
Abstract
Spinocerebellar ataxia (SCA) is a neurodegenerative disorder characterized by ataxia and cerebellar atrophy. A number of different mutations gives rise to different types of SCA with characteristic ages of onset, symptomatology, and rates of progression. SCA type 34 (SCA34) is caused by mutations in ELOVL4 (ELOngation of Very Long-chain fatty acids 4), a fatty acid elongase essential for biosynthesis of Very Long Chain Saturated and Polyunsaturated Fatty Acids (VLC-SFA and VLC-PUFA, resp., ≥28 carbons), which have important functions in the brain, skin, retina, Meibomian glands, testes, and sperm. We generated a rat model of SCA34 by knock-in of the SCA34-causing 736T>G (p.W246G) ELOVL4 mutation. Rats carrying the mutation developed impaired motor deficits by 2 months of age. To understand the mechanism of these motor deficits, we performed electrophysiological studies using cerebellar slices from rats homozygous for W246G mutant ELOVL4 and found marked reduction of long-term potentiation at parallel fiber synapses and long-term depression at climbing fiber synapses onto Purkinje cells. Neuroanatomical analysis of the cerebellum showed normal cytoarchitectural organization with no evidence of degeneration out to 6 months of age. These results point to ELOVL4 as essential for motor function and cerebellar synaptic plasticity. The results further suggest that ataxia in SCA34 patients may arise from a primary impairment of synaptic plasticity and cerebellar network desynchronization before onset of neurodegeneration and progression of the disease at a later age.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipids are fundamental biological molecules that play many important roles in the central nervous system. Errors in lipid metabolism lead to a variety of neurological diseases. Very Long Chain Saturated and Polyunsaturated Fatty Acids (VLC-SFA and VLC-PUFA, resp., ≥28 carbons) are essential for life, but their functions remain largely obscure. The enzyme ELOngation of Very Long chain fatty acids-4 (ELOVL4) catalyzes the first and rate-limiting step in VLC-SFA and VLC-PUFA biosynthesis and is the only fatty acid elongase that serves this function [1,2,3]. ELOVL4 is expressed in only a small number of organs, including the brain, retina, skin, Meibomian glands, and testes [4,5,6], each of which shows a tissue-specific profile of VLC-SFA and VLC-PUFA.
ELOVL4 and its VLC-SFA and VLC-PUFA products are of critical importance to the CNS [7]. Three distinct groups of mutations in the ELOVL4 gene give rise to three distinct CNS diseases. Heterozygous inheritance of several different ELOVL4 mutations in exon 6 that cause early truncation of the ELOVL4 protein and loss of the ER-retention signal in the C-terminus leads to autosomal dominant Stargardt-like macular dystrophy (STGD3), an aggressive juvenile-onset macular degeneration [8,9,10,11]. Patients with STGD3 show no CNS or skin phenotypes. Heterozygous inheritance of several different point mutations in ELOVL4 causes autosomal dominant spinocerebellar ataxia-34 (SCA34) with or without erythrokeratodermia variabilis (EKV, a skin condition) [12,13,14,15,16,17]. SCA34 patients typically show no retinal phenotype, although a recent report identified a point mutation (c.512T>C, p.I171T) in exon 4 of ELOVL4 as a cause of SCA34 that also presents with retinitis pigmentosa in some of the affected family members [17]. Homozygous inheritance of other recessive ELOVL4 mutations that result in truncation of the ELOVL4 protein causes a neuro-ichthyotic syndrome characterized by severe seizures, intellectual disability, spasticity, ichthyosis, and early death [18, 19]. No human patients with homozygous inheritance of STGD3 or SCA34 alleles have been reported.
