
Abstract
Autophagy is a conservative self-degradation system, which includes the two major processes of enveloping abnormal proteins, organelles and other macromolecules, and transferring them into lysosomes for the subsequent degradation. It holds the stability of the intracellular environment under stress. So far, three types of autophagy have been found: microautophagy, chaperone-mediated autophagy and macroautophagy. Many diseases have the pathological process of autophagy dysfunction, such as nervous system diseases. Pyroptosis is one kind of programmed cell death mediated by gasdermin (GSDM). In this process of pyroptosis, the activated caspase-3, caspase-4/5/11, or caspase-1 cleaves GSDM into the N-terminal pore-forming domain (PFD). The oligomer of PFD combines with the cell membrane to form membrane holes, thus leading to pyroptosis. Pyroptosis plays a key role in multiple tissues and organs. Many studies have revealed that autophagy and pyroptosis participate in the nervous system, but the mechanisms need to be fully clarified. Here, we focused on the recent articles on the role and mechanism of pyroptosis and autophagy in the pathological processes of the nervous system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Overview of autophagy
As a conservative intracellular process, autophagy wraps abnormal proteins, organelles and other macromolecules, and then transfers them to lysosomes for the subsequent degradation [1,2,3]. Autophagy can be divided into three types according to the difference in the particularity of goods and the different delivery methods to lysosomes:chaperone-mediated autophagy, microautophagy and macroautophagy [4, 5]. Macroautophagy, which is commonly referred to as autophagy, and one of the most studied, is an evolutionarily conservative stress response process. During macroautophagy, after being isolated in double-membrane vesicles and transported to lysosomes for degradation, the invasive pathogens and abnormal organelles are degraded [6, 7]. In the process of microautophagy, the lysosome membrane is directly invaginated, and then the cell contents are wrapped [8,9,10]. Instead of capturing cargoes with vesicle intermediates, the chaperone-mediated autophagy transports the substrate binding with the chaperone protein directly to the lysosome cavity. To date, chaperone-mediated autophagy has only been described in mammalian cells (Fig. 1) [11,12,13,14]. Autophagy can be activated by a variety of stimuli, including glucose deprivation, nutritional starvation, caloric restriction, transient ischemia and reperfusion, and oxidative stress. Under physiological conditions, autophagy which is at a basic level helps to maintain the integrity of intracellular organelles [15, 16]. However, autophagy disorders have been observed under a wide range of pathological conditions, including obesity, infectious and inflammatory diseases, type 2 diabetes, neurodegenerative diseases and cancers [3, 17]. Neurons are a kind of polarized cells with a large amount of cytoplasm, and accumulation of cell waste will bring a heavy burden to these cells. Therefore, neurons are considered to be particularly vulnerable to autophagic dysfunction. Autophagy is important for maintaining neuronal homeostasis and eliminating protein aggregates, thus preventing neuronal disorder [18]. Autophagy also plays an important role in neuronal activity, plasticity, and memory [19]. Additionally, autophagy dysfunction often occurs in microglia [20] and astrocytes [21]. More and more evidence indicates that abnormal autophagy participates in many nervous system diseases such as Alzheimer’s disease [22], Huntington’s disease [23], and Parkinson’s disease [24]. Up to now, the role and mechanism of autophagy in the nervous system are not completely clarified.

Diagram of three types of autophagy processes
Overview of pyroptosis
Pyroptosis is one kind of programmed cell death mediated by gasdermin and was first discovered in Salmonella-induced macrophage death [25]. Also known as secondary necrosis, pyroptosis is a new nonapoptotic form of programmed cell death and is closely related to the inflammatory reaction [26]. It is mainly touched off by inflammasomes and is executed by the gasdermin (GSDM) family and caspases including caspase-1/- 3/- 4/- 5/- 11 [27,28,29]. Pyroptosis is characterized by cell swelling, the formation of large bubbles on the plasma membrane, and the rupture of the plasma membrane [30]. The GSDM family consists of six members, including DFNB and GSDM-A, -B, -C, -D, -E [31]. During pyroptosis, caspase-1 cuts GSDMD into its N-terminal fragment, which then forms cell membrane holes. Therefore, GSDMD is the main pyroptosis executor [32]. Until now, according to the different triggering modes, three kinds of pyroptosis have been found, including non-canonical pyroptosis pathway, caspase-3-mediated pyroptosis pathway and canonical pyroptosis pathway [33]. The canonical pyroptosis pathway is mediated by caspase-1 and nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3). During this process, NLRP3 inflammasome is activated, resulting in the activated caspase-1, which subsequently cuts GSDMD into its N- and C-terminal fragments. The N-terminal fragments then bind with the cell membrane to form pyroptotic holes [34]. The non-canonical pyroptosis pathway is mediated by human caspases-4 and-5 and mouse caspase-11. The caspase recruitment domain (CARD) of the gram-negative bacteria directly binds to lipopolysaccharide (LPS), triggering non-standard inflammatory pathways. Caspase-4/5/11 can directly cut GSDMD into an N-terminal fragment and a C-terminal fragment. The N-terminal domain then binds with the cell membrane to produce pyroptotic pores, which release interleukin-1beta (IL-1β) and IL-18 out of cells, thus activating the process of non-canonical pyroptosis [35]. In pyroptosis mediated by caspase-3, caspase-3 cleaves GSDME into the GSDME-N domain that binds with the cell membrane to result in pyroptotic pores, thereby triggering pyroptosis [36, 37]. See Fig. 2 for the above three pathways of pyroptosis [38, 39]. The evidence indicates that pyroptosis occurs in multiple types of nervous system cells, including microglia, astrocytes, oligodendrocytes, neurons and peripheral cells [40]. Therefore, pyroptosis is involved in many pathological processes of the nervous system, such as multiple sclerosis, Alzheimer’s disease, stroke, traumatic brain injury (TBI) and spinal cord injury (SCI), resulting in neuroinflammation and neurodegeneration through a variety of mechanisms. The treatment of targeted pyroptosis has shown promise in various preclinical models of nerve injury and diseases [41, 42]. Recently, the evidence indicates that autophagy and pyroptosis are involved in the nervous system, but the mechanisms have not been completely clarified. Hence, we focus on the recent progress in the role and mechanisms of pyroptosis and autophagy in the pathological processes of the nervous system.

