
Abstract
Due to the limitations of the present risk genes in understanding the etiology of amyotrophic lateral sclerosis (ALS), it is necessary to find additional causative genes utilizing novel approaches. In this study, we conducted a two-stage proteome-wide association study (PWAS) using ALS genome-wide association study (GWAS) data (N = 152,268) and two distinct human brain protein quantitative trait loci (pQTL) datasets (ROSMAP N = 376 and Banner N = 152) to identify ALS risk genes and prioritized candidate genes with Mendelian randomization (MR) and Bayesian colocalization analysis. Next, we verified the aberrant expression of risk genes in multiple tissues, including lower motor neurons, skeletal muscle, and whole blood. Six ALS risk genes (SCFD1, SARM1, TMEM175, BCS1L, WIPI2, and DHRS11) were found during the PWAS discovery phase, and SARM1 and BCS1L were confirmed during the validation phase. The following MR (p = 2.10 × 10−7) and Bayesian colocalization analysis (ROSMAP PP4 = 0.999, Banner PP4 = 0.999) confirmed the causal association between SARM1 and ALS. Further differential expression analysis revealed that SARM1 was markedly downregulated in lower motor neurons (p = 7.64 × 10−3), skeletal muscle (p = 9.34 × 10−3), and whole blood (p = 1.94 × 10−3). Our findings identified some promising protein candidates for future investigation as therapeutic targets. The dysregulation of SARM1 in multiple tissues provides a new way to explain ALS pathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a common neurodegenerative disease that damages upper and lower motor neurons, eventually resulting in muscular weakness and paralysis [1]. The pathophysiology of ALS has been shown to be significantly influenced by genetic factors, and the lifetime risk in the general population has a heritability of 52.3% [2, 3]. To study the pathogenic mechanisms underlying the disease, researchers have identified many disease-associated genes, such as SOD1, TDP-43, FUS, and C9ORF72, but there are still some ALS patients whose etiology cannot be explained [4]. Among the current drug treatments, riluzole has long been the only drug to extend the survival of patients with ALS [5]. Moreover, other treatments, such as gene therapies, are still under investigation [5]. However, the present effective therapeutic targets and strategies for ALS are still limited. Therefore, new risk genes remain to be discovered, which will also facilitate the development of new targets for disease treatment.
Previous studies have used genome-wide association studies (GWAS) to identify disease-related genetic variants in case–control studies and have achieved great success in mapping susceptibility genes for ALS [1]. However, GWAS examining genetic variation in isolation to explain the pathogenic causes of ALS is limited, and complex biological processes have been shown to be involved in the pathogenesis of ALS, such as malfunctioning proteins [6, 7]. Previous studies have highlighted the pathology of brain proteins in the development of ALS, such as TDP-43 [8] and FUS [9]. These proteins play crucial roles in regulating RNA metabolism and protein homeostasis, and mutations or misregulation of their activity can lead to neurodegeneration and disease pathology [10]. Therefore, studying the abnormalities in protein abundance in the brain is essential to reveal ALS pathogenesis and to identify reliable biomarkers.
To study genetic causes and etiopathogenesis, researchers have also concentrated on other ALS-related tissues in addition to motor neurons, including skeletal muscle [11] and peripheral blood [12]. Skeletal muscle is one of the main target tissues affected by ALS [13], and studying genetic abnormalities in skeletal muscle biopsies can be correlated with clinical features and disease progression in ALS patients. In addition, changes observed in gene expression patterns in whole blood may relate to the same changes occurring within the brain [14], which may offer insight into the pathogenic mechanisms of motor neuron degeneration in ALS. Therefore, we tried to combine the strengths across tissues and utilize differential expression strategies to verify whether the high-confidence risk genes are consistently dysregulated in multiple ALS-related tissues.