Our lipidomic analyses of mouse, rat, and baboon brains found that the main VLC-fatty acid species in the brain are VLC-SFA (primarily 28:0 and 30:0) that are incorporated into complex sphingolipids [3, 20]. Furthermore, VLC-SFA were enriched specifically in synaptic vesicles prepared from baboon hippocampus and found to modulate presynaptic transmitter release kinetics in cultured mouse hippocampal neurons [3]. In contrast, we have not been able to identify VLC-PUFA in brains of 2-, 12-, and 24-month-old mice. However, VLC-PUFA have been reported in the early postnatal rat brain and the brains of young patients afflicted with Zellweger’s disease, a peroxisomal disorder that disrupts lipid metabolism [21, 22]. Based on these studies, VLC-PUFA appear to be present only at trace levels in the healthy, mature brain. In contrast, the main VLC-fatty acids in the retina are VLC-PUFA incorporated into phosphatidylcholine in the outer segments of rod and cone photoreceptors [23,24,25,26], although VLC-SFA incorporated into non-sialylated sphingolipids also have been found [20]. VLC-PUFA in an amide linkage to sphingolipids are present in testes [27,28,29]. VLC-SFA are also synthesized in the Meibomian glands and skin, where they are incorporated into ω-O-acylceramides and form the water barrier of the tear film and skin, respectively [5, 30,31,32,33]. Our recent studies of W246G ELOVL4 function in the retina and skin showed that the W246G mutation selectively impaired synthesis of VLC-SFA (28:0 decreased to around 60%, and 30:0 decreased to 10% of levels in skin of WT rats), but had no significant effect on VLC-PUFA synthesis [34].
SCAs arise from defects in a large number of different genes, which produce distinct forms of SCA with characteristic times of onset and rates of disease progression [35, 36]. It has been suggested from the published SCA studies that cerebellar dysfunction is caused by altered output from the cerebellar cortex. The cerebellar Purkinje cells (PCs) are the sole output neuron of the cerebellar cortex. Thus, understanding the effects of SCA-causing mutations on PC synaptic inputs and outputs is critical to understanding the underlying disease process. The excitatory inputs to PCs are synapses formed by the parallel fibers (PF) from granule cells and climbing fibers (CF) from inferior olive neurons. Changes in synaptic plasticity at PF and CF inputs to PCs are thought to be critical to motor function and learning. Our previous studies showed that ELOVL4 is prominently expressed in the granule cells, molecular layer interneurons, and Purkinje cells of the cerebellum as well as in the inferior olivary complex [37], which provides the CF input to the cerebellum. This is consistent with the clinical and pathological findings that ELOVL4 mutations cause SCA34, although the effects of these mutations on the cytoarchitectural organization and synaptic plasticity of the cerebellum are unknown.
To better understand the functional role of ELOVL4 and its products in synaptic function and SCA34 pathology, we generated a rodent model of the human SCA34-causing Elovl4 mutation reported by Ozaki et al. [16] using CRISPR-Cas9 gene editing to replace one copy of the rat Elovl4 gene with the c.736T>G (p.W246G) mutation in the Long Evans rat genome [34]. We used a range of behavioral, biochemical, anatomical, and ex vivo electrophysiological studies to evaluate the effect of the W246G ELOVL4 mutation on motor function, cerebellar organization, and synaptic function. This is the first report of an animal model of how mutations in ELOVL4 contribute to age-related cerebellar degeneration and loss of motor coordination in SCA34 patients. Our results also underscore the essential role of ELOVL4 products in neuronal health and aging and may have broader implications for age-related loss of motor coordination independent of ELOVL4 mutations.
Materials and Methods
Animals
A CRISPR/Cas9 gene editing approach was used to generate F0 founder Long-Evans rats with heterozygous knock-in of the c.736T>G, p.W246G mutation in ELOVL4 that causes human SCA34 [16]. These founders were bred to WT Long-Evans rats to establish a breeding colony. Details of the generation of the knock-in rat line are described in Agbaga et al. [34]. All experiments were performed using WT, HET, and MUT rats, with genotype confirmed by PCR. Rats were maintained in a pathogen-free barrier facility on a 12-h light:12-h dark daily light cycle (~25–40 lux at cage level) with food and water available at all times. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Oklahoma Health Sciences Center and conformed to the National Institute of Health Guide for the Care and Use of Laboratory Animals, US Public Health Service guidelines, and the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Research.