The process of three types of pyroptosis
The role of pyroptosis and autophagy in microglia
The role of pyroptosis and autophagy in microglia with hypoxemia
The typical symptom of acute respiratory distress syndrome (ARDS) is persistent hypoxemia, which often requires ventilation to correct hypoxemia. The ventilatory strategies can lead to hypercapnia [43, 44]. It has been reported that in hypoxic adult rats, hypercapnia can induce IL-1β overproduction by activating nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome [45, 46]. However, its mechanism is not completely clear. In addition, mitophagy plays a vital role in the ischemic model of Alzheimer’s disease [47,48,49]. To seek the role and mechanism of hypercapnia and mitophagy in ARDS, Hong guang Ding and colleagues conducted a lot of experiments, and the results showed that hypercapnia reduced the partial pressure of oxygen (PbtO2) levels in the brain tissue of hypoxic rats, and this was further demonstrated by an increase of cerebral oxygen extraction ratio (CERO2) levels. Hypercapnia decreased the expression of microtubule-associated protein light chain-II (LC3-II) and upregulated p62 expression in hypoxic microglia, indicating that hypercapnia downregulated microglia mitophagy. The in-depth research showed that hypercapnia upregulated mitochondrial reactive oxygen species (ROS) production, and activated NLRP3 inflammasome, thus promoting microglia pyroptosis, as evidenced by the increased expression of GSDMD-N in hypoxia-activated microglia, while the scavenging of ROS could suppress microglial pyroptosis and decrease the expressions of IL-18 and IL-1β [50]. Evidence indicates that mitophagy inhibits NLRP3 inflammasome activation by clearing mitochondrial ROS [51, 52]. Therefore, it can be inferred from the above that hypercapnia promotes NLRP3 inflammasome-mediated microglial pyroptosis by suppressing mitophagy via increasing mitochondrial ROS overproduction in hypoxemic adult rats [50]. Parkin RBR E3 ubiquitin-protein ligase (PARKIN)/PTEN-induced kinase 1 (PINK1) pathway has been reported to regulate mitophagy in nervous system diseases [53, 54]. Hence, whether hypercapnia inhibited mitophagy through PINK1/PARKIN remains to be studied.
The role of pyroptosis and autophagy in microglia with pneumococcal infection
The gram-positive bacterium Streptococcus pneumoniae can cause pneumonia, otitis media, meningitis and other diseases with the characteristics of high mortality and high incidence rate [55,56,57]. However, the exact mechanism of its pathogenesis is not completely clarified. Ji Yun Kim and colleagues demonstrated that S. pneumoniae promoted pyroptosis by inducing caspase-1 activation and IL-1β production in murine microglia. NLRP3 siRNA treatment reduced the levels of IL-1β IL-18 and caspase-1 in murine microglia infected by S. pneumoniae. NLRP3 siRNA also decreased lactate dehydrogenase (LDH) release of S. pneumoniae-infected murine microglial cells. These indicated that NLRP3 inflammasome mediated S. pneumoniae-promoted pyroptosis. Moreover, S. pneumoniae infection upregulated autophagy by increasing the expressions of autophagy-related genes and promoting autophagosome formation in the early stage of pneumococcal infection (4 h). The inhibition of autophagy upregulated the expressions of IL-18, caspase-1 and IL-1β in S. pneumoniae-infected murine microglia, and induced the release of LDH, IL-1β and IL-18 of S. pneumoniae-infected murine microglia, indicating that autophagy suppressed S. pneumoniae-induced NLRP3 caspase-1/inflammasome-induced pyroptosis. These suggested that the promotion of autophagy by S. pneumoniae transiently inhibited S. pneumoniae-induced pyroptosis in microglia in the early stage of pneumococcal infection. Furthermore, in murine microglia, S. pneumoniae increased ROS production, while NAC(a ROS scavenger) downregulated the S. pneumoniae-induced ROS production, and inhibited autophagy, suggesting that S. pneumoniae promoted autophagy via increasing ROS. Moreover, pneumococcal infection-induced ROS suppressed the activation of caspase-1 within 4 h after infection. But in the late stage of infection, ROS induced caspase-1-dependent microglia pyroptosis and IL-1β secretion. From the above, it can be deduced that S. pneumoniae-induced ROS upregulated autophagy to inhibit microglia pyroptosis in the early stage of infection (within 4 h). However, S. pneumoniae-induced ROS promoted pyroptosis in the late phase of infection [58]. The increased ROS from mitochondria or other intracellular compartments promotes pyroptosis and autophagy [59]. Therefore, it can be inferred as follows: in the early stage of Streptococcus pneumoniae infection, the increased ROS can activate autophagy to suppress NLRP3 inflammasome-mediated microglia pyroptosis, while the enhanced autophagy can clear ROS. With the extension of infection time, ROS is cleared by autophagy and reduced to a certain extent, and its tendency to activate autophagy greatly decreases, while its role of activating NLRP3 inflammasome-mediated microglia pyroptosis appears, which shows that S. pneumoniae induced microglia pyroptosis [58].
