
Abstract
Several studies have reported separate roles of adenosine receptors and circadian clockwork in major depressive disorder. While less evidence exists for regulation of the circadian clock by adenosine signaling, a small number of studies have linked the adenosinergic system, the molecular circadian clock, and mood regulation. In this article, we review relevant advances and propose that adenosine receptor signaling, including canonical and other alternative downstream cellular pathways, regulates circadian gene expression, which in turn may underlie the pathogenesis of mood disorders. Moreover, we summarize the convergent point of these signaling pathways and put forward a pattern by which Homer1a expression, regulated by both cAMP-response element binding protein (CREB) and circadian clock genes, may be the final common pathogenetic mechanism in depression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is one of the most prevalent forms of mental illness. It is a complex and heterogeneous disorder associated with high individual suffering, increased risk of suicide, and a severe economic burden for society [1]. Several lines of evidence from animal and human studies have shown that disturbances of circadian clockwork are associated with the development of depression. Moreover, different chronotherapies, a variety of strategies that modulate biological clock, such as sleep deprivation and light therapy, are considered as alternative treatments for depression [2]. However, how the circadian clock influences pathophysiology of mood disorders, as well as the molecular and cellular mechanisms of action of the therapeutic interventions targeting circadian rhythm, is not well understood. The identification of the neurobiological substrates mediating the crosstalk between the circadian clock and mood regulation may lead to the development of new strategies for prevention and treatment of depression.
Numerous studies have demonstrated a role of adenosine receptors in the development of depression and antidepressant therapies [3,4,5]. Moreover, adenosinergic signaling is implicated in the regulation of different aspects of the circadian clock [6, 7]. However, the detailed mechanism has not been completely clarified. Recently, we found that the canonical circadian clock genes Per1 and Per2 were involved in the antidepressant action of an adenosine A1 receptor (A1R) agonist [8]. In addition, it has been shown that the expression of the synaptic plasticity protein Homer1a, proposed by our group as an important element mediating antidepressant effects and also a downstream target of adenosine receptor signaling [9,10,11,12], is directly regulated by the circadian clock [13].
In the present article, we review relevant recent advances linking adenosine receptors, circadian clock, and mood and propose that adenosine signaling regulates circadian clockwork and Homer1a, which may be a potential final common mechanism involved in the neurobiology and treatment of depression.
Adenosine Signaling and Mood
There are numerous studies on adenosine signaling and depression, which have been recently reviewed extensively by others [3,4,5]. The cellular effects of adenosine are mediated by four subtypes of G-protein coupled receptors: A1R, A2AR, A2BR, and A3R. In general, it was proposed that A1Rs promote antidepressant-like effects, while A2ARs’ activation enhances depression-like behaviors in rodents [3]. As for the A2B and A3 receptors, at present, we could not find any reports on their role in mood disorders [14, 15].
Several non-pharmacological antidepressant treatments including sleep deprivation (SD), electroconvulsive therapy (ECT), and deep brain stimulation (DBS) enhance A1R signaling [9]. Hines et al. was first to demonstrate that A1Rs are necessary for the antidepressant action of SD and that their activation leads to rapid antidepressant-like effects [16]. Our group utilized a line of transgenic mice conditionally overexpressing A1R in calcium/calmodulin-dependent protein kinase type II (CaMKII) forebrain neurons [9, 11, 17]. Upregulating A1R led to pronounced acute and chronic resilience toward depressive-like behavior in various tests, while A1R knockout mice displayed an increased depressive-like behavior and were resistant to the antidepressant effects of SD [9]. Furthermore, we have shown that the antidepressant effects of A1R activation are mediated by the synaptic plasticity protein Homer1a, which is upregulated by various antidepressant treatments such as SD, imipramine, ketamine, and A1R activation [9, 12]. Using a different transgenic mouse lines with overexpression of A1R in the cortex and hippocampus, we found that depending on the brain region of A1R upregulation, the mice show different resistance to depression-like behavior, and that enhanced Homer1a expression in the hippocampus increases stress vulnerability [11].
However, activation of A1R may elicit also manic or hypomanic episodes in patients with bipolar disorder [18]. It has been reported that peripheral adenosine levels were negatively correlated to the severity of depressive symptoms of bipolar disorder patients [19]. Therefore, peripheral adenosine levels may have a positive relationship with mood, demonstrating the pivotal role of adenosine in mood regulation.
In contrast, it has been reported that rats with A2AR overexpression in hippocampus, cortex, and striatum show increased depression-like behavior [20]. Vice versa, A2AR KO mice exhibit reduced depression-like behaviors, such as decreases in the immobility time in forced swimming test and tail suspension test [21, 22]. The A2AR antagonist istradefylline (KW6002) showed an antidepressant-like action on learned helplessness model rats [23]. However, some contradictory results of relationship between A2AR and mood have also been released. Tsai et al. reported that they did not find any association of A2AR (1976C > T) genetic polymorphism with mood disorders [24]. However, this does preclude the possibility of a role of A2AR in the pathogenesis of mood disorders; rather, other A2AR variants must also be extensively studied. Moreover, A2ARs have also been linked with depression, suicidal behavior, and impulsivity based on indirect evidence at a statistical association level [25]. For instance, Lucas et al. reported a negative association between caffeine consumption and risk of suicide based on cohort studies [26]. The actions of adenosine receptors on depression are summarized below (Table 1).
Circadian Clock and Mood Regulation
Circadian clocks govern a wide range of biochemical, physiological, and behavioral processes. In mammals, the circadian master pacemaker is located in the suprachiasmatic nucleus (SCN) [27]. The circadian oscillation of the intracellular clock is driven by transcription/translation-based feedback/feedforward loops, consisting of a set of clock genes. Positive regulatory elements are brain and muscle ARNT-like 1 (BMAL1) and circadian locomotor output cycles kaput (CLOCK), which form heterodimers and induce the rhythmic transcription of Period (Per1 & Per2) and Cryptochrome (Cry1 & Cry2) genes. The PER and CRY proteins interact and translocate to the nucleus, where they act as negative regulators inhibiting CLOCK/BMAL1 transcription [28]. An additional loop including both activating and repressing regulatory elements is formed by retinoic acid receptor-related orphan receptors (ROR α, β, and γ) and nuclear receptors REV-ERB (α & β) [29, 30].
A large number of studies have demonstrated the relationship between the circadian clock and depression [31,32,33,34,35,36,37,38,39], with a great many reviews to refer to [31, 32, 34, 36, 40,41,42,43,44,45]. Most of these reports have shown correlation between genes, RNAs, proteins, and single nucleotide polymorphisms with the symptoms of MDD or depression-like behaviors [33, 38, 39, 46,47,48]. For example, variants of circadian genes, such as CLOCK, BMAL1, NPAS2, Per3, and NR1D1, play a role in mood disorders, mainly based on statistical analyses [49,50,51,52,53]. In addition, transgenic mice with mutations in certain clock genes have been characterized with depressive-like behavior. However, each mouse model shows a distinct mood/rhythm combination phenotype: similar mood characteristics occur with opposite changes of circadian period, and reduced circadian amplitude leads to different changes in mood behavior, which hinders a clear conclusion/hypothesis [54].