Western Blotting
The lysate of rat cerebellum was prepared by homogenizing the tissue in 20 mM Tris HCl (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, and EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich, St Louis, MO). Protein concentration was determined using a Bicinchonic Acid kit (Sigma-Aldrich, St. Louis, MO) after which 25 μg of total unboiled protein lysates were resolved on 4–20% polyacrylamide gels and transferred to nitrocellulose membrane. ELOVL4 was detected using previously characterized rabbit polyclonal ELOVL4 antibodies [1] that have been confirmed by other labs [38, 39] to recognize ≈32-kDa ELOVL4 protein from mouse, rat, bovine, and porcine ELOVL4-expressing tissues and by specifically block binding by pre-incubation of the antibody with the antigenic peptide [1]. Immunolabeling of nitrocellulose membranes with this and other primary antibodies (Table 1) was performed at room temperature for 2 h in Intercept TBS Blocking Buffer (LI-COR). Following incubation with fluorescent dye-coupled secondary antibodies (Table 1), the membranes were imaged sequentially in 700-nm and 800-nm channels on an Odyssey Fc Imaging System (LI-COR) using the Image Studio acquisition program (LI-COR). The intensity of the bands (summated signal in the selected area) was quantified using Image Studio Lite (LI-COR). The intensity value of the band of interest was normalized to the loading control in the same lane.
Assessment of Locomotor Function
Testing of locomotor function was performed using the rotarod test on WT, HET, and MUT rats at 2 to 6 months of age. All motor testing was performed under constant illumination conditions by the same experimenter, who was masked to the animals’ genotype. To assess motor learning, coordination, and balance, rats were placed on a rotarod apparatus with a roller diameter of 3.75 inches and five separated, equally sized individual animal compartments (IITC Life Science; Woodland Hills, CA), and presented with an accelerating rotation speed of 25–45 rpm for 5 min. Each animal was tested three times in each session and the mean total distance traveled and time to fall/completion were determined (data not shown) for each animal as measures of motor function [40,41,42]. Rats were trained on the accelerating assay over the course of several days to master the task prior to testing.
Histology, Immunolabeling, and Imaging
For histology and immunolabeling, rats were anesthetized by intraperitoneal injection of ketamine (100 mg/kg body weight) and xylazine (5 mg/kg body weight) and perfused through the left ventricle with 0.1 M phosphate buffered saline (PBS, pH 7.2), followed by 4% paraformaldehyde in PBS for 20 min. Following perfusion, the brain was removed from the head, hemisected along the sagittal plane, and immersed in 4% paraformaldehyde in 0.1 M PBS or 0.1 M cacodylate buffer at 4 °C for several days. Following fixation, brains were rinsed extensively in PBS, cryoprotected in 30% sucrose in PBS, and then embedded in Optimal Cutting Temperature medium (OCT; Sakura Tissue Tek; VWR, West Chester, PA) and frozen. Sagittally oriented frozen sections (10- to 15-μm thickness) were prepared on a cryostat, collected onto Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA), and stored at −20 to −30 °C until use.
Sections for morphometric analyses were stained using 1% toluidine blue and imaged using a Leica M205-MFC imaging system. Morphometric measurements were obtained from lobules III, V, and X using sagittal sections that ranged from 0 to 1320 μm from the midline. Images were imported into FIJI (NIH Image) software, image scale was calibrated, and brightness and contrast were adjusted to highlight cortical layering and Purkinje cell bodies. The linear extent of each cortical layer was measured along a perpendicular line extending from the cortical surface to the inner margin of the granule cell layer. Analysis of the linear density of Purkinje cells was performed in the same tissue sections by counting the number of Purkinje cell bodies in well-oriented, linear regions of the Purkinje cell layer, avoiding curved areas of the proximal and distal regions of the lobules.
Immunolabeling was performed as described previously [3, 37]. Antibody information is provided in Table 1. Sections were rehydrated in Hank’s buffered salt solution (HBSS) and non-specific labeling was blocked using a solution containing 2–10% normal goat serum, 5% bovine serum albumin, and 1% fish gelatin in 0.1–0.5% Triton X-100 in HBSS (“blocker”). In some experiments, antigen retrieval in 10 mM citrate buffer (pH 6.0; heated to 95 °C) was performed for 30–60 min prior to blocking. Blocker was removed and one or more primary antibodies were applied overnight at room temperature. Sections were rinsed in HBSS and appropriate secondary antibodies were applied for 60–75 min at room temperature. Sections were rinsed and mounted using Prolong Gold + DAPI or Prolong Diamond + DAPI to retard photobleaching. Specificity of the labeling method was confirmed by omitting primary antibodies and by substituting normal rabbit serum for primary antibody.