Nucleotide-binding oligomerization domain 2(NOD2), a member of the leucine-rich repeat receptor (NLR) family, can detect the specific bacterial peptidoglycans and regulate the expressions of inflammatory factors by activating the nuclear transcription factor-kappa B (NF-kB) pathway [60]. Receptor-interacting protein 2 kinase (RIP2) is activated by intracellular NOD2 and is involved in bacterial infection-induced inflammatory response [61]. It has been reported that the NOD2-RIP2 pathway participates in S. pneumoniae infection [62], but the relevant mechanisms still require to be fully clarified. The studies of Guan Wang et al. revealed that S. pneumoniae upregulated the level of microglial ROS, LDH release and the secretion of IL-18 and IL-1β, strengthened caspase-1 activity, and decreased microglial viability, indicating that S. pneumoniae promoted microglial ROS production and pyroptosis. Furthermore, S. pneumoniae upregulated the levels of autophagy, NOD2, RIP2 and phospho-ULK1 (p-ULK1, an important regulator of autophagy) in microglial. Treatment of microglia with 6-(tert-Butylsulfonyl)-N-(5-fluoro-1H-indazol-3-yl)quinolin-4-amine (GSK583, a RIP2 kinase inhibitor), as well as knockdown of NOD2 or RIP2, decreased the levels of p-ULK1 and autophagy-related proteins, suggesting that NOD2-RIP2 pathway promoted microglia autophagy via phosphorylating ULK1. In Streptococcus pneumoniae-infected microglia, the knockdown of ULK1 increased the ROS production, the secretion of IL-1β, IL-18 and LDH release and caspase-1 activity, and reduced the levels of microglia viability and autophagy-related proteins, indicating that ULK1-regulated autophagy inhibited microglia ROS production and pyroptosis. Similar to the results in vitro, the NOD2-RIP2 pathway promoted autophagy in the brain of the Streptococcus pneumoniae-infected mouse. Furthermore, the inhibition of ULK1 or RIP2 notably promoted microglia pyroptosis of the brain in Streptococcus pneumoniae-infected mice. Collectively, the NOD2-RIP2 pathway inhibited microglia pyroptosis by promoting ULK1-regulated autophagy during the infection of Streptococcus pneumoniae [63]. Evidence indicates that ROS can activate NLRP3 inflammasome that in turn promotes NLRP3 inflammasome-mediated pyroptosis [64]. In the above study, although NLRP3 inflammasome level was not detected, from the result that the NOD2-RIP2 pathway inhibits ROS generation, it can be deduced that autophagy suppresses NLRP3 inflammasome-induced pyroptosis via reducing ROS, which needs further confirmation.
The role of pyroptosis and autophagy in microglia with oxygen–glucose deprivation/reoxygenation
Neonatal hypoxic–ischemic brain damage (HIBD) is the principal reason for diseases, including epilepsy, cerebral palsy and cognitive impairment in children [65, 66]. To date, there is still no effective treatment for HIBD, which has a serious impact on children’s health and life quality. There is an urgent need to seek effective treatment for HIBD [67, 68]. It has been reported that mesenchymal stem-cell-derived exosomes (MSC-exos) can ameliorate neonatal HIBD [69]. But the relevant mechanism needs to be completely clarified. Zhenzhen Hu and colleagues successfully collected MSC-exos from human MSC, as evidenced by identifying the diameter, the shape and the marker protein of exosomes. MSC-exos promoted microglia viability, reduced the levels of caspase-1, NLRP3 and GSDMD-N, and the release of IL-18 and IL-1β in microglia induced by OGD/R, suggesting that MSC-exos suppressed OGD/R-induced microglia pyroptosis. Compared with the medium from OGD/R-exposed and PBS-treated microglia, the medium from OGD/R-exposed and MSC-exos-treated microglia notably upregulated microglia viability and decreased LDH release, suggesting that MSC-exos mitigated OGD/R-induced microglia damage. MSC-exos also increased the levels of COX IV and TOM20, two mitophagy-related proteins, in microglia induced by OGD/R. Meanwhile, mitochondrial division inhibitor-1 and 3-methyladenine(3-MA) alleviated MSC-exos suppression of pyroptosis, suggesting that MSC-exos suppressed OGD/R-induced microglia pyroptosis through upregulating mitophagy. Additionally, MSC-exos notably upregulated the expression of Forkhead box 3a(FOXO3a) in OGD/R-exposed microglia. The siRNA of FOXO3a partially abolished the neuroprotection of MSC-exos and alleviated MSC-exos-mediated suppression of pyroptosis and promotion of mitophagy, suggesting that MSC-exos promoted mitophagy to suppress OGD/R-induced microglia pyroptosis by promoting FOXO3a. Summarily, MSC-exos mitigated ischemia/reperfusion(I/R)-induced microglia pyroptosis by upregulating the expression of FOXO3a to promote mitophagy [70]. Mitophagy maintains intracellular environment stability through selectively eliminating the injured mitochondria [71]. Therefore, mitophagy suppresses NLRP3 inflammasome activation through decreasing the mitochondrial ROS that can activate NLRP3 inflammasome [72]. It can be inferred that mitophagy suppresses microglia pyroptosis via inhibiting NLRP3 inflammasome in microglia with oxygen–glucose deprivation/reoxygenation.
The role of pyroptosis and autophagy in microglia with inflammation
Dimethyl itaconate (DI), a cell-permeable derivative of the endogenous metabolite itaconate, has been regarded as an anti-inflammatory regulator of macrophages [73, 74]. But the effect of DI on inflammasome-mediated pyroptosis remains unknown. Su Yang and colleagues found that lipopolysaccharide (LPS) + ATP promoted NLRP3 inflammasome activation, LDH release and GSDMD cleavage in microglia, which was reversed by DI or NLRP3 siRNA, indicating that DI inhibited NLRP3-dependent microglia pyroptosis [75]. The evidence showed that M1-polarized microglia can generate pro-inflammatory cytokines to promote inflammation. Conversely, M2-polarized microglia can generate anti-inflammatory cytokines to inhibit inflammation [76, 77]. LPS + ATP induced the transition of microglia from M2 polarization to M1 polarization, increased the levels of inflammatory mediators and activated the NF-κB pathway, which was reversed by DI, indicating that DI suppressed LPS + ATP-induced microglia inflammation. Moreover, DI induced the activation of heme oxygenase‑1 (HO‑1)/nuclear factor erythroid 2‑related factor 2 (Nrf‑2) pathway, and the inhibition of Nrf-2/HO-1 pathway abolished DI protection of microglia, suggesting that DI inhibited NLRP3-dependent microglia pyroptosis by activating Nrf-2/HO-1 pathway. Meanwhile, DI also reduced LPS + ATP-induced ROS production in microglia. In addition, DI promoted LPS + ATP-inhibited autophagy, while suppression of autophagy with 3-MA mitigated DI effects on the levels of IL-1β, cleaved GSDMD and NLRP3, indicating that DI played an anti-inflammatory role by inducing autophagy. Collectively, DI inhibited LPS + ATP-promoted and NLRP3 inflammasome-mediated microglia pyroptosis by promoting Nrf-2/HO-1 pathway and autophagy [75]. Evidence indicates that ROS promotes NLRP3 inflammasome-mediated pyroptosis [78, 79]. Autophagy suppresses NLRP3 inflammasome via clearing mitochondrial ROS and degrading NLRP3 inflammasome components [13]. Therefore, autophagy inhibited NLRP3 inflammasome-mediated pyroptosis by clearing ROS and degrading NLRP3 inflammasome components in microglia in the above study [75].