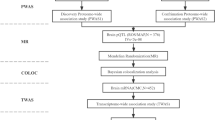
In the current work, we employed proteome-wide association studies (PWAS), a robust technique for investigating the relationship between protein abundance and disease phenotype by combining GWAS and protein quantitative trait loci (pQTL) data [15]. To validate our risk genes from multiple levels, we took a four-step approach to link the proteome and ALS. First, we integrated the large ALS GWAS data [16] and two independent human brain pQTL datasets to conduct a two-stage PWAS. The discovery pQTL data came from the ROSMAP dataset [17, 18], and the confirmatory data came from the Banner dataset [17]. We used 376 and 152 human brain proteomes, which have been the largest available human brain pQTL dataset so far. Next, to further obtain the causal relationship between candidate genes and the disease phenotype, we then applied Mendelian randomization and Bayesian colocalization analysis [19]. Finally, we verified the impact of the dysregulation of susceptibility genes found in human brain on ALS pathogenesis in various tissues using differential expression analysis of lower motor neurons, skeletal muscle, and whole blood. Figure 1 shows the detailed procedures of various analysis stages in the study.

We took a four-step approach to link the proteome and ALS. First, we integrated the large ALS GWAS dataset and two independent human brain pQTL datasets (ROSMAP dataset and Banner dataset) to conduct a two-stage PWAS. Next, we applied Mendelian randomization and Bayesian colocalization analysis to validate the causal association between SARM1 and ALS via their cis-regulated brain protein abundance. Finally, we verified the dysregulation of susceptibility genes found in the PWAS stage using differential expression analysis in lower motor neurons, skeletal muscle, and whole blood. ALS, amyotrophic lateral sclerosis; GWAS, genome-wide association study; pQTL, protein quantitative trait locus; ROSMAP, Religious Orders Study and Rush Memory and Aging Project; PWAS, proteome-wide association study
Materials and Methods
ALS GWAS Data
The main analysis utilized the most recent and largest ALS genome-wide association study by van Rheenen et al. [16]. Briefly, this is a cross-ancestry GWAS meta-analysis. This analysis included 29,612 ALS patients and 122,656 control individuals and consisted of 117 independent cohorts in European ancestries (27,205 ALS patients and 110,881 control individuals) [20, 21] and the summary GWAS statistics of ALS in Asian ancestries (2407 ALS patients and 11,775 control individuals) [22, 23]. All detailed information (e.g., studied subjects, genotyping, quality control, and statistical analyses) and data availability statements can be found in the original article [16].
Human Brain pQTL Data for Discovery PWAS
We obtained the protein abundance, referred to as protein weights, in the human brain from Wingo et al. [17], who estimated the effects of genetic variants on protein weights and generated SNP-protein abundance weights (i.e., pQTL). In addition, 376 subjects with both proteomic and genetic data were included in their PWAS. These patients exhibited different features in clinical diagnosis (including but not limited to dementia, stroke, and depression), cognitive performance, and neuropathologic traits. Furthermore, the proteomic reference dataset went through quality control procedures to identify and control the effects of clinical covariates (i.e., age, sex, and final clinical diagnosis of cognitive status) before estimating protein weights. Please refer to the original papers [17, 18] for further detailed information about proteomic profiling, protein quantification, and quality control.
Human Brain pQTL Data for Confirmation PWAS
The other human brain pQTL data were generated from 152 subjects with both proteomic and genetic data. The human brain protein abundance included 8168 proteins after quality control. All the samples came from the dlPFC of European participants. It also passed the quality control procedures to remove the effects of clinical covariates. More detailed information can be found in the original papers [17, 24].
Proteome-Wide Association Studies
In the discovery phase, we performed the PWAS by integrating ALS GWAS results with human brain proteomes profiled from the dlPFC (ROSMAP dataset [17]) using the FUSION pipeline. Confirmatory PWAS was performed using the same ALS GWAS and an independent set of 152 human brain proteomes profiled from the dlPFC (Banner dataset [17]). The PWAS was performed using FUSION [25] software with default settings.
Mendelian Randomization Analysis
We further used MR analysis to validate the causal association between the identified risk genes and ALS. The brain protein abundance from the ROSMAP dataset was selected as the exposure. The SNPs included in the study robustly and independently (R2 < 0.001) predicted exposures at a genome-wide level (5 × 10−8). The Wald ratio calculated the log odds change in ALS risk per standard deviation change in protein biomarker in relation to the instrumenting SNP’s risk allele. MR analysis was performed using the “TwoSampleMR” package in R 4.1.02 (https://github.com/MRCIEU/TwoSampleMR).