Fluorescence microscopy was performed using an Olympus IX70 inverted fluorescence microscope fitted with a QiCAM CCD camera controlled via QCapture software (QImaging), or a Leica M205-MFC microscope controlled via LasX software. Image scale was calibrated and figures were prepared using Photoshop software. If needed, adjustments to brightness and contrast were applied equally to all pixels in the image to highlight specific labeling.
Electrophysiology
Slice Preparation
Both male and female rats at 3–4 months of age were used. The rats were decapitated using a guillotine and the cerebellum was quickly removed and cooled with ice-cold artificial CSF (ACSF) containing the following: 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 1.0 mM CaCl2, 1.0 mM MgCl2, 2.0 mM pyruvic acid, 0.4 mM ascorbic acid, and 10 mM D-glucose (final pH 7.4, oxygenated with 95% O2/5% CO2). The cerebellum was trimmed and the tissue block containing the cerebellar vermis was glued to the stage of a vibrating tissue slicer. Sagittal slices of about 350 μm were collected in oxygenated cold sucrose slicing solution containing 240 mM sucrose, 25 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 0.4 mM ascorbic acid, 10 mM D-glucose, 10 mM MgCl2, and 2 mM pyruvic acid (final pH 7.4) using a HM650V vibrating microtome (Thermo Scientific, Burlington, ON). The slices were transferred to a holding chamber containing oxygenated ACSF at room temperature and allowed to recover for at least 1 h.
Multi-electrode Array Recordings
For recording network activity and extracellular field potentials, the slice was transferred to and positioned on a multi-electrode array system (Alpha MED Scientific, Inc., Osaka, Japan) with a hexagonal electrode arrangement of 61 electrodes and electrode spacing of 70 μm (MED-P2H07A). Under the dissecting microscope, three layers of the cerebellar cortex, including the molecular layer, Purkinje cell layer, and granule cell layer, and the white matter could readily be identified in each folium. To secure contact between the slice and electrodes and to improve mechanical stability, a piece of nylon mesh and a slice anchor harp were placed on top of the slice. The slice was maneuvered to ensure that the hexagonal array was positioned within a folium and that the electrodes covered all three layers of the cortex. The chamber was perfused with oxygenated ACSF at a rate of 2 mL/min at 32 °C. After the slice had settled in the recording chamber, before applying any stimulation, six hundred (600) traces of network activity were recorded, each for a 1-s duration, under physiological conditions with continuous ACSF perfusion [3]. Network activity data were analyzed as previously described [3]. Briefly, raw MED64 Mobius workflow files were opened and spikes were extracted in Mobius (© WitWerx, Inc.). Positive and negative spike threshold was set to + 0.021 and −0.021 mV, respectively. Spike traces were extracted along with 1 ms of baseline before and after the spike event, without down sampling. Raw data were filtered using a Bessel high-pass (2-pole) with a cutoff frequency of 1000 Hz and a DC filter with a typical spike length set to 1 ms. The resulting file containing all extracted spikes within that slice recording was then processed in Microsoft Excel using a visual basic macro that was coded and validated by us to extract each of the final parameters for statistical comparison.
All evoked responses were obtained by stimulating one electrode in the molecular layer and one electrode in the granular cell layer to evoke responses in the molecular layer. Input/output curves (I/O curves) were generated by applying increasing stimulus currents to the pathway from 5 to 100 μA and recording the responses. The threshold stimulus for generating fEPSPs was determined as 30–40% of the stimulus strength needed to generate the maximum fEPSP amplitude during the I/O curve measurement. The pathway was stimulated once every 30 s until a stable baseline lasting at least 10 min was observed. Long-term potentiation (LTP) was induced in the molecular layer using 600 low-frequency stimulation pulses at 1 Hz applied for 10 min. Long-term depression (LTD) of the CF was induced using 600 low-frequency stimulation pulses at 1 Hz applied for 10 min to an electrode that evoked responses at the boundary of Purkinje and molecular layers. Next, baseline stimulation was resumed and the fEPSPs recorded for at least 60 more minutes. Finally, another I/O curve was recorded and generated as described above. For all recordings, the MED-64 system and Mobius software were used (Alpha MED Scientific, Inc.). LTP/LTD was calculated as the percent increase/decrease of the mean fEPSP descending slope (10–90 section) after 1-Hz stimulation and normalized to the mean fEPSP descending slope of baseline recordings (during 3 min prior to 1-Hz stimulus).