Several brain regions relevant to psychopathology of depression, including the prefrontal cortex, hippocampus, amygdala, lateral habenula (LHb), and nucleus accumbens (NAc), possess an oscillating molecular clock [55,56,57]. Increasing evidence from human and rodents suggests that these region-specific oscillators in limbic areas are instrumental regulators of mood. Indeed, a microarray study demonstrated that the circadian patterns of gene expression in six brain regions (including amygdala, prefrontal cortex, hippocampus, and NAc) are significantly altered in human postmortem subjects with MDD [48]. Moreover, many chronic stress-based animal models of depression show dysregulated circadian rhythms of locomotor activity, body temperature, and corticosterone levels [58], as well as reduced circadian expression amplitude of several canonical circadian clock genes in the SCN and amygdala, but increased amplitude in the NAc [55, 56, 59]. For instance, Christiansen et al. demonstrate effects of chronic mild stress on core circadian genes in rats [46]—the mean peak times of Per2 and Bmal1 expressions in SCN were either phase-delayed or phase-advanced in the chronic stress group. Taken together, these reports suggest that stress and/or MDD might differently affect the circadian clockwork in particular brain areas and that further investigation on region-specific circadian mechanisms are needed.
A potential role has been recently proposed for the circadian clock in the mechanism of rapid antidepressant treatments, like SD and ketamine [60]. Duncan et al. revealed an association between ketamine’s clinical antidepressant response and circadian-related wrist-activity parameters [39], finding that responders showed a phase-advanced activity rhythm and a decreased measure compared with nonresponders at baseline. Orozco-Solis et al. showed downregulation of several canonical clock genes, including Per1, Per2 and Cry2, by rapid antidepressant therapies SD and low-dose ketamine, using comparative transcriptomics analyses [38]. Furthermore, ketamine usually takes its most robust effect on the next day of its treatment [61], a phenomenon probably related to the effect of ketamine on circadian system [43]. Preclinical studies reveal that both SD and ketamine downregulate circadian genes, probably through NMDAR, AMPAR, TrkB, MAPK, mTOR, GSK3β, and CREB [38, 62,63,64,65], but the exact cellular pathway has not been confirmed and needs to be further investigated. Until now, only a few studies have revealed signaling pathways that act directly on the molecular circadian clock and mediate the pathogenesis of major depression or depression-like behaviors [8, 66].
The role of circadian rhythm in mood regulation is bidirectional, affecting both depression and mania [67,68,69,70,71,72,73,74,75]. For example, phase advance during manic episodes and phase delay during depressive episodes were found in the patients with bipolar disorder [76,77,78,79]. The CLOCKΔ19 and Per2Brdm1 mice exhibit hyperdopaminergic state and mania-like phenotypes [80,81,82], while in contrast, Per1 knockout mice show depression-like behavior in forced swim test [83], directly demonstrating that the circadian clock influences monoamine oxidase A and mood. In addition, Olejniczak et al. revealed that light affects depression-like behavior through Per1 in the LHb [83]. Therefore, our focus should not be restricted to only one axis of investigation. For instance, while A1R agonism shows antidepressant-like effects, it may potentially induce manic or hypomanic episodes and vice versa [18, 84]. As a result, this side effect must be avoided when exploring novel antidepressants or mood stabilizers. Recently, Hinton et al. reported that administration of caffeine during adolescence in mice could induce circadian-dependent changes in mood fluctuations in adulthood, including depression and mania [85]. However, the exact cellular pathway underlying this phenomenon needs to be investigated further. In future, elucidation of the pathogenesis of trans-phase may be an important research field.
Taken together, these reports support a causal relationship between the circadian system and mood. However, alternative hypotheses have been proposed, and whether the disruption of circadian clocks are causes or consequences of mood disorders remains undetermined. Accordingly, Lazzerini Ospri et al. provide a model suggesting that mood may be an output of circadian rhythm by probability [86]. However, this hypothesis also needs to be further verified.
The Role of Adenosine Receptors in Circadian Clock Modulation
Light is the most potent resetting stimuli of the circadian clock. In addition to glutamate, adenosine appears to be a strong candidate for modulating SCN activity [87]. Indeed, application of adenosine attenuates light-induced phase shifts, while A1R antagonism can reverse this effect [88, 89]. Adenosine is known to increase during SD [90] and accordingly it has been shown in rodents and humans that SD also reduces the photic resetting of circadian activity [91, 92]. Likewise, in response to acute SD, a subset of circadian clock genes behave as immediate early genes and are transcriptionally responsive within hours of treatment [93, 94]. Conversely, longer SD suppresses 80% of rhythmic genes in the mouse brain [95, 96]. Moreover, the adenosine receptor antagonist caffeine modulates different aspects of the circadian rhythms including behavioral rhythm and the molecular clock [87, 97, 98]. It increases the light-entraining activity rhythm and lengthens the period of hPer2 and mBmal1 [97, 99]. In human-cultured cells, caffeine produced its effect on the circadian clock through adenosine receptor-cAMP signaling [100].
Adenosine A1 R and A2AR Signaling Pathways as Regulators of the Molecular Circadian Clock and Mood
In the following chapter, we will review and discuss A1R and A2AR downstream signaling, including classical pathways and some alternative cascades, implicated in the regulation of the cellular circadian system and mood. Moreover, transcriptional factor CREB phosphorylation and induction of the synaptic protein Homer1a appear to be a convergent point of various pathways [101], and play a critical role both in the regulation of circadian rhythm [102, 103] and in the pathogenesis of depression [101].
Canonical Adenosine Signaling
ERK MAPK Pathway
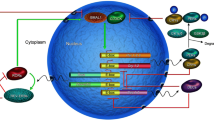
A1R activates the phospholipase C (PLC)β—inositol triphosphate (IP3) pathway in order to induce the release of calcium from endoplasmic reticulum and subsequently activates extracellular regulated protein kinase (ERK) [10, 104]. After ERK is activated, it can consequently activate the downstream part of the MAPK signaling pathway. CREB is the endpoint of the pathway, which can enter the nucleus and bind to the CRE sites in the promoter regions of Homer1a, Per1, and Per2 genes to regulate their transcription (Fig. 1) [10, 38, 44, 103, 105,106,107]. Moreover, it has been reported that adenosine A1R-ERK1/2 signaling pathway in the prefrontal cortex and hippocampus region of mice was involved in the anti-menopausal depressant-like effect of Jiao-Tai-Wan [108]. Additionally, there have been numerous references showing that the ERK MAPK pathway plays a critical role in MDD [109,110,111,112,113]. Moreover, ERK-CREB signaling in the hippocampus and prefrontal cortex was revealed as the downstream pathway of inosine to produce its antidepressant-like effect [114]. Thus, the ERK MAPK pathway is both important in circadian systems and mood regulation.