Colocalization Analysis
We investigated colocalization using a Bayesian colocalization method, COLOC. Colocalization was performed using the “coloc” R package [26] (http://cran.r-project.org/web/packages/coloc) and FUSION software [25]. The posterior probabilities for a shared causal variant between a pQTL and ALS significant genes in the discovery PWAS were calculated, and a high PP4 probability indicates that there is a single variant that affects both the protein expression and ALS. To confirm that the risk proteins discovered in PWAS share a common causal variant with ALS, the evidence was defined as a posterior probability of hypothesis 4 (PPH4) of 0.8 or greater [26].
Expression Analysis of Significant PWAS Genes
To further verify the dysregulation of the significant PWAS genes in other ALS-related tissues, we collected gene expression datasets from lower motor neurons [27], skeletal muscle [28], and whole blood [29]. The original studies carried out strict quality control of the sample sources, and the expression levels of genes were normalized during the analysis process. In this study, differential expression analysis was performed in R (http://cran.r-project.org) using a two-sample t test. A p value < 0.05 was considered statistically significant.
Gene Expression Analysis from Lower Motor Neurons
The lower motor neuron dataset was obtained from Highley et al. [27]. In their study, RNA was extracted from lower motor neurons that were isolated from the anterior horns of the postmortem cervical spinal cord. In total, there were 6 sporadic ALS patients with a mean age of 60.2 years and 6 healthy control individuals with a mean age of 61.7 years. Samples were hybridized to GeneChip Human Exon 1.0 ST Arrays, and the Partek Genomics Suite was used for normalization [27].
Gene Expression Analysis from Skeletal Muscle
Bakay et al. [28] studied 125 human muscle biopsies from 13 diagnostic groups, including ALS, fascioscapulohumeral muscular dystrophy, Becker muscular dystrophy, and so on. Affymetrix Human Genome U133A and U133B Array were used for this gene expression analysis [28]. Here, the gene expression profile, including 9 ALS patients and 18 control individuals, and the annotation platform GPL97 were used in our analysis. Characteristics of the patient and normal volunteer samples used for mRNA profiling can be found in the original article [28].
Gene Expression Analysis in Whole Blood
Wouter et al. [29] conducted a two-stage transcriptome-wide study, and with the corresponding whole blood gene expression profile, including 397 Netherlands ALS patients and 645 control subjects, they identified 2943 differentially expressed transcripts. The quality of isolated RNA was assessed using the Agilent 2100 Bioanalyzer system, and samples were hybridized to Illumina’s HumanHT-12 version 3 and version 4 BeadChips according to the manufacturer’s protocol (Illumina, Inc., San Diego, CA, USA) [29]. All the probes were aligned to the NCBI reference genome build 36, and expression heterogeneity was eliminated by applying surrogate variable analysis (SVA) [29]. The last dataset we used in differential expression analysis included 233 ALS patients and 508 matched control individuals.
Results
PWAS Identified 6 Candidate Genes Associated with ALS Using Human Brain pQTL
In the discovery phase, we performed a PWAS by integrating ALS GWAS and ROSMAP pQTL data. As a result, 6 genes (SCFD1, SARM1, TMEM175, BCS1L, WIPI2, and DHRS11) whose brain protein levels were associated with ALS were identified. Next, to validate the results, we conducted a replication study using the Banner dataset, and 2 (SARM1 and BCS1L) of these 6 proteins were replicated (as shown in Table 1).
MR Verified 3 Genes Associated with ALS
Using the ROSMAP dataset, MR analysis of brain pQTL and ALS GWAS verified that SARM1 (OR = 0.33, p = 2.10 × 10−7), SCFD1 (OR = 4.54, p = 3.72 × 10−13), and DHRS11 (OR = 0.61, p = 7.40 × 10−5) provided evidence of a causal association with ALS.
Colocalization Between ALS Risk Genes and pQTL in Human Brain
To further explore the probability that there are the same shared causal variants driving the ALS GWAS and pQTL signals, we performed colocalization analysis. The ROSMAP dataset and Banner dataset were separately used in the analysis. Two (SCFD1 and SARM1) of six genes provided evidence based on PPH4 > 80% in the ROSMAP dataset, and only one gene (SARM1) passed replication analysis in the Banner dataset (as shown in Table 2). In particular, SARM1 showed the most causal association with ALS in two datasets (ROSMAP PP4 = 0.999, Banner PP4 = 0.999), which indicates the potential role SARM1 plays in ALS risk.