Results
Generation of the SCA34 Rat Model
We used the CRISPR-Cas9 gene editing approach to knock in the c.736T>G (p.W246G) mutation that causes human SCA34 [16] into exon 6 of the rat Elovl4 gene (Ensembl: ENSRNOG00000009773) to generate heterozygous Long-Evans F0 founder rats (Fig. 1A). See Agbaga et al. (2020) for a more detailed description [34]. The characterization of the model is reproduced (34) in Supplemental Figures 1-6. The F0 founder rats were bred to wild-type (WT) Long-Evans rats to establish heterozygote breeders. For consistency and clarity, this mutation is referred to as the “W246G ELOVL4 mutation.” Editing of a single copy of the WT Elovl4 allele to the c.736T>G (p.W246G) mutant allele was confirmed by genotyping and by Sanger and whole-genome DNA sequencing, which did not find any aberrant indels, insertions, or off-target editing effects [34]. Heterozygous W246G ELOVL4 knock-in rats were fertile and passed the 736T>G allele to their offspring to produce heterozygote (HET) and homozygote (MUT) offspring. MUT rats were viable and fertile, in contrast to mice with Elovl4 knockout or homozygous expression of STGD3 alleles, which die from dehydration shortly after birth due to loss of the skin water barrier [30,31,32,33]. Phenotypically, MUT rats presented with EKV characterized by reddened skin and patchy loss of hair [34], consistent with the finding of EKV in some human SCA34 patients [14, 15]. Age-matched female rats showed no differences in weight across genotypes from 2 to 6 months of age. In contrast, male rats exhibited a sex-related difference in weight, with male MUT rats being significantly smaller than age-matched male WT and HET rats from 2 to 6 months of age (Fig. 1B). Immunoblotting showed no differences in ELOVL4 levels in the WT, HET, and MUT cerebellum (Fig. 1C, D) using a well-characterized antibody that recognizes WT and full-length MUT ELOVL4 [1]. Finally, assessment of the effect of the W246G ELOVL4 mutation on VLC-SFA and VLC-PUFA synthesis showed that the mutation significantly reduced, but did not eliminate, VLC-SFA production in the skin (remaining 28:0 + 30:0 levels around 35% compared with WT), and had no significant effect on VLC-PUFA synthesis in the retina (34; Supplemental Figs. 5 and 6).

Knock-in of the W246G ELOVL4 mutation, physical differences, and ELOVL4 expression in WT, HET, and MUT rats. A Cartoon depicting the overall structure and functional domains of ELOVL4 and the location of the W246G ELOVL4 mutation. B The W246G ELOVL4 mutation does not affect the size of female rats at any age between 2 months and 6 months of age. Male MUT rats are significantly smaller than age-matched WT and HET rats from 3 months of age (females, 2 months: n = 26 WT, 22 HET, 24 MUT; 3 months: n = 17 WT, 13 HET, 17 MUT; 4 months: n = 9 WT, 9 HET, 10 MUT; 6 months: n = 17 WT, 14 HET, 10 MUT. Males, 2 months: n = 5 WT, 8 HET, 4 MUT; 3 months: n = 5 WT, 8 HET, 4 MUT; 4 months: n = 4 WT, 7 HET, 3 MUT; 6 months: n = 7 WT, 10 HET, 4 MUT. Data are shown as mean ± SD. 1-way ANOVA + Bonferroni post-hoc test. *p< 0.05; **p<0.01; ***p<0.001; ****p<0.0001). C Western blot of cerebellum from WT, HET, and MUT rats (n=3 each). D Quantification of ELOVL4 levels (normalized to Actin loading control) showed no differences across genotypes. Analysis performed on samples from 3 different rats of each genotype (data are shown as mean ± SEM. Statistical analysis by 1-way ANOVA with Tukey’s post-hoc test)
The W246G ELOVL4 mutation did not cause overt impairment of motor function, as male and female HET and MUT rats could walk, rear on their hind legs, and breed, consistent with the relatively late onset and slow progression of SCA34 in humans with the W246G ELOVL4 mutation. To better assess effects on motor performance, cohorts of female WT, HET, and MUT rats were tested at 2, 3, 4, and 6 months of age using the rotarod assay. The rotarod test revealed significant motor impairment in female HET and MUT rats compared with WT rats at all ages examined (Fig. 2), indicating that the deficits observed at 2 months were not transient. Interestingly, HET and MUT female rats performed equally poorly in the test. These studies indicate that the W246G ELOVL4 mutation caused persistent motor impairment in HET and MUT rats starting from early ages. We also tested the performance of male rats on the rotarod task at 2 and 6 months of age. Male rats of all genotypes showed poor performance at both ages (Supplemental Fig. 7). Statistical analysis showed no significant differences across male rats regardless of genotype. The reason for this sex difference in rotarod performance is unclear; however, the difference in motor behavior between male and female rats was striking and consistent.

