral blood samples of all participants using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The quality and quantity of DNA samples of patients were assessed using fluorometric Denovix Qubit™ dsDNA BR Assay Kit (ThermoFisher, Waltham, MA, USA). DNA samples were sequenced by using the Twist Human Core Exome Plus kit (Twist Bioscience, San Francisco, CA, USA) and Illumina NovaSeq 6000 system (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. Libraries were prepared in paired-end mode (2 × 100 bp) for an output of 6 GB per sample, and an average coverage of 50 × . Sequencing reads were demultiplexed using Illumina Genes 2022, 13, 1056 4 of 24 bcl2fastq (2.20), and adapter sequences were trimmed using Skewer (version 0.2.2) [12]. The quality of the generated FASTQ files was analyzed with FastQC software (version 0.11.5; Illumina, San Diego, CA, USA). Variant Annotation and Filtration PhenoDB tool were used to annotate Vcf files using ANNOVAR [13]. Variants were filtered based on the depth of coverage and minor allele frequencies (MAF) (less than 1% MAF) in large population databases, including dbSNP [14], 1000 Genomes Project [15], and the Genome Aggregation Database (gnomAD v2.1.1) [16].
Variant Segregation
Sanger sequencing was used to confirm that prioritized variants segregated consistently among parents and available family members according to the predicted mode of inheritance. We designed primers targeting exons that harbor the filtered variants of interest using the Primer3 tool [17]. PCR was carried out as previously described. Reactions were sequenced according to the manufacturer’s recommendation using the Big Dye Termination kit (Applied Biosystems, Waltham, MA, USA) and ABI Prism 3500 Genetic Analyzer (Applied Biosystems, Waltham, MA, USA). Variants were named based on Human Genome Variation Society nomenclature recommendations [18]. The standards of the American College of Medical Genetics and Genomics (ACMG) were used to classify the level of variant pathogenicity, i.e., pathogenic, likely pathogenic, variant of unknown significance (VUS), benign, or likely benign [19].
Multi-scale Computational Analysis
The possible biological effect of all missense variants was done using multi-scale computational analysis tools considering all probable pathogenicity relevant aspects. Multi-scale computational analysis approach supports multiple entry variants for annotation and analysis permitting the closest true pathogenicity implication to be concluded. To explore the functional network among all variants harboring genes in this study, the functional enrichment and protein-protein enrichment analyses were carried out using GeneMania and STRING servers.
All the variants were described using chromosomal reference sequences according to HGVS recommendations and were checked by LUMC mutalyzer v. 3.0.4 according to GRCH38 human genome assembly. All mentioned genes were described according to HGNC nomenclature.
Results
Clinical Features and Phenotyping
Table 1 displays the patient’s anthropometric measurements, demographic information, and siblings who are also affected.
Patient 1 presented with global developmental delay (GDD), language impairment, seizures, progressive loss of vision with poor ocular fixation, progressive spastic quadriplegia, and scoliosis. A maternal history of recurrent spontaneous abortion was recorded. The ancillary tests-ERG showed retinal dysfunction, EEG showed interictal generalized epileptiform activity, and brain MRI showed cerebral and cerebellar atrophy and hypoplastic corpus callosum (Fig. 2A, B).

Brain MRI findings of the studied patients. P1 (A, B) cerebral and cerebellar atrophy and hypoplastic corpus callosum; P2 (C, D) cerebellar vermis hypoplasia; P3 (E) cerebellar atrophy
Patient 2 presented with dysmorphic facial features including “synophrys, short philtrum, thick lips, micrognathia and low set ears,” poor ocular fixation and oculomotor apraxia, delayed motor development, spasticity, generalized tonic-clonic seizures, atrial septal defect (ASD), scoliosis, and bilateral hip dislocation. The family history of a similarly affected sister was reported. The ancillary tests-EEG was normal, and brain MRI showed cerebellar vermis hypoplasia (Fig. 2C, D).