The role of pyroptosis and autophagy in diabetes nervous system diseases
The role of pyroptosis and autophagy in diabetes-induced brain injury
Diabetes is a vital risk factor for ischemic stroke. The prognosis of diabetes patients after ischemic stroke is poor [80, 81]. The evidence from animal models of diabetes shows that neuronal death is a vital factor leading to brain injury [82, 83], and caspase-1 inhibition mitigates brain ischemic injury-induced neuronal death [84, 85]. Hui Che and colleagues found that melatonin treatment notably inhibited neuronal death in both streptozotocin (STZ)-treated diabetic mice and high glucose (HG)-induced neuronal cells. Furthermore, melatonin suppressed neuronal pyroptosis and enhanced autophagy induced by diabetes through reducing the levels of Beclin1, NLRP3, ATG12, caspase-1, GSDMD-N, and LC3 in vivo and in vitro. The in-depth research revealed that MicroRNA-214-3p (miR-214-3p) level was downregulated in HG-induced neuronal cells and diabetic mice, which was reversed by melatonin. MiR-214-3p overexpression decreased the levels of IL-1β, caspase-1 and GSDMD-N, while the inhibition of miR-214-3p had the opposite effects on HG-induced neuronal cells, indicating that miR-214-3p suppressed pyroptosis. In addition, the inhibition of miR-214-3p abolished the melatonin suppression of pyroptosis and autophagy induced by diabetes in vitro. Summarily, melatonin played a neuroprotective role via suppressing neuronal autophagy and pyroptosis induced by diabetes by promoting miR-214-3p [86]. Melatonin may be a vital candidate drug for the treatment of brain injury with diabetes. The relationship between autophagy and pyroptosis in diabetes nervous system diseases needs to be clarified.
The role of pyroptosis and autophagy in diabetes cerebral ischaemia diseases
Hypothermia is considered one of the neuroprotective strategies, which can significantly improve cerebral ischemia injury in non-diabetic animal models [87, 88]. However, whether hypothermia can improve diabetes-aggravated cerebral ischemia injury is not completely clear. To clarify this, Yanling Tu et al. introduced permanent middle cerebral artery occlusion (pMCAO) into the rat model of type 2 diabetes that was constructed by a high-fat diet combined with intraperitoneal injection of STZ in vivo. Moreover, the HG stimulation and OGD/R were used to establish the cell model of cerebral ischemia with diabetes in vitro. The subsequent studies revealed that diabetes aggravated brain edema and cerebral infarction, and worsened the nerve defect of cerebral ischemia, which was improved by hypothermia. Diabetes increased the blood–brain barrier (BBB) permeability through upregulating matrix metalloproteinase 9 (MMP-9) expression and promoting the degradation of tight junction proteins, and hypothermia reversed this phenomenon. Diabetes upregulated the expressions of GSDMD, caspase-1, NLRP3 and p62, and downregulated microtubule-associated protein 1 light chain 3B (LC3B) II/I ratio in the rat model of cerebral I/R injury with diabetes, which were abolished by hypothermia, suggesting that hypothermia increased autophagy and downregulated pyroptosis. Furthermore, 3-MA reduced the LC3 II/I ratio and increased the levels of p62, caspase-1, NLRP3 and GSDM-N, suggesting that hypothermia inhibited pyroptosis via inducing autophagy. Baf (an autophagy inhibitor) significantly increased the levels of GSDM-N, NLRP3 and caspase-1 inhibited by hypothermia, indicating that hypothermia could suppress pyroptosis by promoting autophagy via promoting the fusion of lysosome and autophagosome. Summarily, diabetes-aggravated cerebral I/R injury was ameliorated via hypothermia by inhibiting pyroptosis and inducing autophagy, which needed to be further verified [89]. Contrary to some of the above conclusions, the upregulation of autophagy can aggravate cerebral ischemia injury [90]. The reason may be related to the duration of cerebral ischemia, which requires to be further study. The study shows that the activation of autophagy protects neurons from cerebral I/R injury by clearing damaged mitochondria [91]. Therefore, it can be deduced that autophagy can suppress cell pyroptosis via clearing mitochondrial ROS, which needs to be further verified [89].
Contrary to the above result that the activation of autophagy plays a protective role against cerebral I/R injury, the suppression of autophagy also improves cerebral I/R injury. Spautin-1 is an autophagy inhibitor and can induce the degradation of the Vps34 PI3 complex by suppressing USP10 and USP13 (two ubiquitin-specific peptidases) [92]. Hui Liu et al. established the model rats of cerebral I/R injury in vivo through middle cerebral artery occlusion for 60 min and reperfusion for 24 h, and the models of cerebral I/R injury in vitro models with PC12 cell through OGD/R. The results revealed that spautin-1 alleviated cerebral I/R injury through reducing infarct size and ameliorating cerebral I/R-induced neurological impairment in vivo. In OGD/R-induced PC12 cells, spautin-1 also increased cell viability, and decreased ROS production and the number of autophagic microsomes. The in-depth research showed that spautin-1 suppressed NLRP3 inflammasome-mediated pyroptosis and autophagy induced by cerebral I/R through downregulating the expression levels of NLRP3, Beclin 1, USP13 and GSDMD-N in vivo and in vitro. However, the overexpression of USP13 counteracted spautin-1 improvement of cerebral I/R injury and significantly abolished spautin-1 suppression of autophagy and NLRP3 inflammasome-dependent pyroptosis. Collectively, spautin-1 protected against cerebral I/R injury by suppressing autophagy/pyroptosis via the inhibition of USP13 [93]. The relationship between pyroptosis and autophagy in the above study needs to be elucidated.