Pattern of a potential final common mechanism of antidepressant action. Acute or chronic SD increases the adenosine levels in the brain and activates A1Rs and A2ARs. Subsequently, ARs act on various signaling pathways, including cAMP, PKA, Ca2 + , and MAPK in the cytoplasm, and converge on the phosphorylation of the transcriptional factor CREB. The phosphorylated CREB enters the nucleus and binds with the CRE sites on Homer1 and Per1/2 promoters to regulate their transcription. Simultaneously, these genes are transcriptionally regulated by CLOCK/BMAL1 via E-box elements on their promoters. Concurrently, PER form complexes with CRY in the cytoplasm, which in turn compete with CLOCK/BMAL1 complexes and block their transcription, as well as Homer1a expression. SD, sleep deprivation; AC, adenylate cyclase; PLCb, phospholipase C beta; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; IP3, inositol triphosphate; PKA, protein kinase A; ER, endoplasmic reticulum. Arrows indicate activation: the thicker arrow indicates that adenosine may have a preferential activation effect on A1R during acute SD. In parallel, the red lines indicate inhibition, where the thicker line indicates greater inhibition of A2AR by caffeine
cAMP Signaling Pathway
cAMP is also a classical downstream signaling pathway of both A1R and A2AR and plays a key role in the mammalian circadian clock [100, 105, 115]. The A1R can suppress the cAMP pathway through inhibiting adenylate cyclase (AC) via its Gi; contrarily, the adenosine A2A receptor can stimulate cAMP pathway through activating AC via its Gs (Fig. 1) [3]. Burke et al. found that the intracellular mechanism of caffeine-induced regulation of the circadian rhythm is via the adenosine A1 receptor-cAMP signaling pathway in human cells in vitro [100]. In addition, it was revealed that the cAMP-protein kinase A (PKA)-CREB pathway in rat hippocampal neurons was involved in the antidepressant-like effect of serum [116]. However, no interaction was identified of this pathway with circadian genes. As CREB is the endpoint of various cellular pathways, including the cAMP pathway, it has been suggested that CRE sites on the Per1 or Per2 genes might be the potential target (Fig. 1) [103, 105, 106]. Therefore, cAMP is another downstream signaling pathway that plays critical roles in both regulation of circadian genes and mood.
Ca 2+ Signaling Pathway
Studies have revealed that both L-type calcium channels and calcium-induced calcium release can induce post-synaptic adenosine elevation [117] and that calcium signaling acts upon Per1/2 genes directly via CREB in mammalian cells [103, 115, 118]. In addition, A1R can also inhibit L-type calcium channels via its Gi [3]. Besides this, the ERK/MAPK signaling pathway is calcium-dependent. It has also been reported that the Gq-Ca2+ axis controls the circadian clock in the SCN [119], involved in both input and output of circadian systems [115, 120]. Furthermore, the cAMP/Ca2+ signaling pathway determines properties of the circadian system, including phase, amplitude, and period; in turn, cAMP/Ca2+ signaling is regulated by circadian system and rhythmically expressed [115].
In conclusion, ERK MAPK, cAMP, and Ca2+ signaling pathways are the major downstream pathways of adenosine, which in parallel can regulate circadian molecular clock (Fig. 1). Pertinently, it has been demonstrated that levels of cAMP, Ca2+, ERK, and CREB were decreased in postmortem patients with MDD [121]. Moreover, levels of these molecules were oppositely altered in patients with bipolar disorder treated with mood stabilizers compared to MDD patients administered antidepressants [121], demonstrating their roles in mood regulation.
Other Potential Alternative Downstream Cellular Pathways of Adenosine Receptors
In addition to the canonical cellular pathways, there are also some recently explored downstream signaling pathways of A1R, which have not been demonstrated to be involved in mood regulation but may suggest new further research directions.
Recently, Jagannath et al. revealed that adenosine could regulate the circadian clock through activating the adenosine A1/A2A receptor, and their downstream Ca2+-ERK-AP-1 and CREB/cAMP-regulated transcriptional coactivators (CRTC1)-CRE signaling pathways to modulate the expression of Per1 and Per2 genes in mice [122]. They found that these signaling pathways were also stimulated by light [122]. Thus, adenosine can alter the circadian time by integrating signals from light and sleep. Furthermore, Trautmann et al. showed that caffeine acts on mood through the elevation of phosphorylated Thr75-DARPP-32, which can bind to CLOCK and inhibit the CLOCK/BMAL1 complex interaction, consequently modulating the expression of circadian genes and potentially linking adenosine, circadian systems, and mood [66].
Adenosine receptors are also involved in the modulation of other neurotransmitter systems. For example, the A2AR is colocalized postsynaptically in dopamine areas, including the striatum and NAc [123]. Indeed, it has been demonstrated that there is a functional interaction between dopamine D2Rs and A2ARs, which converge on the same signal transduction pathways in an antagonistic way [124]. Likewise, A1R and D1Rs antagonistically interact [125]. The dopaminergic system plays an important role in the control of reward and motivation-oriented behavior, which is severely affected in MDD. Since dopamine synthesis and particularly its limiting enzyme tyrosine hydroxylase (TH) are under circadian regulation, this interaction between adenosinergic and dopaminergic system represents another potential signaling pathway involved in mood regulation [126].
Convergent Points of Adenosine Receptor Signaling
CREB
CREB is a convergent point of various pathways in the pathogenesis of MDD and is the downstream effector molecule of adenosine signaling [101]. The role of CREB in MDD varies with different brain regions [101]. For example, overexpression of CREB in the dentate gyrus of the hippocampus produced an antidepressant-like effect in rats [127], while overexpression of CREB in either the CA1 pyramidal cell layer of the hippocampus or the prefrontal cortex did not show this effect [127]. Conversely, overexpression of CREB in the basolateral amygdala or in the NAc produced a pro-depressive-like effect [128, 129]. Meanwhile, the acting points of CREB on the circadian genes Per1/2 have been elucidated (Fig. 1) [8, 106, 122]. Phosphorylated CREB is one of the transcriptional factors regulating Per1/2. Besides this, AP-1 is another transcriptional factor that can also bind with AP-1 sites in the promoter regions of Per genes. In the Per2 gene, AP-1 REs are putative and conserved, while in contrast are not well conserved in Per1 [122]. Moreover, it has been reported that the sequences of CRE (TGACGTCA) and 12–0-tetradecanoylphorbol-13-acetate-responsive element (TRE) (TGACTCA) are very similar, and that the nuclear factors of CREB, CRE modulator (CREM), and Jun were also very similar in structure [130, 131]. This may lead to transcriptional cross-talk and potential competitive effects. Therefore, we deduce that this physiological process may be involved in the interaction between Per1, Per2, and Homer1a genes, and may play a key role in supplement to the traditional feedback loops of the molecular circadian clock.
CREB conduction signals can also be regarded as an intrinsic part of clock oscillations, modulating acute alterations in the circadian clock and transcription-translation feedback loops [118, 132].
Apart from circadian genes Per1/2, there are hundreds of genes that have CRE sequences in their promoter regions that can be bound with pCREB/CREB. Therefore, Per1/2 might not be the only final common targets of antidepressants and other genes, such as Homer1a, might have an interaction with these circadian genes (see below).
Homer1a
Homer1a is a member of the Homer family of postsynaptic scaffolding proteins, which is rhythmically expressed and acts as neuronal activity-inducible modulator of glutamatergic signaling [10, 13, 133]. It has been shown that Homer1a induction, as a downstream effect of A1R signaling, may be a convergent point of several non-pharmaceutical treatments of MDD [9,10,11, 133]. Homer1a has been subsequently proposed as a final common pathway of various antidepressant therapies, including ECT, TMS, SD, and ketamine, as well as for classical treatments, such as imipramine and fluoxetine [10]. In addition, it has been shown that metabotropic glutamate receptor 5 (mGlu5) and α-amino-3-hydroxy-5-methyl-4-isoxazole-propionicacid receptor (AMPAR) might be the potential targets for Homer1a to act and exhibit its antidepressant effect [12]. Recently, Sato et al. revealed that the Homer1 gene is bimodally regulated by CREB via the CRE site and by the CLOCK/BMAL1 complex via E-box, demonstrating an important crosstalk between CREB and the circadian clock, and thus showing a pivotal role of Homer1a in integrating signals from both adenosine signaling and circadian rhythms [13]. Therefore, this may be the most promising final common pattern in the pathogenesis of depression and the mechanism of antidepressants. Thus, we propose that Homer1 and Per genes, receiving signals from both CREB and the CLOCK/BMAL1 complex, which is inhibited by PERs, may be a potential common mechanism of various antidepressant therapies (Fig. 1).