Patient 3 presented with GDD, dysmorphic facial features “course facies, narrow forehead, thick eyebrows, broad bulbous nose, short philtrum, thick lips, and large ears,” oculomotor apraxia, and seizures. He had multiple skeletal deformities “arachnodactyly, bilateral hyperextensibility of the interphalangeal joints, bilateral low inserted thumb, toes camptodactyly, bilateral prominent heel, severe scoliosis and joint contractures” and bilateral fungal infection of both feet (Fig. 3). The ancillary tests-ERG showed retinal dysfunction, EEG was abnormal, and brain MRI showed cerebellar atrophy (Fig. 2E).

Patient 3—phenotypic dysmorphic features, scoliosis, skeletal deformities, and fungal feet infection. X-ray chest PA view showing severe scoliosis and ribs crowding (E), MRI brain (F, G) showed cerebellar atrophy
Patient 4 presented with intellectual disability, progressive ataxia “started at the age of 5 years,” intension tremors, and oculomotor apraxia. He had also aortic regurgitation with a thickened valve. The ancillary tests-ERG showed retinal dysfunction, VEP showed bilateral optic nerve dysfunction, and brain MRI showed cerebral and cerebellar atrophy and hypoplastic corpus callosum (Fig. 2G, H).
Patients 5 and 6 (siblings) both presented with progressive NDR, intellectual disability, and seizures (photosensitive epilepsy). They were also noted to have progressive visual decline, poor visual hand-motor coordination, and abnormal clumsy gait. Patient 5 had focal to bilateral seizures with loss of awareness that were preceded by seeing different colors. His EEG showed a single burst of sharply contoured sharp waves over the right parasagittal area. Patient 6 had scoliosis and seizures “absence and generalized tonic and clonic” that were triggered by intense light. Her EEG showed generalized, fragmented, and focal rhythmic epileptiform discharges arising mainly from the right hemisphere. Their seizures were initially well controlled on levetiracetam, but later on, they showed a refractory course. Both patients had abnormal ERG/VEP as shown in Table 2. Both patients had cerebellar atrophy as shown in brain MRI findings in patient 5 (Fig. 2I, J).
Patient 7 presented with GDD, speech difficulties, and impaired cognition. He had feeding difficulties, strabismus, and oculomotor apraxia. He had a left-hand preference and poor fine motor skills. He had never been able to walk independently and exhibited bilateral knee contractures and bilateral tightness of the Achilles tendon. The ancillary tests-ERG/VEP showed bilateral optic nerve dysfunction, and brain MRI showed cerebellar atrophy (Fig. 2K).
The Genetic Spectrum of NDs Patients
Exome analyses of the seven studied patients descending from six unrelated Egyptian families identified six different disease-causing variants in five genes; TPP1 (NM_000391.4), MFSD8 (NM_001371596.2), AGTPBP1 (NM_001330701.2), APTX (NM_001195248.2), and PCDHGC4 (NM_018928.3) genes as displayed in Tables 2 and 3. All of these variants were missense except one defined as a splicing variant and according to ACMG classification criteria were classified as pathogenic variants. As well, the analysis confirmed that among these, three variants were not found in dbSNP, TGP, gnomAD exome, and ExAC databases or in our in-house database of 55 Egyptian exomes.
Sanger sequencing was performed to confirm that prioritized variants segregated consistently among parents and available family members according to the predicted mode of inheritance. The chromatograph for available patients who completed the follow-up was described in Fig. 4.

Sanger sequencing chromatograms of three patients (P1, P3, P4)
Patient 1 had a homozygous splicing variant in TPP1 gene (C.1145 + 2 T > G). This variant is predicted to disrupt the highly conserved donor splice site of exon 9. Together with the clinical information and biochemical results, it is classified as pathogenic (class 1) according to the ACMG. The result is consistent with the genetic diagnosis of AR NCL type 2 (Fig. 4).
Patient 2 had a homozygous missense variant in the PCDHGC4 gene (c.1463C > T; p.(Ala488Val). It is a likely pathogenic variant according to ACMG. Defects in PCDHGC4 have been associated with NEDGS.