Expression Analysis of PWAS Genes in Lower Motor Neurons, Skeletal Muscle, and Whole Blood
We conducted differential expression analysis using 3 datasets, and the tissue sources where RNA was extracted separately came from lower motor neurons, skeletal muscle, and whole blood. According to the t test, SARM1 was downregulated in ALS patients compared with control individuals and matched the directionality of PWAS associations in lower motor neurons, skeletal muscle, and whole blood (as shown in Fig. 2). In addition, TMEM175 (p = 6.42 × 10−12), BCS1L (p = 2.02 × 10−7), and DHRS11 (p = 2.60 × 10−10) were also downregulated in whole blood with the same directionality in PWAS. Table 3 displays the consolidated results for all six genes.

Differential expression analysis for SARM1 validated dysregulation of risk genes at transcription level in multiple tissues. The boxplot shows the differential expression analysis results of SARM1 between ALS patients and healthy control individuals in lower motor neurons, skeletal muscle, and whole blood. The p value was obtained from the t test. a NALS = 6 and NControl = 6. SARM1 was downregulated in lower motor neurons (p = 7.64 × 10−3). b NALS = 9 and NControl = 18. SARM1 was downregulated in skeletal muscle (p = 9.34 × 10−3). c NALS = 233 and NControl = 508. SARM1 was downregulated in whole blood (p = 1.94 × 10−3). ALS, amyotrophic lateral sclerosis. #p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
In the present work, we used a two-stage approach of PWAS, MR, Bayesian colocalization, and differential expression analysis to thoroughly identify ALS risk genes. First, we identified 6 high-risk proteins (SCFD1, SARM1, TMEM175, BCS1L, WIPI2, and DHRS11) linked to ALS in the human brain. In two distinct PWAS, SARM1 and BCS1L were shown to be significant. Second, we verified the causal association of SARM1 with ALS via their cis-regulated brain protein abundance in MR analysis. Colocalization analysis also corroborated the clear causal relationship between SARM1 and ALS. Finally, we confirmed the dysregulation of SARM1 at the transcriptome level in three distinct tissues (lower motor neurons, skeletal muscle, and whole blood) with the same directionality as in PWAS. In addition, BCS1L, TMEM175, and DHRS11 were verified in whole blood. Future research on ALS pathophysiology and treatment strategies could focus on these genes, particularly SARM1.
In the current work, we primarily focused on the ALS risk gene SARM1 [21, 30]. SARM1, Sterile Alpha and TIR Motif Containing 1, encodes a protein with critical NADase activity and has been shown to be a key mediator of axon degeneration in early animal experiments [31]. Axon degeneration can be caused by activated SARM1 due to the depletion of the axon survival factor NMNAT2 induced by injury or disease [32]. At present, preclinical studies of SARM1 inhibitors and gene therapies targeting SARM1 for neurodegenerative diseases are still being conducted [33, 34]. Recently, literature regarding the ALS PWAS-significant genes also indicated the abnormal expression of SARM1 in ALS [35]. We obtained the similar results utilizing a different comprehensive analytical framework, showing that the SARM1 risk gene has high confidence in populations of different origins. In addition, we further emphasized the tissue specificity of SARM1 and verified its downregulation in lower motor neurons, skeletal muscle, and whole blood. Similarly, it has been shown that the loss of SARM1 cannot slow ALS disease progression or improve ALS-associated axon degeneration in the SOD1 mouse model of ALS [36,37,38]. In addition, in the mutant TDP-43 mouse model of ALS, deleting SARM1 can significantly attenuate motor axon and motor neuron cell body degeneration [38, 39], which indicates that the downregulation of SARM1 could be a result of the body’s compensatory mechanisms to protect neurons from cell death. In addition to the brain and motor neurons, our study first reported the dysregulation of SARM1 in skeletal muscle and whole blood. The downregulation of SARM1 may affect energy metabolism by inhibiting the clearance of abnormal mitochondria in skeletal muscle and promoting the clinical manifestation of muscle weakness in ALS patients. Maintenance of appropriate NAD + levels is known to be important for mitochondrial function [40]. Additionally, SARM1, encoding a protein with critical NADase activity, may impair mitochondrial function by affecting the level of NAD+ in skeletal muscle, causing muscle damage in ALS patients. Furthermore, the dysregulation of SARM1 in the peripheral circulation of ALS patients provided new evidence for its efficacy as a biomarker for disease monitoring. Our results suggested that SARM1 was dysregulated in a number of ALS core affected tissues and that it may be involved in a variety of pathophysiological events contributing to the development of ALS. These results at transcription level validated our PWAS findings, which prioritized proteins that were stably differentially expressed in multiple peripheral tissues, with important implications for the identification of future drug targets. Therefore, even in its early stages, SARM1 is very promising and has important ramifications for future studies to determine the function of SARM1 in the onset of ALS.