Conclusions and Future Directions
Acute SD is known to elicit rapid antidepressant effects, while chronic sleep restriction is considered as a risk factor for depression [134]. However, adenosine is accumulated in the brain after both acute and chronic sleep loss and acts as modulatory neurotransmitter regulating brain homeostasis via modulation of sleep and homeostatic plasticity, circadian clockwork, and mood [3, 9, 135,136,137,138].
We deduce that, on one hand, this conflicting effect of adenosine might be due to the preferential activation of its receptors, since A1R and A2AR signaling have contrasting effects on mood. Perhaps during the acute SD phase, adenosine has a greater effect on A1R [9], whereas, during chronic sleep loss, there may be a counterbalancing effect and possibly more action on the A2AR with an opposing effect on downstream signaling (Serchov et. 2020). At the same time, caffeine, an antagonist of adenosine receptors, may have a stronger antagonistic effect on A2AR than A1R, resulting in an antidepressant effect [21, 22, 139]. On the other hand, pCREB may also have a biased or counterbalancing effect on Homer1 and Per genes, and the final Homer1a protein expression level may depend on the probability of circadian output, which may match the alternative hypothesis model provided by Lazzerini Ospri et al. [86].
As discussed above, circadian gene expression is differentially affected by chronic stress, depression, or antidepressant treatments in different brain regions. Thus, the different effects of adenosine signaling on the circadian clock, Homer1a expression, and mood might also be brain region-specific [11, 133].
Taken together, we summarize that adenosine A1R/A2AR signaling converges on the transcriptional factor CREB. After CREB is activated, it can bind to both CRE/AP-1 sites on Per1/2 gene promoters, or the CRE site on the Homer1 promoter, thus modulating their expression. In turn, Per/Cry complexes translocate to the nucleus and inhibit BMAL1 activity. Since Homer1 expression is bimodally regulated by BMAL1 and CREB, we deduce that Homer1a expression might be inhibited by Per indirectly (Fig. 1). Thus, Homer1a is potentially a final common pathway in the pathogenesis and treatment of depression, which links adenosine signaling, circadian clock, and neuro-plasticity together, mediating both the antidepressant effects of acute SD and the detrimental action on mood of chronic sleep loss.
Additionally, the synthesis of dopamine is also regulated in a circadian manner, through the time-dependent expression of TH by promoter occupancy of CLOCK, NAD + -dependent sirtuin 1 (SIRT1), and CREB [107]. This pathway links the metabolic system, circadian rhythms, and neurotransmitter system together, important in the regulation of many physiological processes and psychiatric diseases. In the future, we shall investigate this common mechanism from several other perspectives: neuro-inflammation, systems of monoamine and glutamatergic signaling, the hypothalamic–pituitary–adrenal (HPA) axis, brain-gut axis, metabolic peptide signal transduction, and mitochondrial function, with the aim of exploring the common pathophysiology of depression from a cellular to systemic level.
Above all, identification of the common pathogenesis of MDD will help us to better understand the underlying pathogenesis of mood disorders and to explore novel antidepressants or mood stabilizers with fewer side effects.
References
Mrazek F, Onderkova J, Szotkowski T et al (2014) Somatic mutation in acute myelogenous leukemia cells imitate novel germline HLA-A allele: a case report. Tissue Antigens 83:414–417. https://doi.org/10.1111/tan.12362
Dallaspezia S, Suzuki M, Benedetti F (2015) Chronobiological therapy for mood disorders. Curr Psychiatry Rep 17:95. https://doi.org/10.1007/s11920-015-0633-6
van Calker D, Biber K, Domschke K et al (2019) The role of adenosine receptors in mood and anxiety disorders. J Neurochem 151:11–27. https://doi.org/10.1111/jnc.14841
Gomes JI, Farinha-Ferreira M, Rei N et al (2021) Of adenosine and the blues: the adenosinergic system in the pathophysiology and treatment of major depressive disorder. Pharmacol Res 163:105363. https://doi.org/10.1016/j.phrs.2020.105363
Szopa A, Socala K, Serefko A et al (2021) Purinergic transmission in depressive disorders. Pharmacol Ther 224:107821. https://doi.org/10.1016/j.pharmthera.2021.107821
Reichert CF, Maire M, Schmidt C, Cajochen C (2016) Sleep-wake regulation and its impact on working memory performance: the role of adenosine. Biology (Basel) 5(1). https://doi.org/10.3390/biology5010011
Lindberg D, Andres-Beck L, Jia YF et al (2018) Purinergic signaling in neuron-astrocyte interactions, circadian rhythms, and alcohol use disorder. Front Physiol 9:9. https://doi.org/10.3389/fphys.2018.00009
Wang XL LX, Yuan K, Han Y, Xue YY, Meng SQ, Li SX (2021) Clock genes Period1 and Period2 in the hippocampal CA1 mediate depression-like behaviors and rapid antidepressant response. BioRxiv. https://doi.org/10.1101/2021.08.14.456364
Serchov T, Clement HW, Schwarz MK et al (2015) Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of Homer1a. Neuron 87:549–562. https://doi.org/10.1016/j.neuron.2015.07.010
Serchov T, Heumann R, van Calker D et al (2016) Signaling pathways regulating Homer1a expression: implications for antidepressant therapy. Biol Chem 397:207–214. https://doi.org/10.1515/hsz-2015-0267
Serchov T, Schwarz I, Theiss A et al (2020) Enhanced adenosine A1 receptor and Homer1a expression in hippocampus modulates the resilience to stress-induced depression-like behavior. Neuropharmacology 162:107834. https://doi.org/10.1016/j.neuropharm.2019.107834
Holz A, Mulsch F, Schwarz MK et al (2019) Enhanced mGlu5 signaling in excitatory neurons promotes rapid antidepressant effects via AMPA receptor activation. Neuron 104(338–352):e337. https://doi.org/10.1016/j.neuron.2019.07.011
Sato S, Bunney BG, Vawter MP et al (2020) Homer1a undergoes bimodal transcriptional regulation by CREB and the circadian clock. Neuroscience 434:161–170. https://doi.org/10.1016/j.neuroscience.2020.03.031
Pedata F, Dettori I, Coppi E et al (2016) Purinergic signalling in brain ischemia. Neuropharmacology 104:105–130. https://doi.org/10.1016/j.neuropharm.2015.11.007
Moidunny S, Vinet J, Wesseling E et al (2012) Adenosine A2B receptor-mediated leukemia inhibitory factor release from astrocytes protects cortical neurons against excitotoxicity. J Neuroinflammation 9:198. https://doi.org/10.1186/1742-2094-9-198