Patient 3 had a novel homozygous missense variant in the AGTPBP1 gene (c.2650A > C; p.(Thr884Pro) substituting threonine residue for proline at position 884. It is a likely pathogenic variant according to ACMG. Pathogenic variants in the AGTPBP1 gene are associated with AR CONDCA (Fig. 4).
Patient 4 had a novel homozygous missense variant in the APTX gene (c.635C > T; p.(Ala212Val) in exon 6 substituting alanine for valine at position 212. The pathogenic variant has previously been described as disease-causing AOA1. It is classified as a variant of uncertain significance (class 3) according to ACMG (Fig. 4).
Patients 5 and 6 are affected siblings, both of whom had a novel homozygous missense variant in MFSD8 gene (c.638C > A; p.(Pro213Gln) substituting proline residue for glutamine at position 213. The homozygous state of this variant has been confirmed by parental targeted testing. It is classified as a variant of uncertain significance (class 3) according to ACMG. Pathogenic variants in the MFSD8 gene are associated with AR NCL type 7.
Patient 7 had a homozygous missense variant in the AGTPBP1 gene (c.1534A > G; p. (Thr512Ala) causing an amino acid change from Thr to Ala at position 512. It is classified as a variant of uncertain significance (class 3) according to the recommendations of Centogene and ACMG. Pathogenic variants in the AGTPBP1 gene are associated with AR CONDCA.
Computational Analysis Implications
All the used computational tools and the corresponding implications are shown in Supplementary Table 1. The functional enrichment analysis is shown in (Supplementary Fig. 1).
Discussion
In this study, we described the detailed phenotypic, radiological, and molecular findings of seven Egyptian patients presenting with neurological Neurodevelopmental disorders or neurodegenerative disorders disorder. All parents were first-degree cousins, highlighting the impact of consanguineous marriage on the increased rate of AR genetic disorders reported in our country [20].
Genetic diagnostic testing based on exome sequencing revealed three novel variants in “MFSD8 (NM_001371596.2), AGTPBP1 (NM_001330701.2), and APTX (NM_001195248.2)” genes, and three variants have been previously reported in “TPP1 (NM_000391.4), AGTPBP1(NM_001330701.2), and PCDHGC4 (NM_018928.3)” genes. These genes are associated with AR NCL type 2, AR NCL type 7, AR CONDCA, AOA1, and NEDGS, respectively [21]. Three patients in our cohort were diagnosed with NCL: patient 1 (NCL type 2) and patients 5 and 6—siblings (NCL type 7). To date, 131 variants have been reported in the CLN2 gene which is distributed over the whole protein structure. These include missense variants (63, 48%) followed by frameshift (21, 16%) and nonsense (17, 13%) variants. Two known common pathogenic variants, c.509–1 G > C and c.622 C > T p.(Arg208*), occur in 60% of affected individuals with NCL2 [22]. WES analysis revealed a splice site variant in exon 9 of the TPP1 gene in patient 1. Our patient presented at the age of 3 years with GDD which was followed by progressive visual impairment, motor disability, spasticity, and scoliosis. NCL type 2 typically presents with seizures and/or ataxia in the late-infantile period by the age of 2–4 years, often in combination with a history of speech delay, followed by progressive childhood dementia, motor and visual impairment, and early death [23]. Our findings agree with previous reports that studied the clinical characteristics of NCL2 patients [24,25,26]. However, our patient had progressive spastic quadriplegia and scoliosis so there was a clinical overlap with other conditions such as hereditary spastic paraplegia [27].
A novel (c.638C > A; p.Pro213Gln) in the MFSD8 gene in a homozygous state was detected in patients 5 and 6. Stogmann et al. (2009) reported an Egyptian family with late-infantile seizures, deterioration, and loss of psychomotor skills 1 year after the seizures’ onset. Additionally, they had aggressive behavior, memory impairment, and language abnormalities with substantial loss of speech function [28]. In our study, we have observed marked intra-familial disease variability as both patients (P5 and 6) had different age of onset. Also, one of the two probands (P6) presented with scoliosis. Several previously reported studies showed inter and intrafamilial phenotypic variability for the same genotype in different forms of NCLs which agrees with our study [29]. The clinical heterogeneity may be related to the profoundly different disease mechanisms, the presence of modifier genes, other environmental factors, or lifestyle. Modifier genes could influence gene expression levels and post-translational processing [30]. The current computational analysis showed that Pro213Gln is highly conserved with potential pathogenicity impact on the transportation function of MFSD8 protein accumulating the diseased harmful compound.