The W246G ELOVL4 mutation impaired motor function assessed by rotarod test. (A–D) Female HET and MUT rats showed consistent impairment of motor performance on the rotarod test compared to age-matched WT female rats from 2 to 6 months of age. Distance traveled variable shown. (E) Data from panels (A–D) re-plotted to show performance over time (2 months: n = 26 WT, 22 HET, 24 MUT. 3 months: n = 17 WT, 13 HET, 17 MUT. 4 months: n = 9 WT, 9 HET, 10 MUT. 6 months: n = 17 WT, 14 HET, 10 MUT; data are shown as mean ± SEM. 1-way ANOVA + Bonferroni post-hoc test. *p< 0.05; **p<0.01; ***p<0.001)
The W246G ELOVL4 Mutation Does Not Disrupt ELOVL4 Distribution or Cerebellar Cytoarchitecture Out to At Least 6 Months of Age
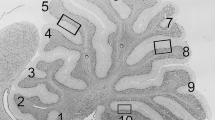
Cerebellar atrophy is a common feature of SCA34 [14, 16]. Loss of Purkinje cells in particular is a prominent feature of many forms of SCA [43]. To assess whether the W246G ELOVL4 mutation caused neurodegeneration or disrupted cerebellar cytoarchitecture, we conducted a detailed morphological assessment of the cerebellar cortex. Gross cerebellar structure was not affected by the W246G ELOVL4 mutation (Fig. 3(A–I)). Lobule organization was similar in WT, HET, and MUT rats through at least 6 months of age. The cerebellar cortex showed the normal three-layered structure comprised of a thick, outer molecular layer housing interneurons and synapses, a well-organized monolayer of Purkinje cells, and a densely packed granule cell layer adjacent to the deep white matter of the arbor vitae (Fig. 3(J–L)). Analysis of cortical layer thickness in lobules 3, 5, and 10 showed no thinning of the molecular or granule cell layers at 3 or 6 months (Fig. 3(M–R)). The linear density of Purkinje cells also showed no differences across ages or genotypes (Fig. 3(S, T)), indicating that there was no substantial loss of Purkinje cells over this time frame.