Hines DJ, Schmitt LI, Hines RM et al (2013) Antidepressant effects of sleep deprivation require astrocyte-dependent adenosine mediated signaling. Transl Psychiatry 3:e212. https://doi.org/10.1038/tp.2012.136
Serchov T, Atas HC, Normann C et al (2012) Genetically controlled upregulation of adenosine A(1) receptor expression enhances the survival of primary cortical neurons. Mol Neurobiol 46:535–544. https://doi.org/10.1007/s12035-012-8321-6
Lewis KS, -Smith KG, Forty L, et al (2017) Sleep loss as a trigger of mood episodes in bipolar disorder: individual differences based on diagnostic subtype and gender. Br J Psychiatry 211:169–174. https://doi.org/10.1192/bjp.bp.117.202259
Gubert C, Jacintho Moritz CE, Vasconcelos-Moreno MP et al (2016) Peripheral adenosine levels in euthymic patients with bipolar disorder. Psychiatry Res 246:421–426. https://doi.org/10.1016/j.psychres.2016.10.007
Coelho JE, Alves P, Canas PM et al (2014) Overexpression of adenosine A2A receptors in rats: effects on depression, locomotion, and anxiety. Front Psychiatry 5:67. https://doi.org/10.3389/fpsyt.2014.00067
El Yacoubi M, Ledent C, Parmentier M et al (2001) Adenosine A2A receptor antagonists are potential antidepressants: evidence based on pharmacology and A2A receptor knockout mice. Br J Pharmacol 134:68–77. https://doi.org/10.1038/sj.bjp.0704240
El Yacoubi M, Costentin J, Vaugeois JM (2003) Adenosine A2A receptors and depression. Neurology 61:S82-87. https://doi.org/10.1212/01.wnl.0000095220.87550.f6
Yamada K, Kobayashi M, Shiozaki S et al (2014) Antidepressant activity of the adenosine A2A receptor antagonist, istradefylline (KW-6002) on learned helplessness in rats. Psychopharmacology 231:2839–2849. https://doi.org/10.1007/s00213-014-3454-0
Tsai SJ, Hong CJ, Hou SJ et al (2006) Association study of adenosine A2a receptor (1976C>T) genetic polymorphism and mood disorders and age of onset. Psychiatr Genet 16:185. https://doi.org/10.1097/01.ypg.0000218627.26622.eb
Bartoli F, Clerici M, Carra G (2020) Purinergic system and suicidal behavior: exploring the link between adenosine A2A receptors and depressive/impulsive features. Mol Psychiatry 25:512–513. https://doi.org/10.1038/s41380-018-0057-x
Lucas M, O’Reilly EJ, Pan A et al (2014) Coffee, caffeine, and risk of completed suicide: results from three prospective cohorts of American adults. World J Biol Psychiatry 15:377–386. https://doi.org/10.3109/15622975.2013.795243
Albrecht U (2012) Timing to perfection: the biology of central and peripheral circadian clocks. Neuron 74:246–260. https://doi.org/10.1016/j.neuron.2012.04.006
Reppert SM, Weaver DR (2002) Coordination of circadian timing in mammals. Nature 418:935–941. https://doi.org/10.1038/nature00965
Preitner N, Damiola F, Lopez-Molina L et al (2002) The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110:251–260. https://doi.org/10.1016/s0092-8674(02)00825-5
Sato TK, Panda S, Miraglia LJ et al (2004) A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron 43:527–537. https://doi.org/10.1016/j.neuron.2004.07.018
McClung CA (2013) How might circadian rhythms control mood? Let me count the ways. Biol Psychiatry 74:242–249. https://doi.org/10.1016/j.biopsych.2013.02.019
Ketchesin KD, Becker-Krail D, McClung CA (2020) Mood-related central and peripheral clocks. Eur J Neurosci 51:326–345. https://doi.org/10.1111/ejn.14253
Wang XL, Wang DQ, Jiao FC et al (2021) Diurnal rhythm disruptions induced by chronic unpredictable stress relate to depression-like behaviors in rats. Pharmacol Biochem Behav 204:173156. https://doi.org/10.1016/j.pbb.2021.173156
Mendoza J (2019) Circadian insights into the biology of depression: Symptoms, treatments and animal models. Behav Brain Res 376:112186. https://doi.org/10.1016/j.bbr.2019.112186
Chellappa SL (2020) Circadian misalignment: a biological basis for mood vulnerability in shift work. Eur J Neurosci 52:3846–3850. https://doi.org/10.1111/ejn.14871
Mendoza J, Vanotti G (2019) Circadian neurogenetics of mood disorders. Cell Tissue Res 377:81–94. https://doi.org/10.1007/s00441-019-03033-7
Gutiérrez-Zotes A, Díaz-Peña R, Costas J et al (2020) Interaction between the functional SNP rs2070951 in NR3C2 gene and high levels of plasma corticotropin-releasing hormone associates to postpartum depression. Arch Womens Ment Health 23:413–420. https://doi.org/10.1007/s00737-019-00989-x
Orozco-Solis R, Montellier E, Aguilar-Arnal L et al (2017) A circadian genomic signature common to ketamine and sleep deprivation in the anterior cingulate cortex. Biol Psychiatry 82:351–360. https://doi.org/10.1016/j.biopsych.2017.02.1176
Duncan WC, Jr., Slonena E, Hejazi NS, Brutsche N, Yu KC, Park L, et al (2017) Motor-activity markers of circadian timekeeping are related to ketamine’s rapid antidepressant properties. Biol Psychiatry 82:361–369. https://doi.org/10.1016/j.biopsych.2017.03.011
Huhne A, Welsh DK, Landgraf D (2018) Prospects for circadian treatment of mood disorders. Ann Med 50:637–654. https://doi.org/10.1080/07853890.2018.1530449
McClung CA (2007) Circadian genes, rhythms and the biology of mood disorders. Pharmacol Ther 114:222–232
McClung CA (2011) Circadian rhythms and mood regulation: insights from pre-clinical models. Eur Neuropsychopharmacol 21:S683–S693
Kohtala S, Alitalo O, Rosenholm M, Rozov S, Rantamaki T (2021) Time is of the essence: coupling sleep-wake and circadian neurobiology to the antidepressant effects of ketamine. Pharmacol Ther 221:107741. https://doi.org/10.1016/j.pharmthera.2020.107741
Wang XL, Yuan K, Zhang W et al (2020) Regulation of circadian genes by the MAPK pathway: implications for rapid antidepressant action. Neurosci Bull 36:66–76. https://doi.org/10.1007/s12264-019-00358-9
Logan RW, McClung CA (2019) Rhythms of life: circadian disruption and brain disorders across the lifespan. Nat Rev Neurosci 20:49–65. https://doi.org/10.1038/s41583-018-0088-y
Christiansen SL, Bouzinova EV, Fahrenkrug J et al (2016) Altered expression pattern of clock genes in a rat model of depression. Int J Neuropsychopharmacol 19.https://doi.org/10.1093/ijnp/pyw061
Li SX, Liu LJ, Xu LZ et al (2013) Diurnal alterations in circadian genes and peptides in major depressive disorder before and after escitalopram treatment. Psychoneuroendocrinology 38:2789–2799. https://doi.org/10.1016/j.psyneuen.2013.07.009
Li JZ, Bunney BG, Meng F et al (2013) Circadian patterns of gene expression in the human brain and disruption in major depressive disorder. Proc Natl Acad Sci U S A 110:9950–9955. https://doi.org/10.1073/pnas.1305814110
Etain B, Milhiet V, Bellivier F et al (2011) Genetics of circadian rhythms and mood spectrum disorders. Eur Neuropsychopharmacol 21(Suppl 4):S676-682. https://doi.org/10.1016/j.euroneuro.2011.07.007