The role of pyroptosis and autophagy in traumatically injured spinal cord
Spinal cord injury (SCI) is a destructive neuropathological disease that can lead to major motor, sensory, and autonomic dysfunction [94, 95]. The evidence indicates that pyroptosis plays an important role in SCI [96,97,98]. Yu Xu and colleagues found that the growth differentiation factor 11 (GDF-11) significantly optimized functional recovery of SCI by decreasing the glial scars and increasing the number of SYN-positive synapses on neurons and the expression of neuronal microtubule-associated protein-2 (MAP2). GDF-11 inhibited pyroptosis by reducing the levels of pyroptosis-associated markers after SCI, including NLRP3, caspase-1, apoptosis-associated speck-like protein (ASC), IL-1β, IL-18 and GSDMD. Moreover, GDF-11 upregulated the levels of Beclin1, LC3II, CTSD (an autolysosome-related marker), and VPS34 (an autophagosomal marker) in neurons, but downregulated p62 level, indicating that GDF-11 activated autophagy. The co-treatment GDF-11 with 3-MA downregulated the levels of autophagy-related markers, and upregulated the levels of pyroptosis-associated markers after SCI, and abolished the conducive influences of GDF-11 on SCI, suggesting that autophagy mediated GDF-11 effects. Mechanism research revealed that GDF-11 upregulated TFE3 expression in neurons, while TFE3 siRNA reversed GDF-11 effects on autophagy and pyroptosis, indicating that GDF-11 promoted autophagy and suppressed pyroptosis by activating TFE3. In addition, GDF-11 activated the AMPK-TRPML1-Calcineurin pathway after SCI, while compound C (an AMPK inhibitor) inhibited the AMPK-TRPML1-Calcineurin pathway and TFE3, and abolished the above GDF-11 effects on autophagy, pyroptosis and improvements of SCI, indicating that AMPK-TRPML1-Calcineurin pathway mediated GDF-11 effects on TFE3, autophagy, pyroptosis and improvement of SCI. Summarily, GDF-11 ameliorated SCI by inhibiting neuron pyroptosis through promoting TFE3-mediated autophagy via activating the AMPK-TRPML1-Calcineurin pathway [99]. Autophagy suppresses pyroptosis by inhibiting NLRP3 inflammasome, and GDF-11-targeting autophagy/pyroptosis may be a new therapeutic method for SCI.
Conclusion
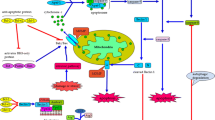
The increasing evidence shows that pyroptosis and autophagy play a vital role in the pathological processes of the nervous system. Here, we focused on the role of pyroptosis and autophagy in the pathological processes of the nervous system as follows: (1)hypercapnia promotes NLRP3 inflammasome-mediated microglial pyroptosis by inhibiting mitophagy through increasing mitochondrial ROS production in hypoxemic adult rats; (2)Streptococcus pneumoniae inhibits microglia pyroptosis by upregulating autophagy via ROS in the early stage of infection; (3)NOD2-RIP2 pathway suppresses microglial pyroptosis by promoting ULK1-regulated autophagy during streptococcus pneumonia infection; (4) MSC-exos alleviates microglial pyroptosis induced by I/R through upregulating the expression of FOXO3a to promote mitophagy; (5) dimethyl itaconate suppressed NLRP3 inflammasome-mediated and LPS + ATP-induced microglia pyroptosis via inducing autophagy and activating Nrf-2/HO-1 pathway; (6) melatonin inhibits neuronal pyroptosis and autophagy induced by diabetes through promoting miR-214-3p; (7) hypothermia improves diabetes-promoted cerebral I/R injury via inhibiting pyroptosis through inducing autophagy; (8) spautin-1 improves cerebral I/R injury via suppressing autophagy/pyroptosis through suppressing inhibition of USP13; (9) the growth differentiation factor 11(GDF-11) ameliorates SCI by inhibiting neuronal pyroptosis through promoting TFE3-mediated autophagy via activating AMPK-TRPML1-Calcineurin pathway (Table 1). It can be concluded from the above that the signaling pathways, including the NOD2-RIP2 pathway, Nrf-2/HO-1 pathway, and AMPK-TRPML1-Calcineurin pathway, are involved in the role of pyroptosis and autophagy in pathological processes of the nervous system. Whether there are other signal pathways involved in autophagy/pyroptosis in the nervous system remains to be further studied. There are two mechanisms for autophagy to negatively regulate pyroptosis: one is that autophagy suppresses pyroptosis through clearing pathogen-associated molecular patterns (PAMPs) and damaging associated molecular patterns (DAMPs). The other is that autophagy suppresses pyroptosis through suppressing pyroptosis components [100]. In this review, autophagy inhibits NLRP3 inflammasome-mediated neuronal pyroptosis by clearing mitochondrial ROS. Whether the other mechanism for autophagy to negatively regulate pyroptosis is involved in the role of pyroptosis and autophagy in pathological processes of the nervous system needs to be studied. Furthermore, in the above-cited literature, it is clarified that ROS can also activate autophagy, and regulate autophagy/pyroptosis, and ROS is an important node connecting autophagy and pyroptosis (Fig. 3 by Figdraw). How ROS regulates autophagy/pyroptosis in the nervous system requires to be further clarified. In addition to the NLRP3/caspase-1/GSDM pathway, whether autophagy can regulate neuronal pyroptosis through 3/- 4/- 5/- 11/GSDM is worthy of future research.

Reactive oxygen species (ROS) regulates autophagy and pyroptosis in nervous system
In this review, in most cases, autophagy and pyroptosis are mutually exclusive, but in a few cases, it is the opposite. Pyroptosis and autophagy can both repel each other and coexist in different nervous system cases. The reason may be related to different pathological processes and is required to be clarified.
It is believed that autophagy and pyroptosis will become new targets for the treatment of the nervous system.
In macroautophagy, the invasive pathogens and abnormal organelles are degraded after being isolated and transported to lysosomes in double-membrane vesicles (autophage) for degradation. In microautophagy, the direct invagination of lysosome membrane and then the encapsulation of cell contents. Companion-mediated autophagy does not capture goods with vesicular intermediates, but directly transports the substrate bound with chaperone protein to lysosomal cavity.