Several NCL disease-specific therapies have been identified depending on the unique subtype identified. These therapies range from several options in the CLN2 subtype such as enzyme replacement therapies, gene therapies, stem cell therapies, and pharmacological drugs to no available options in the CLN7 subtype rendering the identification of each type of particular importance [31].
Patient 2 was diagnosed with NEDGS due to a pathogenic homozygous missense variant in the PCDHGC4 gene (c.1463C > T; p.Ala488Val). This syndrome was first described in a cohort of 19 patients from nine unrelated families originating from Iran, Iraq, Turkey, Morocco, Pakistan, Saudi Arabia, Lebanon, Sudan, and Jordan. It is characterized by the presence of dysmorphic features, neurodevelopmental delay, microcephaly, short stature, seizures, hypotonia, spasticity, strabismus, and skeletal anomalies [32]. Our patient had a similar presentation apart from cardiac anomalies (ASD) that was not previously reported. Five nonsense, frameshift, or splice site mutations were predicted to result in premature termination and a loss of function, and three missense mutations at highly conserved residues were reported. To our knowledge, this is the 2nd study that has detected a pathogenic variant in the PCDHGC4 gene. Both patients 3 and 7 had novel homozygous mutations in AGTPBP1. A gene that was first described by Shashi et al. [33] in 13 individuals from 10 unrelated families with abnormal eye movements, GDD, microcephaly, tongue fasciculation, hypotonia, muscle atrophy, feeding difficulties, and failure to thrive. All patients had cerebellar atrophy; hence, it was subsequently recognized as CONDCA [34, 35]. Another study reported a similar phenotypic presentation but without any eye movement abnormalities [36]. Our computational studies showed a potential pathogenicity effect of the Gly884Arg variant concluding it might lead to altered protein conformation and inhibit deglutamylation of tubulin and non-tubulin target proteins.
To our knowledge, this is the third clinical study reporting patients with CONDCA. Our patients had similar presentation but patient (3) had additional unique phenotypic features “dysmorphic features, seizures and skeletal deformities” that were not previously reported. Patient (7) had areflexia in the lower extremities due to axonal motor neuropathy as confirmed by nerve conduction and electromyography studies possibly due to Purkinje cell degeneration and motor neuropathy that has been described in this disorder. Brain MRI in our patients showed cerebellar atrophy which is the hallmark of this disorder that should be carefully interpreted given their clinical presentation to avoid confusion with other disorders such as pontocerebellar diseases that are associated with cerebellar hypoplasia [33]. The current computational analysis showed that p.Gly884Arg might lead to altered protein conformation inhibiting deglutamylation of tubulin and non-tubulin target proteins.
APTX variant, identified in patient 4, was first described by Aicardi et al. [37] as the cause of a syndrome mimicking ataxia telangiectasia that was named AOA1. Anheim et al. found that AOA1 was the fourth most common cause of AR cerebellar ataxia [38]. The p.Pro206Leu variant was the most frequent variant described worldwide [39]. Oculomotor apraxia was not a constant finding in all reported patients. Our result was in agreement with the previous studies regarding most of the clinical manifestations; however, our patient had congenital heart disease and poor retinal function that were not previously reported in APTX gene mutations thus expanding its phenotypic spectrum.
To our knowledge, this is the first genetic study of its kind from North Africa “Egypt” exploring the possible molecular defects underlying the overlapping NDs phenotypes. We identified three novel pathogenic mutations and expanded the phenotypic spectrum with newly associated clinical phenotypic findings in the studied patients.
In conclusion, this study highlights the importance of genetic testing for patients presenting with ND disorders where phenotypic characterization might not be sufficient for proper classification and early disease identification, especially for potentially treatable ones.