The present study has several strengths. First, we used two datasets independently in each PWAS and colocalization analysis, which not only verified the preliminary outcomes but also shed light on the causal relationship through the latter analysis. The alteration of protein abundance in ALS patients’ brains implicated the involvement of novel proteins in pathological pathways as functional molecules, suggesting the effectiveness of PWAS. Second, we validated the risk genes in multiple tissues, which enhanced the reliability of our results. This provided several available tissues to identify future biomarkers and emphasized the tissue specificity of gene expression in ALS.
This study has several limitations. First, the relatively small sample size limited the output of protein abundance; thus, the pQTL data size in PWAS was limited, which explained the modest number of important PWAS genes revealed. Second, the sample size varied among the different datasets utilized in the differential expression analysis. Third, tissue sources were restricted, which may lead to a one-sided view of disease mechanisms, with the causes manifesting in multiple dimensions. (1) As a neurodegenerative illness, ALS affects the entire brain network, while the brain tissue used in PWAS only included the dlPFC. (2) We only used three peripheral tissues in the differential expression study, and other helpful tissues, such as higher motor neurons, should be investigated further. Finally, future exploration of the pathogenesis of ALS in SARM1 cell models and animal studies is still needed.
Conclusion
At the human brain proteome level, we discovered six ALS risk genes (SCFD1, SARM1, TMEM175, BCS1L, WIPI2, and DHRS11). Notably, SARM1 was the most promising biomarker for ALS and was verified in multiple ALS-related tissues (lower motor neurons, skeletal muscle, and whole blood). SARM1-mediated axon degeneration may serve as a therapeutically targeted pathway for ALS. However, more studies are needed to confirm our results in the future.
References
Al-Chalabi A, van den Berg LH, Veldink J (2017) Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 13(2):96–104. https://doi.org/10.1038/nrneurol.2016.182
Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, Feldman EL (2022) Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol 21(5):465–479. https://doi.org/10.1016/S1474-4422(21)00414-2
Du Y, Wen Y, Guo X, Hao J, Wang W, He A, Fan Q, Li P et al (2018) A genome-wide expression association analysis identifies genes and pathways associated with amyotrophic lateral sclerosis. Cell Mol Neurobiol 38(3):635–639. https://doi.org/10.1007/s10571-017-0512-2
Cicardi ME, Marrone L, Azzouz M, Trotti D (2021) Proteostatic imbalance and protein spreading in amyotrophic lateral sclerosis. EMBO J 40(10):e106389. https://doi.org/10.15252/embj.2020106389
Yang X, Ji Y, Wang W, Zhang L, Chen Z, Yu M, Shen Y, Ding F et al (2021) Amyotrophic lateral sclerosis: molecular mechanisms, biomarkers, and therapeutic strategies. Antioxidants (Basel) 10(7):1012. https://doi.org/10.3390/antiox10071012
Mandrioli J, Mediani L, Alberti S, Carra S (2020) ALS and FTD: where RNA metabolism meets protein quality control. Semin Cell Dev Biol 99:183–192. https://doi.org/10.1016/j.semcdb.2019.06.003
Zhang S, Cooper-Knock J, Weimer AK et al (2022) Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 110(6):992-1008.e11. https://doi.org/10.1016/j.neuron.2021.12.019
Keating SS, San Gil R, Swanson MEV, Scotter EL, Walker AK (2022) TDP-43 pathology: from noxious assembly to therapeutic removal. Prog Neurobiol 211:102229. https://doi.org/10.1016/j.pneurobio.2022.102229
Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK (2019) Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 14(12):25. https://doi.org/10.3389/fnmol.2019.00025