Autophagy inhibits ROS activation of NLRP3 inflammasome-mediated pyroptosis by clearing ROS. Autophagy also inhibits pyroptosis by inhibiting NLRP3 inflammasome. The elevated ROS activates autophagy and NLRP3 inflammasome-mediated pyroptosis.
References
Wong SQ et al (2020) Autophagy in aging and longevity. Hum Genet 139(3):277–290
Sciarretta S et al (2018) The role of autophagy in the heart. Annu Rev Physiol 80:1–26
Miller DR, Thorburn A (2021) Autophagy and organelle homeostasis in cancer. Dev Cell 56(7):906–918
Racanelli AC et al (2018) Autophagy and inflammation in chronic respiratory disease. Autophagy 14(2):221–232
Antunes F et al (2018) Autophagy and intermittent fasting: the connection for cancer therapy? Clinics (Sao Paulo) 73(suppl 1):e814s
Galluzzi L et al (2017) Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17(2):97–111
Levine B, Kroemer G (2019) Biological functions of autophagy genes: a disease perspective. Cell 176(1–2):11–42
Sieńko K, Poormassalehgoo A, Yamada K, Goto-Yamada S (2020) Microautophagy in plants: consideration of its molecular mechanism. Cells 9(4):887. https://doi.org/10.3390/cells9040887
Krause GJ et al (2022) Reduced endosomal microautophagy activity in aging associates with enhanced exocyst-mediated protein secretion. Aging Cell 21(10):e13713
Oku M, Sakai Y (2018) Three distinct types of microautophagy based on membrane dynamics and molecular machineries. BioEssays 40(6):e1800008
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24(1):92–104
Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19(6):365–381
Madrigal-Matute J, Cuervo AM, Sluimer JC (2022) Chaperone-mediated autophagy protects against atherosclerosis. Autophagy 18(10):2505–2507
Assaye MA, Gizaw ST (2022) Chaperone-mediated autophagy and its implications for neurodegeneration and cancer. Int J Gen Med 15:5635–5649
Dong Y et al (2019) Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J Mol Cell Cardiol 136:27–41
Belgrad J, De Pace R, Fields RD (2020) Autophagy in myelinating glia. J Neurosci 40(2):256–266
Allaire M et al (2019) Autophagy in liver diseases: time for translation? J Hepatol 70(5):985–998
Park H, Kang JH, Lee S (2020) Autophagy in neurodegenerative diseases: a hunter for aggregates. Int J Mol Sci 21(9):3369. https://doi.org/10.3390/ijms21093369
Stavoe AKH, Holzbaur ELF (2019) Autophagy in neurons. Annu Rev Cell Dev Biol 35:477–500
Eshraghi M, Adlimoghaddam A, Mahmoodzadeh A, Sharifzad F, Yasavoli-Sharahi H, Lorzadeh S, Albensi BC, Ghavami S (2021) Alzheimer's disease pathogenesis: role of autophagy and mitophagy focusing in microglia. Int J Mol Sci 22(7):3330. https://doi.org/10.3390/ijms22073330
Sung K, Jimenez-Sanchez M (2020) Autophagy in astrocytes and its implications in neurodegeneration. J Mol Biol 432(8):2605–2621
Reddy PH, Oliver DM (2019) Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer's disease. Cells 8(5):488. https://doi.org/10.3390/cells8050488
Lizama BN, Chu CT (2021) Neuronal autophagy and mitophagy in Parkinson’s disease. Mol Aspects Med 82:100972
Croce KR, Yamamoto A (2019) A role for autophagy in Huntington’s disease. Neurobiol Dis 122:16–22
Mónaco A et al (2022) Inflammasome activation, NLRP3 engagement and macrophage recruitment to tumor microenvironment are all required for Salmonella antitumor effect. Cancer Immunol Immunother 71(9):2141–2150
Silva MT (2010) Bacteria-induced phagocyte secondary necrosis as a pathogenicity mechanism. J Leukoc Biol 88(5):885–896
Ruan J, Wang S, Wang J (2020) Mechanism and regulation of pyroptosis-mediated in cancer cell death. Chem Biol Interact 323:109052
Chen S et al (2018) Gasdermin family: a promising therapeutic target for stroke. Transl Stroke Res 9(6):555–563
Al Mamun A et al (2021) Pyroptosis in diabetic nephropathy. Clin Chim Acta 523:131–143
Galluzzi L et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25(3):486–541
Bourdonnay E, Henry T (2022) Transcriptional and epigenetic regulation of gasdermins. J Mol Biol 434(4):167253
Rao Z et al (2022) Pyroptosis in inflammatory diseases and cancer. Theranostics 12(9):4310–4329
Knorr J, Wree A, Feldstein AE (2022) Pyroptosis in steatohepatitis and liver diseases. J Mol Biol 434(4):167271
Yang F et al (2022) Pyroptosis and pyroptosis-inducing cancer drugs. Acta Pharmacol Sin 43(10):2462–2473
Huang C, Li J, Zhang C (2022) What role does pyroptosis play in cancer? Mol Metab 65:101587
Wang Y et al (2018) GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem Biophys Res Commun 495(1):1418–1425
Zhang Z et al (2021) Caspase-3-mediated GSDME induced pyroptosis in breast cancer cells through the ROS/JNK signalling pathway. J Cell Mol Med 25(17):8159–8168
Zhao H, Yang Y, Si X, Liu H, Wang H (2022) the role of pyroptosis and autophagy in ischemia reperfusion injury. Biomolecules 12(7):1010. https://doi.org/10.3390/biom12071010
Zhao H, Liu H, Yang Y, Wang H (2022) The role of autophagy and pyroptosis in liver disorders. Int J Mol Sci 23(11):6208. https://doi.org/10.3390/ijms23116208
Hu Y et al (2022) Pyroptosis, and its role in central nervous system disease. J Mol Biol 434(4):167379
McKenzie BA, Dixit VM, Power C (2020) Fiery cell death: pyroptosis in the central nervous system. Trends Neurosci 43(1):55–73
Yang B et al (2022) Emerging mechanisms and targeted therapy of pyroptosis in central nervous system trauma. Front Cell Dev Biol 10:832114