Gross cerebellar structure and morphometric analysis. (A–I) Gross cerebellar structure in WT, HET, and MUT rats at postnatal day 45 (P45), 3 months, and 6 months of age is comparable. Scale bar = 1 mm for each row. (J–L) Lamination and cellular distribution in the cerebellar cortex is normal in WT, HET, and MUT rats. Six months of age shown, scale bar = 100 μm. (M–T) Morphometric analysis of cortical layer thickness in WT, HET, and MUT rats indicates no substantial reduction in any cortical layer at 3 months or 6 months, although a significant difference in Purkinje cell layer width between WT and MUT rats was noted in lobule 3 at 6 months (3 months: n = 3–5 animals/genotype. 6 months: n = 4–8 animals/genotype. Data are shown as mean ± SEM. 1-way ANOVA with Tukey’s post-hoc test. *p< 0.05). (U, V) Linear density of Purkinje cells shows no difference among WT, HET, and MUT rats (3 months: n = 5 animals/genotype. 6 months: n = 7 animals/genotype. Data are shown as mean ± SEM. 1-way ANOVA with Tukey’s post-hoc test)
To further assess cerebellar integrity, immunolabeling for ELOVL4 and a variety of well-known cell- and synapse-specific markers was performed (Fig. 4). Notably, as the ELOVL4 antibody recognizes both wild-type and mutant protein, we were able to detect the distribution of WT and W246G mutant ELOVL4 protein using the same antibody (see Fig. 1). The W246G ELOVL4 mutation did not alter ELOVL4 distribution in the cerebellum. ELOVL4 was present in granule cells, inhibitory interneurons in the molecular layer, and Purkinje cells in the cerebellum of rats of all genotypes (Fig. 4(A–C)), similar to ELOVL4 distribution in the mouse cerebellum [37]. Purkinje and granule cells labeled for calbindin and NeuN, respectively, showed normal morphology and distribution in WT, HET, and MUT rats at all ages examined (Fig. 4(D–I); Supplemental Figs. 8, 9). Labeling for VGluT1, a marker for mossy and parallel fiber synapses, and synaptotagmin 2, a marker for GABAergic synapses, both showed numerous synapses throughout the molecular layer and in the synaptic glomeruli of the granule cell layer in all genotypes at all ages, as appropriate (Fig. 4(J–O); Supplemental Figs. 10, 11). Labeling for VGluT2, a specific marker for climbing fiber synapses onto Purkinje cells, confirmed that climbing fiber synapses were present in the molecular layer of WT, HET, and MUT cerebellar cortex, as expected (Fig. 5). The distribution of mGluR1 [44], which is critical to Purkinje cell synaptic plasticity [45], was unaffected by the W246G ELOVL4 mutation (Fig. 5). Similarly, Protein Kinase C gamma (PKCɣ) and Inositol Phosphate 3 Receptor (IP3R), which are also important to Purkinje cell synaptic plasticity [46, 47], showed no substantial differences in distribution or expression levels in the cerebellum (Supplemental Figs. 12, 13). Labeling for astrocytes and microglia showed the normal distribution and morphology of these cell populations across genotypes of all ages, with no sign of overt neuroinflammation (Supplemental Figs. 14, 15).

Distribution of ELOVL4 and cell- and synapse-specific markers is normal in WT, HET, and MUT rat cerebellum. (A–C) Distribution of ELOVL4 in the cerebellar cortex of WT, HET, and MUT rats is comparable. Labeling is present in granule cells in the granule cell layer (GCL), Purkinje cells (arrows) in the Purkinje cell layer (PCL), and inhibitory neurons in the molecular layer (ML). (D–F) Purkinje cells identified by labeling for calbindin are organized in a monolayer in the Purkinje cell layer (PCL) and show similar structure in the WT, HET, and MUT cerebellum. (G–I) Distribution of NeuN, a marker for granule cells in the granule cell layer (GCL), is comparable across all genotypes. A small population of NeuN-positive cells is also present in the molecular layer (ML), as appropriate. (J–L) Distribution of Vesicular Glutamate Transporter 1 (VGluT1), a marker for excitatory parallel fiber and mossy fiber synapses, is comparable across all genotypes. (M–O) Distribution of synaptotagmin 2 (Syt2), a marker for inhibitory synapses, is comparable across all genotypes. Scale bars = 200 μm for each row

Distribution of VGluT2 and mGluR1 in WT, HET, and MUT rat cerebellum. (A–C) Distribution of VGluT2, a marker for climbing fiber synapses, is comparable in the cerebellar cortex of WT, HET, and MUT rats. Labeling is present in climbing fiber synapses onto Purkinje cell dendrites in the molecular layer (ML) and in the synaptic glomeruli of the granule cell layer (GCL). PCL, Purkinje cell layer. (D–F) Distribution of mGluR1 is comparable in the WT, HET, and MUT cerebellum. PCL, Purkinje cell layer. Scale bars = 100 μm for panels (A–C); 200 μm for panels (D–F)
To further assess the effects of the W246G ELOVL4 mutation at the synaptic level, western blot analysis of several important synaptic proteins associated with glutamatergic and GABAergic synapses was performed (Fig. 6). We examined the following proteins: excitatory presynaptic markers—VGluT1, VGluT2; AMPA glutamate receptors—GluA1, GluA2, and the postsynaptic density protein PSD-95; and inhibitory synaptic markers—Syt2, GAD65, and GAD67. No significant differences were present among WT, HET, and MUT cerebellum for any of the glutamatergic and GABAergic synaptic proteins examined.