Lee KY, Song JY, Kim SH et al (2010) Association between CLOCK 3111T/C and preferred circadian phase in Korean patients with bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 34:1196–1201. https://doi.org/10.1016/j.pnpbp.2010.06.010
Soria V, Martinez-Amoros E, Escaramis G et al (2010) Differential association of circadian genes with mood disorders: CRY1 and NPAS2 are associated with unipolar major depression and CLOCK and VIP with bipolar disorder. Neuropsychopharmacology 35:1279–1289. https://doi.org/10.1038/npp.2009.230
Mansour HA, Talkowski ME, Wood J et al (2009) Association study of 21 circadian genes with bipolar I disorder, schizoaffective disorder, and schizophrenia. Bipolar Disord 11:701–710. https://doi.org/10.1111/j.1399-5618.2009.00756.x
Partonen T, Treutlein J, Alpman A et al (2007) Three circadian clock genes Per2, Arntl, and Npas2 contribute to winter depression. Ann Med 39:229–238. https://doi.org/10.1080/07853890701278795
Landgraf D, McCarthy MJ, Welsh DK (2014) The role of the circadian clock in animal models of mood disorders. Behav Neurosci 128:344–359. https://doi.org/10.1037/a0036029
Landgraf D, Long JE, Welsh DK (2016) Depression-like behaviour in mice is associated with disrupted circadian rhythms in nucleus accumbens and periaqueductal grey. Eur J Neurosci 43:1309–1320. https://doi.org/10.1111/ejn.13085
Logan RW, Edgar N, Gillman AG et al (2015) Chronic stress induces brain region-specific alterations of molecular rhythms that correlate with depression-like behavior in mice. Biol Psychiat 78:249–258. https://doi.org/10.1016/j.biopsych.2015.01.011
Salaberry NL, Hamm H, Felder-Schmittbuhl MP et al (2019) A suprachiasmatic-independent circadian clock(s) in the habenula is affected by Per gene mutations and housing light conditions in mice. Brain Struct Funct 224:19–31. https://doi.org/10.1007/s00429-018-1756-4
Meerlo P, van den Hoofdakker RH, Koolhaas JM et al (1997) Stress-induced changes in circadian rhythms of body temperature and activity in rats are not caused by pacemaker changes. J Biol Rhythms 12:80–92. https://doi.org/10.1177/074873049701200109
Savalli G, Diao W, Schulz S, Todtova K, Pollak DD (2014) Diurnal oscillation of amygdala clock gene expression and loss of synchrony in a mouse model of depression. Int J Neuropsychopharmacol 18(5). https://doi.org/10.1093/ijnp/pyu095
Sato S, Bunney B, Mendoza-Viveros L et al (2022) Rapid-acting antidepressants and the circadian clock. Neuropsychopharmacology 47:805–816. https://doi.org/10.1038/s41386-021-01241-w
Coyle CM, Laws KR (2015) The use of ketamine as an antidepressant: a systematic review and meta-analysis. Hum Psychopharmacol-Clin Exp 30:152–163. https://doi.org/10.1002/hup.2475
Bellet MM, Vawter MP, Bunney BG et al (2011) Ketamine influences CLOCK:BMAL1 function leading to altered circadian gene expression. PLoS ONE 6:e23982. https://doi.org/10.1371/journal.pone.0023982
Bunney BG, Li JZ, Walsh DM et al (2015) Circadian dysregulation of clock genes: clues to rapid treatments in major depressive disorder. Mol Psychiatry 20:48–55. https://doi.org/10.1038/mp.2014.138
Bunney BG, Bunney WE (2012) Rapid-acting antidepressant strategies: mechanisms of action. Int J Neuropsychopharmacol 15:695–713. https://doi.org/10.1017/S1461145711000927
Wisor JP, Pasumarthi RK, Gerashchenko D et al (2008) Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains (vol 28, pg 7193, 2008). J Neurosci 28:7929–7929
Trautmann C, Burek D, Hubner CA et al (2020) A regulatory pathway linking caffeine action, mood and the diurnal clock. Neuropharmacology 172:108133. https://doi.org/10.1016/j.neuropharm.2020.108133
Robillard R, Hermens DF, Lee RS et al (2016) Sleep-wake profiles predict longitudinal changes in manic symptoms and memory in young people with mood disorders. J Sleep Res 25:549–555. https://doi.org/10.1111/jsr.12413
Fang L, Yu Q, Yin F, Yu J, Zhang Y, Zhang Y, et al (2021) Combined cortisol and melatonin measurements with detailed parameter analysis can assess the circadian rhythms in bipolar disorder patients. Brain Behav 11(7):e02186. https://doi.org/10.1002/brb3.2186
McCarty R, Josephs T, Kovtun O et al (2021) Enlightened: addressing circadian and seasonal changes in photoperiod in animal models of bipolar disorder. Transl Psychiatry 11:373. https://doi.org/10.1038/s41398-021-01494-5
Gonzalez R, Tohen M (2018) Circadian rhythm and the prediction of relapse in bipolar disorder. J Clin Psychiatry 79(1). https://doi.org/10.4088/JCP.17com11821
Wehr TA (2018) Bipolar mood cycles associated with lunar entrainment of a circadian rhythm. Transl Psychiatry 8:151. https://doi.org/10.1038/s41398-018-0203-x
Rosenthal SJ, Josephs T, Kovtun O et al (2020) Seasonal effects on bipolar disorder: a closer look. Neurosci Biobehav Rev 115:199–219. https://doi.org/10.1016/j.neubiorev.2020.05.017
Gonzalez R (2014) The relationship between bipolar disorder and biological rhythms. J Clin Psychiatry 75:E323–E331. https://doi.org/10.4088/JCP.13r08507
Rosenthal SJ, McCarty R (2019) Switching winter and summer photoperiods in an animal model of bipolar disorder. Neuropsychopharmacology 44:1677–1678. https://doi.org/10.1038/s41386-019-0337-4
Porcu A, Gonzalez R, McCarthy MJ (2019) Pharmacological manipulation of the circadian clock: a possible approach to the management of bipolar disorder. CNS Drugs 33:981–999. https://doi.org/10.1007/s40263-019-00673-9
Novakova M, Prasko J, Latalova K et al (2015) The circadian system of patients with bipolar disorder differs in episodes of mania and depression. Bipolar Disord 17:303–314. https://doi.org/10.1111/bdi.12270
Moon JH, Cho CH, Son GH et al (2016) Advanced circadian phase in mania and delayed circadian phase in mixed mania and depression returned to normal after treatment of bipolar disorder. EBioMedicine 11:285–295. https://doi.org/10.1016/j.ebiom.2016.08.019
Inder ML, Crowe MT, Porter R (2016) Effect of transmeridian travel and jetlag on mood disorders: evidence and implications. Aust N Z J Psychiatry 50:220–227. https://doi.org/10.1177/0004867415598844
Duarte Faria A, Cardoso Tde A, Campos Mondin T et al (2015) Biological rhythms in bipolar and depressive disorders: a community study with drug-naive young adults. J Affect Disord 186:145–148. https://doi.org/10.1016/j.jad.2015.07.004
Kristensen M, Nierenberg AA, Ostergaard SD (2018) Face and predictive validity of the ClockDelta19 mouse as an animal model for bipolar disorder: a systematic review. Mol Psychiatry 23:70–80. https://doi.org/10.1038/mp.2017.192