Nin N, Angulo M, Briva A (2018) Effects of hypercapnia in acute respiratory distress syndrome. Ann Transl Med 6(2):37
Shimoda T et al (2021) Removal of a catheter mount and heat-and-moisture exchanger improves hypercapnia in patients with acute respiratory distress syndrome: a retrospective observational study. Medicine (Baltimore) 100(36):e27199
Ding HG et al (2018) Hypercapnia induces IL-1β overproduction via activation of NLRP3 inflammasome: implication in cognitive impairment in hypoxemic adult rats. J Neuroinflammation 15(1):4
Ding H et al (2020) Hypercapnia exacerbates the disruption of the blood-brain barrier by inducing interleukin-1β overproduction in the blood of hypoxemic adult rats. Int J Mol Med 46(2):762–772
Ułamek-Kozioł M et al (2017) Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol Rep 69(6):1289–1294
Ułamek-Kozioł M et al (2016) Dysregulation of autophagy, mitophagy, and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer’s disease. J Alzheimers Dis 54(1):113–121
Ułamek-Kozioł M et al (2019) Dysregulation of autophagy, mitophagy, and apoptosis genes in the CA3 region of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. J Alzheimers Dis 72(4):1279–1286
Ding HG et al (2020) Hypercapnia promotes microglial pyroptosis via inhibiting mitophagy in hypoxemic adult rats. CNS Neurosci Ther 26(11):1134–1146
Hseu YC et al (2022) Coenzyme Q(0) inhibits NLRP3 inflammasome activation through mitophagy induction in LPS/ATP-stimulated macrophages. Oxid Med Cell Longev 2022:4266214
Molagoda IMN, Athapaththu AMGK, Choi YH, Park C, Jin CY, Kang CH, Lee MH, Kim GY (2021) Fisetin inhibits NLRP3 inflammasome by suppressing TLR4/MD2-mediated mitochondrial ROS production. Antioxidants (Basel) 10(8):1215. https://doi.org/10.3390/antiox10081215
Wu X et al (2021) NLRP3 deficiency protects against intermittent hypoxia-induced neuroinflammation and mitochondrial ROS by promoting the PINK1-Parkin pathway of mitophagy in a murine model of sleep apnea. Front Immunol 12:628168
Barazzuol L, Giamogante F, Brini M, Calì T (2020) PINK1/Parkin mediated mitophagy, Ca2+ signalling, and ER-mitochondria contacts in Parkinson's disease. Int J Mol Sci 21(5):1772. https://doi.org/10.3390/ijms21051772
Chen YY et al (2021) Bacterial factors required for Streptococcus pneumoniae coinfection with influenza A virus. J Biomed Sci 28(1):60
Adriani KS et al (2007) Community-acquired recurrent bacterial meningitis in adults. Clin Infect Dis 45(5):e46-51
Valente C et al (2021) Intra-species interactions in Streptococcus pneumoniae biofilms. Front Cell Infect Microbiol 11:803286
Kim JY et al (2015) Streptococcus pneumoniae induces pyroptosis through the regulation of autophagy in murine microglia. Oncotarget 6(42):44161–44178
Chung SD et al (2012) Activating Nrf-2 signaling depresses unilateral ureteral obstruction-evoked mitochondrial stress-related autophagy, apoptosis and pyroptosis in kidney. PLoS One 7(10):e47299
Hwang HS et al (2019) NOD2 signaling pathway is involved in fibronectin fragment-induced pro-catabolic factor expressions in human articular chondrocytes. BMB Rep 52(6):373–378
Gong Q et al (2018) Structural basis of RIP2 activation and signaling. Nat Commun 9(1):4993
Zheng Y et al (2018) NOD2-RIP2 contributes to the inflammatory responses of mice in vivo to Streptococcus pneumoniae. Neurosci Lett 671:43–49
Wang G et al (2022) NOD2-RIP2 signaling alleviates microglial ROS damage and pyroptosis via ULK1-mediated autophagy during Streptococcus pneumonia infection. Neurosci Lett 783:136743
Dominic A, Le NT, Takahashi M (2022) Loop between NLRP3 inflammasome and reactive oxygen species. Antioxid Redox Signal 36(10–12):784–796
Tetorou K et al (2021) Current therapies for neonatal hypoxic-ischaemic and infection-sensitised hypoxic-ischaemic brain damage. Front Synaptic Neurosci 13:709301
Ogawa Y et al (2021) Brain damage caused by neonatal hypoxia-ischemia and the effects of hypothermia in severe combined immunodeficient (SCID) mice. Exp Neurol 337:113577
Zhang L et al (2021) Overexpression of long noncoding RNA H19 inhibits cardiomyocyte apoptosis in neonatal rats with hypoxic-ischemic brain damage through the miR-149-5p/LIF/PI3K/Akt axis. Biopreserv Biobank 19(5):376–385
Peeples ES, Genaro-Mattos TC (2022) Ferroptosis: A Promising Therapeutic Target for Neonatal Hypoxic-Ischemic Brain Injury. Int J Mol Sci 23(13):7420. https://doi.org/10.3390/ijms23137420
Shu J et al (2022) Human bone marrow mesenchymal stem cells-derived exosomes protect against nerve injury via regulating immune microenvironment in neonatal hypoxic-ischemic brain damage model. Immunobiology 227(3):152178
Hu Z et al (2021) Human umbilical cord mesenchymal stem cell-derived exosomes attenuate oxygen-glucose deprivation/reperfusion-induced microglial pyroptosis by promoting FOXO3a-dependent mitophagy. Oxid Med Cell Longev 2021:6219715
Lou G et al (2020) Mitophagy and neuroprotection. Trends Mol Med 26(1):8–20
Duffner PK, Cohen ME, Parker MS (1988) Prospective intellectual testing in children with brain tumors. Ann Neurol 23(6):575–579
Swain A et al (2020) Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat Metab 2(7):594–602
Vidal I, Fernández-Florido E, Marrero AD, Castilla L, Quesada AR, Martínez-Poveda B, Medina MÁ (2022) The immunomodulator dimethyl itaconate inhibits several key steps of angiogenesis in cultured endothelial cells. Int J Mol Sci 23(24):15972. https://doi.org/10.3390/ijms232415972