Data availability
Availability of variant data during the current study has been submitted to the LOVD database under the following links;
NM_001371596.2:c.638C > A (MFSD8; p.Pro213Gln; Novel variant)
Data available at https://databases.lovd.nl/shared/individuals/00433029.
NM_018928.3:c.1463C > T (PCDHGC4; p.Ala488Val; Reported variant; rs775104626) ClinVar; pathogenic, and citation = 0
Data available at https://databases.lovd.nl/shared/individuals/00433030
NM_001195248.2:c.635C > T (APTX; p.Ala212Val; Novel variant)
Data available at https://databases.lovd.nl/shared/individuals/00433031.
NM_001330701.2:c.2650G > C (AGTPBP1; p.Gly884Arg; Novel variant)
REPORTED PREVIOUSLY AS NM_001330701.2:c.2650G > A; p.Gly884Arg) with ClinVar = NA, and citation = 0
Data available at https://databases.lovd.nl/shared/individuals/00433032.
NM_000391.4: c.1145 + 2 T > G (TPP1; c.1145 + 2 T > G; Reported variant; COSV100196937); splice_donor_variant (Int. 9)
Data available at https://databases.lovd.nl/shared/individuals/00433033.
NM_001330701.2: c.1534A > G (AGTPBP1; p.Thr512Ala; reported variant rs1375829417) ClinVar; NA, and citation = 0
Data available at https://databases.lovd.nl/shared/individuals/00433034
References
Temtamy S, Aglan M (2012) Consanguinity and genetic disorders in Egypt. Middle East J Med Genet 1:12–17. https://doi.org/10.1097/01.MXE.0000407744.14663.d8
Lee H, Nelson SF (2020) The frontiers of sequencing in undiagnosed neurodevelopmental diseases. Curr Opin Genet Dev 65:76–83. https://doi.org/10.1016/j.gde.2020.05.003
Chérot E, Keren B, Dubourg C et al (2018) Using medical exome sequencing to identify the causes of neurodevelopmental disorders: experience of 2 clinical units and 216 patients. Clin Genet 93(3):567–576. https://doi.org/10.1111/cge.13102
Mastrangelo M (2019) Clinical approach to neurodegenerative disorders in childhood: an updated overview. Acta Neurol Belg 119(4):511–521. https://doi.org/10.1007/s13760-019-01160-0
Córdoba M, Rodriguez-Quiroga SA, Vega PA et al (2018) Whole exome sequencing in neurogenetic odysseys: an effective, cost- and time-saving diagnostic approach. PLoS ONE 13(2):e0191228. https://doi.org/10.1371/journal.pone.0191228
Johnston M (2016) Neurodegenerative disorders of childhood. In: Behrman RE, Kliegman RM, Jenson HB, Nelson textbook of pediatrics (20th edn.) Elsevier, Philadelphia, ISBN: 978–1–4557–7566–8
Jan MM (2002) Clinical approach to children with suspected neurodegenerative disorders. Neurosciences 7:2–6 (Riyadh)
Blaser SI, Steinlin M, Al-Maawali A et al (2016) The Pediatric Cerebellum in inherited neurodegenerative disorders: a pattern-recognition approach. Neuroimaging Clin N Am 26(3):373–416. https://doi.org/10.1016/j.nic.2016.03.007
Stephenson J, Nutma E, van der Valk P et al (2018) Inflammation in CNS neurodegenerative diseases. Immunology 154:204–219. https://doi.org/10.1111/imm.12922
Tanner JM, Hiernaux J, Jarman S (1969) Growth and physique studies. In: Human Biology, A Guide to Field Methods (Weiner JS, Lourie JA, eds). IBP Handbook No. 9. Oxford: Blackwell, International Biological Programme, pp 2–71
Verma RS, Babu A (1995) Human chromosomes: principles and techniques McGraw-Hill, New York.https://doi.org/10.1002/mrd.1996.1080430105
Jiang H, Lei R, Ding SW et al (2014) Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinforma 15:182. https://doi.org/10.1186/1471-2105-15-182
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164. https://doi.org/10.1093/nar/gkq603
Sherry ST, Ward MH, Kholodov M et al (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311. https://doi.org/10.1093/nar/29.1.308