Logan RW, McClung CA (2016) Animal models of bipolar mania: the past, present and future. Neuroscience 321:163–188. https://doi.org/10.1016/j.neuroscience.2015.08.041
Hampp G, Ripperger JA, Houben T et al (2008) Regulation of monoamine oxidase A by circadian-clock components implies clock influence on mood. Curr Biol 18:678–683. https://doi.org/10.1016/j.cub.2008.04.012
Olejniczak I, Ripperger JA, Sandrelli F et al (2021) Light affects behavioral despair involving the clock gene Period 1. PLoS Genet 17:e1009625. https://doi.org/10.1371/journal.pgen.1009625
Baldessarini RJ, Tondo L, Vazquez GH (2019) Pharmacological treatment of adult bipolar disorder. Mol Psychiatry 24:198–217. https://doi.org/10.1038/s41380-018-0044-2
Hinton DJ, Andres-Beck LG, Nett KE et al (2019) Chronic caffeine exposure in adolescence promotes diurnal, biphasic mood-cycling and enhanced motivation for reward in adult mice. Behav Brain Res 370:111943. https://doi.org/10.1016/j.bbr.2019.111943
Lazzerini Ospri L, Prusky G, Hattar S (2017) Mood, the circadian system, and melanopsin retinal ganglion cells. Annu Rev Neurosci 40:539–556. https://doi.org/10.1146/annurev-neuro-072116-031324
Antle MC, Steen NM, Mistlberger RE (2001) Adenosine and caffeine modulate circadian rhythms in the Syrian hamster. NeuroReport 12:2901–2905. https://doi.org/10.1097/00001756-200109170-00029
Elliott KJ, Todd Weber E, Rea MA (2001) Adenosine A1 receptors regulate the response of the hamster circadian clock to light. Eur J Pharmacol 414:45–53. https://doi.org/10.1016/s0014-2999(01)00786-5
Sigworth LA, Rea MA (2003) Adenosine A1 receptors regulate the response of the mouse circadian clock to light. Brain Res 960:246–251. https://doi.org/10.1016/s0006-8993(02)03896-9
Porkka-Heiskanen T, Strecker RE, McCarley RW (2000) Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience 99:507–517. https://doi.org/10.1016/s0306-4522(00)00220-7
Burgess HJ (2010) Partial sleep deprivation reduces phase advances to light in humans. J Biol Rhythms 25:460–468. https://doi.org/10.1177/0748730410385544
van Diepen HC, Lucassen EA, Yasenkov R et al (2014) Caffeine increases light responsiveness of the mouse circadian pacemaker. Eur J Neurosci 40:3504–3511. https://doi.org/10.1111/ejn.12715
Wisor JP, Pasumarthi RK, Gerashchenko D et al (2008) Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. J Neurosci 28:7193–7201. https://doi.org/10.1523/JNEUROSCI.1150-08.2008
Zhang B, Gao Y, Li Y et al (2016) Sleep Deprivation influences circadian gene expression in the lateral habenula. Behav Neurol 2016:7919534. https://doi.org/10.1155/2016/7919534
Maret S, Dorsaz S, Gurcel L et al (2007) Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A 104:20090–20095. https://doi.org/10.1073/pnas.0710131104
Thompson CL, Wisor JP, Lee CK et al (2010) Molecular and anatomical signatures of sleep deprivation in the mouse brain. Front Neurosci 4:165. https://doi.org/10.3389/fnins.2010.00165
Oike H, Kobori M, Suzuki T et al (2011) Caffeine lengthens circadian rhythms in mice. Biochem Biophys Res Commun 410:654–658. https://doi.org/10.1016/j.bbrc.2011.06.049
Jha PK, Bouaouda H, Gourmelen S et al (2017) Sleep deprivation and caffeine treatment potentiate photic resetting of the master circadian clock in a diurnal rodent. J Neurosci 37:4343–4358. https://doi.org/10.1523/JNEUROSCI.3241-16.2017
Ruby CL, Verbanes NM, Palmer KN et al (2018) Caffeine delays light-entrained activity and potentiates circadian photic phase-resetting in mice. J Biol Rhythms 33:523–534. https://doi.org/10.1177/0748730418789236
Burke TM, Markwald RR, McHill AW et al (2015) Effects of caffeine on the human circadian clock in vivo and in vitro. Sci Transl Med 7:305ra146. https://doi.org/10.1126/scitranslmed.aac5125
Blendy JA (2006) The role of CREB in depression and antidepressant treatment. Biol Psychiatry 59:1144–1150. https://doi.org/10.1016/j.biopsych.2005.11.003
Maurer C, Winter T, Chen S et al (2016) The CREB-binding protein affects the circadian regulation of behaviour. FEBS Lett 590:3213–3220. https://doi.org/10.1002/1873-3468.12336
Travnickova-Bendova Z, Cermakian N, Reppert SM et al (2002) Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc Natl Acad Sci U S A 99:7728–7733. https://doi.org/10.1073/pnas.102075599
Jajoo S, Mukherjea D, Kumar S et al (2010) Role of beta-arrestin1/ERK MAP kinase pathway in regulating adenosine A1 receptor desensitization and recovery. Am J Physiol Cell Physiol 298:C56-65. https://doi.org/10.1152/ajpcell.00190.2009
O’Neill JS, Maywood ES, Chesham JE et al (2008) cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science 320:949–953. https://doi.org/10.1126/science.1152506
Ikegami K, Nakajima M, Minami Y et al (2020) cAMP response element induces Per1 in vivo. Biochem Biophys Res Commun 531:515–521. https://doi.org/10.1016/j.bbrc.2020.07.105
Logan RW, Parekh PK, Kaplan GN et al (2019) NAD+ cellular redox and SIRT1 regulate the diurnal rhythms of tyrosine hydroxylase and conditioned cocaine reward. Mol Psychiatry 24:1668–1684. https://doi.org/10.1038/s41380-018-0061-1
Xiang L, Feng Y, Hu Q et al (2020) Jiao-Tai-Wan ameliorates depressive-like behavior through the A1R pathway in ovariectomized mice after unpredictable chronic stress. Biomed Res Int 2020:1507561. https://doi.org/10.1155/2020/1507561
Qi H, Mailliet F, Spedding M et al (2009) Antidepressants reverse the attenuation of the neurotrophic MEK/MAPK cascade in frontal cortex by elevated platform stress; reversal of effects on LTP is associated with GluA1 phosphorylation. Neuropharmacology 56:37–46. https://doi.org/10.1016/j.neuropharm.2008.06.068
Duric V, Banasr M, Licznerski P et al (2010) A negative regulator of MAP kinase causes depressive behavior. Nat Med 16:1328–1332. https://doi.org/10.1038/nm.2219
Zhang L, Xu T, Wang S et al (2012) Curcumin produces antidepressant effects via activating MAPK/ERK-dependent brain-derived neurotrophic factor expression in the amygdala of mice. Behav Brain Res 235:67–72. https://doi.org/10.1016/j.bbr.2012.07.019
Di Benedetto B, Radecke J, Schmidt MV et al (2013) Acute antidepressant treatment differently modulates ERK/MAPK activation in neurons and astrocytes of the adult mouse prefrontal cortex. Neuroscience 232:161–168. https://doi.org/10.1016/j.neuroscience.2012.11.061
Wang JQ, Mao L (2019) The ERK pathway: molecular mechanisms and treatment of depression. Mol Neurobiol 56:6197–6205. https://doi.org/10.1007/s12035-019-1524-3
Yuan S, Jiang X, Zhou X et al (2018) Inosine alleviates depression-like behavior and increases the activity of the ERK-CREB signaling in adolescent male rats. NeuroReport 29:1223–1229. https://doi.org/10.1097/WNR.0000000000001101