Yang S, Zhang X, Zhang H, Lin X, Chen X, Zhang Y, Lin X, Huang L, Zhuge Q (2021) Dimethyl itaconate inhibits LPS induced microglia inflammation and inflammasome mediated pyroptosis via inducing autophagy and regulating the Nrf 2/HO 1 signaling pathway. Mol Med Rep 24(3):672. https://doi.org/10.3892/mmr.2021.12311
Zeng H et al (2019) Lentivirus-mediated downregulation of α-synuclein reduces neuroinflammation and promotes functional recovery in rats with spinal cord injury. J Neuroinflammation 16(1):283
Chauhan P, Sheng WS, Hu S, Prasad S, Lokensgard JR (2021) Differential cytokine-induced responses of polarized microglia. Brain Sci 11(11):1482. https://doi.org/10.3390/brainsci11111482
Hurtado-Navarro L, Angosto-Bazarra D, Pelegrín P, Baroja-Mazo A, Cuevas S (2022) NLRP3 inflammasome and pyroptosis in liver pathophysiology: the emerging relevance of Nrf2 inducers. Antioxidants (Basel). 11(5):870. https://doi.org/10.3390/antiox11050870
Sok SPM et al (2021) 1’-Acetoxychavicol acetate inhibits NLRP3-dependent inflammasome activation via mitochondrial ROS suppression. Int Immunol 33(7):373–386
Maida CD, Daidone M, Pacinella G, Norrito RL, Pinto A, Tuttolomondo A (2022) Diabetes and ischemic stroke: an old and new relationship an overview of the close interaction between these diseases. Int J Mol Sci. 23(4):2397. https://doi.org/10.3390/ijms23042397
Jensen T et al (2021) Insulin-treated versus noninsulin-treated diabetes and risk of ischemic stroke in patients with atrial fibrillation. Vascul Pharmacol 136:106809
Wang C et al (2020) Diabetic encephalopathy causes the imbalance of neural activities between hippocampal glutamatergic neurons and GABAergic neurons in mice. Brain Res 1742:146863
Hussain S et al (2014) Evidence for cortical neuronal loss in male type 2 diabetic Goto-Kakizaki rats. J Alzheimers Dis 41(2):551–560
Zhang D et al (2019) Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J Neurosci Res 97(6):645–660
Zhao N et al (2019) Amentoflavone suppresses amyloid β1-42 neurotoxicity in Alzheimer’s disease through the inhibition of pyroptosis. Life Sci 239:117043
Che H et al (2020) Melatonin exerts neuroprotective effects by inhibiting neuronal pyroptosis and autophagy in STZ-induced diabetic mice. Faseb j 34(10):14042–14054
Byun JC, Lee SR, Kim CS (2021) Effects of carnosine and hypothermia combination therapy on hypoxic-ischemic brain injury in neonatal rats. Clin Exp Pediatr 64(8):422–428
Colls Garrido C, Riquelme Gallego B, Sánchez García JC, Cortés Martín J, Montiel Troya M, Rodríguez BR (2021) The effect of therapeutic hypothermia after cardiac arrest on the neurological outcome and survival-a systematic review of RCTs published between 2016 and 2020. Int J Environ Res Public Health. 18(22):11817. https://doi.org/10.3390/ijerph182211817
Tu Y et al (2019) Mild hypothermia alleviates diabetes aggravated cerebral ischemic injury via activating autophagy and inhibiting pyroptosis. Brain Res Bull 150:1–12
Wen YD et al (2008) Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 4(6):762–769
Zhang X et al (2013) Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 9(9):1321–1333
Liu J et al (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147(1):223–234
Liu H et al (2021) Inhibition of autophagy-dependent pyroptosis attenuates cerebral ischaemia/reperfusion injury. J Cell Mol Med 25(11):5060–5069
Anjum A, Yazid MD, Fauzi Daud M, Idris J, Ng AMH, Selvi Naicker A, Ismail OHR, Athi Kumar RK, Lokanathan Y (2020) Spinal cord injury: pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int J Mol Sci. 21(20):7533. https://doi.org/10.3390/ijms21207533
Eli I, Lerner DP, Ghogawala Z (2021) Acute traumatic spinal cord injury. Neurol Clin 39(2):471–488
Al Mamun A et al (2021) Role of pyroptosis in spinal cord injury and its therapeutic implications. J Adv Res 28:97–109
Wu C et al (2021) Betulinic acid inhibits pyroptosis in spinal cord injury by augmenting autophagy via the AMPK-mTOR-TFEB signaling pathway. Int J Biol Sci 17(4):1138–1152
Mortezaee K et al (2018) Inflammasome: its role in traumatic brain and spinal cord injury. J Cell Physiol 233(7):5160–5169
Xu Y et al (2021) GDF-11 protects the traumatically injured spinal cord by suppressing pyroptosis and necroptosis via TFE3-mediated autophagy augmentation. Oxid Med Cell Longev 2021:8186877
Guo R, Wang H, Cui N (2021) Autophagy regulation on pyroptosis: mechanism and medical implication in sepsis. Mediators Inflamm 2021:9925059
Funding
This work was supported by the grants from the key scientific and technological projects in Henan Province, China (Grant No. 202102310153).
Author information
Authors and Affiliations
Contributions
Honggang Wang devised, wrote and funded with the main manuscript; Huijie Zhao wrote and funded with the main manuscript; Yanting Zhang, Xiaodi Fu and Chaoran Chen wrote the main manuscript. All authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Consent to participate
All authors consent to participate.
Consent for publication
All authors consent for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Huijie Zhao and Xiaodi Fu are co-first authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, H., Fu, X., Zhang, Y. et al. The Role of Pyroptosis and Autophagy in the Nervous System. Mol Neurobiol 61, 1271–1281 (2024). https://doi.org/10.1007/s12035-023-03614-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03614-2