Mazumder S, Kiernan MC, Halliday GM, Timmins HC, Mahoney CJ (2022) The contribution of brain banks to knowledge discovery in amyotrophic lateral sclerosis: a systematic review. Neuropathol Appl Neurobiol 48(7):e12845. https://doi.org/10.1111/nan.12845
Pikatza-Menoio O, Elicegui A, Bengoetxea X, Naldaiz-Gastesi N, López de Munain A, Gerenu G, Gil-Bea FJ, Alonso-Martín S (2021) The skeletal muscle emerges as a new disease target in amyotrophic lateral sclerosis. J Pers Med 11(7):671. https://doi.org/10.3390/jpm11070671
Swindell WR, Kruse CPS, List EO, Berryman DE, Kopchick JJ (2019) ALS blood expression profiling identifies new biomarkers, patient subgroups, and evidence for neutrophilia and hypoxia. J Transl Med 17(1):170. https://doi.org/10.1186/s12967-019-1909-0
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z et al (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17071. https://doi.org/10.1038/nrdp.2017.71
Sullivan PF, Fan C, Perou CM (2006) Evaluating the comparability of gene expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet 141B(3):261–8. https://doi.org/10.1002/ajmg.b.30272
Brandes N, Linial N, Linial M (2020) PWAS: proteome-wide association study-linking genes and phenotypes by functional variation in proteins. Genome Biol 21(1):173. https://doi.org/10.1186/s13059-020-02089-x
van Rheenen W, van der Spek RAA, Bakker MK et al (2021) Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet 53(12):1636–1648. https://doi.org/10.1038/s41588-021-00973-1
Wingo AP, Liu Y, Gerasimov ES, Gockley J, Logsdon BA, Duong DM, Dammer EB, Robins C et al (2021) Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat Genet 53(2):143–146. https://doi.org/10.1038/s41588-020-00773-z
Wingo AP, Fan W, Duong DM, Gerasimov ES, Dammer EB, Liu Y, Harerimana NV, White B et al (2020) Shared proteomic effects of cerebral atherosclerosis and Alzheimer’s disease on the human brain. Nat Neurosci 23(6):696–700. https://doi.org/10.1038/s41593-020-0635-5
Kibinge NK, Relton CL, Gaunt TR, Richardson TG (2020) Characterizing the causal pathway for genetic variants associated with neurological phenotypes using human brain-derived proteome data. Am J Hum Genet 106(6):885–892. https://doi.org/10.1016/j.ajhg.2020.04.007
Nicolas A, Kenna KP, Renton AE et al (2018) Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97(6):1268-1283.e6. https://doi.org/10.1016/j.neuron.2018.02.027
van Rheenen W, Shatunov A, Dekker AM et al (2016) Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48(9):1043–1048. https://doi.org/10.1038/ng.3622
Benyamin B, He J, Zhao Q et al (2017) Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat Commun 8(1):611. https://doi.org/10.1038/s41467-017-00471-1
Nakamura R, Misawa K, Tohnai G et al (2020) A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun Biol 3(1):526. https://doi.org/10.1038/s42003-020-01251-2
Beach TG, Adler CH, Sue LI et al (2015) Arizona study of aging and neurodegenerative disorders and brain and body donation program. Neuropathology 35(4):354–389. https://doi.org/10.1111/neup.12189
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, Jansen R, de Geus EJ et al (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48(3):245–252. https://doi.org/10.1038/ng.3506
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10(5):e1004383. https://doi.org/10.1371/journal.pgen.1004383
Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR, Higginbottom A, Raman R et al (2014) Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol 40(6):670–685. https://doi.org/10.1111/nan.12148
Bakay M, Wang Z, Melcon G, Schiltz L, Xuan J, Zhao P, Sartorelli V, Seo J et al (2006) Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain 129(Pt 4):996–1013. https://doi.org/10.1093/brain/awl023
van Rheenen W, Diekstra FP, Harschnitz O, Westeneng HJ, van Eijk KR, Saris CGJ, Groen EJN, van Es MA et al (2018) Whole blood transcriptome analysis in amyotrophic lateral sclerosis: a biomarker study. PLoS One 13(6):e0198874. https://doi.org/10.1371/journal.pone.0198874