O’Neill JS, Reddy AB (2012) The essential role of cAMP/Ca2+ signalling in mammalian circadian timekeeping. Biochem Soc Trans 40:44–50. https://doi.org/10.1042/BST20110691
Wang XL, Gao J, Wang XY et al (2018) Treatment with Shuyu capsule increases 5-HT1AR level and activation of cAMP-PKA-CREB pathway in hippocampal neurons treated with serum from a rat model of depression. Mol Med Rep 17:3575–3582. https://doi.org/10.3892/mmr.2017.8339
Yamashiro K, Fujii Y, Maekawa S et al (2017) Multiple pathways for elevating extracellular adenosine in the rat hippocampal CA1 region characterized by adenosine sensor cells. J Neurochem 140:24–36. https://doi.org/10.1111/jnc.13888
Hastings MH, Maywood ES, O’Neill JS (2008) Cellular circadian pacemaking and the role of cytosolic rhythms. Curr Biol 18:R805–R815. https://doi.org/10.1016/j.cub.2008.07.021
Brancaccio M, Maywood ES, Chesham JE et al (2013) A Gq-Ca2+ axis controls circuit-level encoding of circadian time in the suprachiasmatic nucleus. Neuron 78:714–728. https://doi.org/10.1016/j.neuron.2013.03.011
Aguilar-Roblero R, Mercado C, Alamilla J et al (2007) Ryanodine receptor Ca2+-release channels are an output pathway for the circadian clock in the rat suprachiasmatic nuclei. Eur J Neurosci 26:575–582. https://doi.org/10.1111/j.1460-9568.2007.05679.x
Yuan P, Zhou R, Wang Y et al (2010) Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J Affect Disord 124:164–169. https://doi.org/10.1016/j.jad.2009.10.017
Jagannath A, Varga N, Dallmann R et al (2021) Adenosine integrates light and sleep signalling for the regulation of circadian timing in mice. Nat Commun 12:2113. https://doi.org/10.1038/s41467-021-22179-z
Johansson B, Georgiev V, Fredholm BB (1997) Distribution and postnatal ontogeny of adenosine A2A receptors in rat brain: comparison with dopamine receptors. Neuroscience 80:1187–1207. https://doi.org/10.1016/s0306-4522(97)00143-7
Fuxe K, Ferre S, Genedani S et al (2007) Adenosine receptor-dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol Behav 92:210–217. https://doi.org/10.1016/j.physbeh.2007.05.034
Ferre S (2008) An update on the mechanisms of the psychostimulant effects of caffeine. J Neurochem 105:1067–1079. https://doi.org/10.1111/j.1471-4159.2007.05196.x
Radwan B, Liu H, Chaudhury D (2019) The role of dopamine in mood disorders and the associated changes in circadian rhythms and sleep-wake cycle. Brain Res 1713:42–51. https://doi.org/10.1016/j.brainres.2018.11.031
Chen AC, Shirayama Y, Shin KH et al (2001) Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol Psychiatry 49:753–762
Pliakas AM, Carlson RR, Neve RL et al (2001) Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci 21:7397–7403
Wallace TL, Stellitano KE, Neve RL et al (2004) Effects of cyclic adenosine monophosphate response element binding protein overexpression in the basolateral amygdala on behavioral models of depression and anxiety. Biol Psychiatry 56:151–160. https://doi.org/10.1016/j.biopsych.2004.04.010
Lamph WW, Dwarki VJ, Ofir R et al (1990) Negative and positive regulation by transcription factor camp response element-binding protein is modulated by phosphorylation. Proc Natl Acad Sci USA 87:4320–4324. https://doi.org/10.1073/pnas.87.11.4320
Masquilier D, Sassone-Corsi P (1992) Transcriptional cross-talk: nuclear factors CREM and CREB bind to AP-1 sites and inhibit activation by Jun. J Biol Chem 267:22460–22466
Fiore P, Gannon RL (2003) Expression of the transcriptional coactivators CBP and p300 in the hamster suprachiasmatic nucleus: possible molecular components of the mammalian circadian clock. Brain Res Mol Brain Res 111:1–7. https://doi.org/10.1016/s0169-328x(02)00663-0
van Calker D, Serchov T, Normann C et al (2018) Recent insights into antidepressant therapy: distinct pathways and potential common mechanisms in the treatment of depressive syndromes. Neurosci Biobehav Rev 88:63–72. https://doi.org/10.1016/j.neubiorev.2018.03.014
Dallaspezia S, Benedetti F (2015) Sleep deprivation therapy for depression. Curr Top Behav Neurosci 25:483–502. https://doi.org/10.1007/7854_2014_363
Elmenhorst D, Basheer R, McCarley RW et al (2009) Sleep deprivation increases A(1) adenosine receptor density in the rat brain. Brain Res 1258:53–58. https://doi.org/10.1016/j.brainres.2008.12.056
Kim Y, Elmenhorst D, Weisshaupt A et al (2015) Chronic sleep restriction induces long-lasting changes in adenosine and noradrenaline receptor density in the rat brain. J Sleep Res 24:549–558. https://doi.org/10.1111/jsr.12300
Dias RB, Rombo DM, Ribeiro JA et al (2013) Adenosine: setting the stage for plasticity. Trends Neurosci 36:248–257. https://doi.org/10.1016/j.tins.2012.12.003
Diering GH, Nirujogi RS, Roth RH et al (2017) Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 355:511–515. https://doi.org/10.1126/science.aai8355
Kaster MP, Rosa AO, Rosso MM et al (2004) Adenosine administration produces an antidepressant-like effect in mice: evidence for the involvement of A1 and A2A receptors. Neurosci Lett 355:21–24. https://doi.org/10.1016/j.neulet.2003.10.040
Yamada K, Kobayashi M, Kanda T (2014) Involvement of adenosine A2A receptors in depression and anxiety. Int Rev Neurobiol 119:373–393. https://doi.org/10.1016/B978-0-12-801022-8.00015-5
Funding
Open Access funding enabled and organized by Projekt DEAL. Work of the authors mentioned in this review article was funded by grants from National Natural Science Foundation of China (NSFC81873796; NSFC82071513) to Shu-Yan Yu, the Natural Science Foundation of Shandong Province (ZR2021QH282), and the Fundamental Research Funds of Shandong University (2020GN095) to Xin-Ling Wang and Medical Research Foundation (FRM) (AJE201912009450), University of Strasbourg Institute of Advance Studies (USIAS) (2020–035), and Centre National de la Recherche Scientifique (CNRS UPR3212) to Tsvetan Serchov.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. The first draft of the manuscript was written by Xing-Ling Wang and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interests
Tsvetan Serchov has honoraria consulting Primetime Life Sciences, LLC. All other authors declare that they have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, XL., Gardner, W., Yu, SY. et al. A Pattern to Link Adenosine Signaling, Circadian System, and Potential Final Common Pathway in the Pathogenesis of Major Depressive Disorder. Mol Neurobiol 59, 6713–6723 (2022). https://doi.org/10.1007/s12035-022-03001-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-03001-3