Auton A, Abecasis GR, Altshuler DM et al (2015) A global reference for human genetic variation. Nature 526:68–74. https://doi.org/10.1038/nature15393
Karczewski KJ, Francioli LC, Tiao G et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Untergasser A, Cutcutache I, Koressaar T et al (2012) Primer3-New capabilities and interfaces. Nucleic Acids Res 40:e115. https://doi.org/10.1093/nar/gks596
Sen Dunnen JT, Dalgleish R, Maglott DR et al (2016) HGVS recommendations for the description of sequence variants: 2016 Update. Hum Mutat 37(6):564–569. https://doi.org/10.1002/humu.22981
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–423. https://doi.org/10.1038/gim.2015.30
Shawky RM, El-Awady MY, Elsayed SM et al (2011) Consanguineous matings among Egyptian population. Egypt J Med Hum Genet 12(2):157–163. https://doi.org/10.1016/j.ejmhg.2011.07.001
Ren XT, Wang XH, Ding CH et al (2019) Next-generation sequencing analysis reveals novel pathogenic variants in four Chinese siblings with late-infantile neuronal ceroid lipofuscinosis. Front Genet 25(10):370. https://doi.org/10.3389/fgene.2019.00370
Gardner E, Bailey M, Schulz A et al (2019) Mutation update: review of TPP1 gene variants associated with neuronal ceroid lipofuscinosis CLN2 disease. Hum Mutat 40(11):1924–1938. https://doi.org/10.1002/humu.23860
Fietz M, AlSayed M, Burke et al (2016) Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab 119(1–2):160–167. https://doi.org/10.1016/j.ymgme.2016.07.011
Mole S, Gardner E, Schulz A et al (2018) Molecular basis of CLN2 disease: a review and classification of TPP1 gene variants reported worldwide. Mol Genet Metab 123(2):S97. https://doi.org/10.1016/j.ymgme.2017.12.255
Nickel M, Jacoby D, Lezius S et al (2016) Natural history of CLN2 disease: quantitative assessment of disease characteristics and rate of progression. Neuropediatrics 47(S 01). https://doi.org/10.1055/s-0036-1583730
Nickel M, Simonati A, Jacoby D et al (2018) Disease characteristics and progression in patients with late-infantile neuronal ceroidlipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health 2(8):582–590. https://doi.org/10.1016/S2352-4642(18)30179-2
Kara E, Tucci A, Manzoni C et al (2016) Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 139(Pt 7):1904–1918. https://doi.org/10.1093/brain/aww111
Stogmann E, El Tawil S, Wagenstaller J et al (2009) A novel mutation in the MFSD8 gene in late infantile neuronal ceroidlipofuscinosis. Neurogenetics 10:73–77. https://doi.org/10.1007/s10048-008-0153-1
Weisschuh N, Mazzola P, Heinrich T et al (2020) First submicroscopic inversion of the OPA1 gene identified in dominant optic atrophy—a case report. BMC Med Genet 21:236. https://doi.org/10.1186/s12881-020-01166-z
Butz ES, Chandrachud U, Mole SE et al (1866) (2020) Moving towards a new era of genomics in the neuronal ceroidlipofuscinoses. Biochim Biophys Acta Mol Basis Dis 9:165571. https://doi.org/10.1016/j.bbadis.2019.165571
Rosenberg JB, Chen A, Kaminsky SM et al (2019) Advances in the treatment of neuronal ceroid lipofuscinosis. Expert Opin Orphan Drugs 7(11):473–500. https://doi.org/10.1080/21678707.2019.1684258
Iqbal M, Maroofian R, Çavdarlı B et al (2021) Biallelic variants in PCDHGC4 cause a novel neurodevelopmental syndrome with progressive microcephaly, seizures, and joint anomalies. Genet Med 23:2138–2149. https://doi.org/10.1038/s41436-021-01260-4
Shashi V, Magiera MM, Klein D et al (2018) Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration. EMBO J 37(23). https://doi.org/10.15252/embj.2018100540.