Fogh I, Ratti A, Gellera C et al (2013) A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum Mol Genet 23(8):2220–31. https://doi.org/10.1093/hmg/ddt587
Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA et al (2012) dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337(6093):481–4. https://doi.org/10.1126/science.1223899
Bloom AJ, Mao X, Strickland A, Sasaki Y, Milbrandt J, DiAntonio A (2022) Constitutively active SARM1 variants that induce neuropathy are enriched in ALS patients. Mol Neurodegener 17(1):1. https://doi.org/10.1186/s13024-021-00511-x
Geisler S, Huang SX, Strickland A, Doan RA, Summers DW, Mao X, Park J, DiAntonio A et al (2019) Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J Exp Med 216(2):294–303. https://doi.org/10.1084/jem.20181040
Coleman MP, Höke A (2020) Programmed axon degeneration: from mouse to mechanism to medicine. Nat Rev Neurosci 21(4):183–196. https://doi.org/10.1038/s41583-020-0269-3
Gu XJ, Su WM, Dou M, Jiang Z, Duan QQ, Wang H, Ren YL, Cao B et al (2023) Identifying novel genes for amyotrophic lateral sclerosis by integrating human brain proteomes with genome-wide association data. J Neurol. https://doi.org/10.1007/s00415-023-11757-4
Peters OM, Lewis EA, Osterloh JM, Weiss A, Salameh JS, Metterville J, Brown RH, Freeman MR (2018) Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet 27(21):3761–3771. https://doi.org/10.1093/hmg/ddy260
Collins JM, Atkinson RAK, Matthews LM, Murray IC, Perry SE, King AE (2022) Sarm1 knockout modifies biomarkers of neurodegeneration and spinal cord circuitry but not disease progression in the mSOD1G93A mouse model of ALS. Neurobiol Dis 172:105821. https://doi.org/10.1016/j.nbd.2022.105821
Gilley J, Jackson O, Pipis M, Estiar MA, Al-Chalabi A, Danzi MC, van Eijk KR, Goutman SA et al (2021) Enrichment of SARM1 alleles encoding variants with constitutively hyperactive NADase in patients with ALS and other motor nerve disorders. Elife 10:e70905. https://doi.org/10.7554/eLife.70905
White MA, Lin Z, Kim E, Henstridge CM, Pena Altamira E, Hunt CK, Burchill E, Callaghan I et al (2019) Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathol Commun 7(1):166. https://doi.org/10.1186/s40478-019-0800-9
Hikosaka K, Yaku K, Okabe K, Nakagawa T (2021) Implications of NAD metabolism in pathophysiology and therapeutics for neurodegenerative diseases. Nutr Neurosci 24(5):371–383. https://doi.org/10.1080/1028415X.2019.1637504
Acknowledgements
We appreciate Dr. van Rheenen for sharing ALS GWAS data. Special thanks to participants of ROS, MAP, and Banner Sun Health Research Institute Brain and Body Donation Program. Dr. Wingo’s provision of human brain weights is acknowledged. We’re also grateful to Dr. Highley, Dr. Bakay, and Dr. van Rheenen for publicly available gene expression datasets.
Funding
This work was partly funded by Key R&D Projects of Science and Technology Department of Sichuan Province (2021YFS0248); Postdoctoral Foundation of West China Hospital (2020HXBH163); and College Students’ Innovation and Entrepreneurship Training Program (20221174L).
Author information
Authors and Affiliations
Contributions
All authors are grateful for participation in our research. CZ made substantial contributions to the conception and design of the work; YM, TJ, FQ, YH, and FH completed formal analysis and visualization; YM and TJ composed the draft manuscript; CZ revised the manuscript and approved the version to be published; CZ provided funding. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yanni Ma and Tingting Jia contributed equally to this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, Y., Jia, T., Qin, F. et al. Abnormal Brain Protein Abundance and Cross-tissue mRNA Expression in Amyotrophic Lateral Sclerosis. Mol Neurobiol 61, 510–518 (2024). https://doi.org/10.1007/s12035-023-03587-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03587-2