Sheffer R, Gur M, Brooks R et al (2019) Biallelic variants in AGTPBP1, involved in tubulin deglutamylation, are associated with cerebellar degeneration and motor neuropathy. Eur J Hum Genet 27:1419–1426. https://doi.org/10.1038/s41431-019-0400-y
Baltanás FC, Berciano MT, Santos E et al (2021) The childhood-onset neurodegeneration with cerebellar atrophy (CONDCA) disease caused by AGTPBP1 gene mutations: the Purkinje cell degeneration mouse as an animal model for the study of this human disease. Biomedicines 9(9):1157. https://doi.org/10.3390/biomedicines9091157
Karakaya M, Paketci C, Altmueller J et al (2019) Biallelic variant in AGTPBP1 causes infantile lower motor neuron degeneration and cerebellar atrophy. Am J Med Genet A 179(8):1580–1584. https://doi.org/10.1002/ajmg.a.61198
Aicardi J, Barbosa C, Andermann E et al (1988) Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann Neurol 24:497–502. https://doi.org/10.1002/ana.410240404
Anheim M, Fleury M, Monga et al (2010) Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics 11:1–12. https://doi.org/10.1007/s10048-009-0196-y
Ababneh NA, Ali D, Al-Kurdi B et al (2020) Identification of APTX disease-causing mutation in two unrelated Jordanian families with cerebellar ataxia and sensitivity to DNA damaging agents. PLoS ONE 15(8):e0236808. https://doi.org/10.1371/journal.pone.0236808
Acknowledgements
The authors would like to thank the patients and their parents for kindly participating in this study. Our gratitude was extended to the late Professor Mona El Ruby, our mentor, who passed away before the completion of this manuscript for her participation in the diagnosis and establishing the study design.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
- Engy A. Ashaat and Nesma M. Elaraby: designed and conceptualized the study.
- Hoda A. Ahmed, Engy A. Ashaat, Dina Amin Saleh: wrote the first draft of the manuscript.
- Ammal M. Metwally, Neveen A. Ashaat, Dina Amin Saleh: referred cases.
- Engy A. Ashaat, Rasha Moheb Elhossini, Heba Ahmed ElAwady, Dina Amin Saleh, Mohamed Ahmed Al Kersh: examined the patient, and took the family and medical histories.
- Randa H. A. Abdelgawad: performed ophthalmological examination.
- Nesma M. Elaraby, Hoda A. Ahmed: collected DNA samples, interpreted patient data, and performed Sanger sequencing data analysis.
- Alaaeldin Fayez: carried out the bioinformatics analysis, and variants registration in the LOVD database.
- Mona K. Mekkawy, Dalia Farouk Hussen: performed cytogenetic analysis.
- Mohamed Ahmed Al Kersh: performed the radiological investigation.
- Mona El Gammal, Engy A. Ashaat, Dina Amin Saleh: revised and edited the final version of the manuscript.
All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The study was approved by the Medical Research Ethics Committee of the National Research Centre (NRC), Cairo, Egypt (Ethical Approval Number: 932702021). Informed and written consent was obtained from the parents involved in the study.
Consent for Publication
Informed consent for publication was obtained from the patient’s guardians.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12035_2023_3866_MOESM1_ESM.jpg
Supplementary file1 (JPG 43 KB) Supplementary Fig. 1 Functional Enrichment analysis representing (A) GeneMANIA results showed the significant functional relationship among the affected gene with false detective rate (FDR) scores, below each gene listed the interacted partner genes physically. The genes with dark green shadow represent a set of genes that share the non-recombination repair process, while the genes with brown shadow represent a set of genes that share the double-strand break repair process. (B) PPI enrichment network among the affected genes showed that two human phenotypes were significantly enriched in 4 out of 5 analyzed proteins, the highlighted color corresponded with each phenotype.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Ashaat, E.A., Ahmed, H.A., Elaraby, N.M. et al. The Diagnostic Value of Whole-Exome Sequencing in a Spectrum of Rare Neurological Disorders Associated with Cerebellar Atrophy. Mol Neurobiol 61, 4949–4961 (2024). https://doi.org/10.1007/s12035-023-03866-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03866-y