The W246G ELOVL4 mutation does not significantly alter levels of synaptic proteins in the WT, HET, and MUT cerebellum. Immunoblots for A VGluT1, B VGluT2, C GluA1, D GluA2, E PSD-95, F Syt 2, and G GAD-65 and GAD-67 are shown along with their quantification (normalized to loading control: Actin or GAPDH). Quantitation was performed on samples obtained from 3 different rats of each genotype (data are shown as mean ± SEM. Statistical analysis by 1-way ANOVA with Tukey’s post-hoc test)
Together, these data indicate that the motor deficits observed in HET and MUT rats did not arise from any gross disruption of the cellular or synaptic organization of the cerebellum or neurodegeneration.
The W246G ELOVL4 Mutation Alters Neuronal Network Communication in the Cerebellar Cortex
Cerebellar cortical function is essential for motor learning, and it is thought to involve fine tuning of the extensive PF-Purkinje cell synaptic network in coordination with the “error reporting” of CF-Purkinje cell input [48]. To better understand the origins of the motor impairment associated with the W246G ELOVL4 mutation, we assessed neuronal network properties in the cerebellum using multi-electrode array electrophysiological recording from cerebellar slices [49] prepared from WT and MUT rats (Fig. 7, see Methods for details). Briefly, an array of 61 electrodes arranged hexagonally at 70-μm intervals was carefully positioned under the slice to cover all cerebellar cortical layers. The overall spontaneous network activity was recorded for 10 min under resting conditions [3]. Assessing entire 10-min-long recordings and plotting the events per second for all 61 electrodes revealed two major differences in network activity in WT and MUT slices (Fig. 7A). First, slices generally showed either very low activity (less than average of 1 event per sec, upper panels) or high activity (lower panels on Fig. 7A). There was no significant difference between WT and MUT slices in the distribution of low to high activity (20/9 for WT and 15/10 for MUT, chi-square value is 0.4732, p=0.491511). However, there was a striking failure of PF synchronization in MUT slices compared with the almost uniform synchronization of spontaneous activity in WT slices recorded on all electrodes simultaneously (Fig. 7A, lower two panels). Interestingly, in many MUT slices, we detected robust, persistent activity (Fig. 7A, lower two panels). Next, we analyzed spike frequency, amplitude, and interspike interval (ISI) from these recordings. Although average spike frequency did not differ in the cerebellar cortex between WT and MUT slices (Fig. 7B left panel), further analysis revealed an increased average frequency of positive spikes (Fig. 7B middle panel) and a reduced frequency of negative spikes in the MUT cerebellum (Fig. 7B right panel). In addition, the amplitude of network events was significantly decreased in MUT cerebellar slices compared with WT (Fig. 7C, negative spike traces and positive spike traces; and Fig. 7D). The overall distribution of events as shown by the ISI histogram was similar in WT and MUT slices (Fig. 7E, p=0.219, Wilcoxon ranked test), with both showing peak activity at two frequencies of 0.5 Hz and 0.26 Hz. We noticed more activity at the highest frequencies in the WT slices and selectively analyzed the 1 Hz or higher activity signals (Fig. 7F). Indeed, MUT cerebellar slices had a marked reduction in activity above 20 Hz (ISI <0.05 s; Fig. 7F; p<0.0001, Mann-Whitney test). Next, to determine whether PF activity was altered under resting conditions, we analyzed the spontaneous activity from the same electrodes that were used later to record PF LTP. Activity in MUT PF differed from WT PF: the difference was mostly in the high >20-Hz frequencies, but interestingly, the 0.6–4-Hz activity corresponding to delta band was also reduced (Fig. 7G; p<0.0001, Mann-Whitney test). These observations from the spontaneous network activity indicate that W246G ELOVL4 mutation alters and desynchronizes neuronal network communication in the cerebellar cortex.

Decreased amplitude and frequency domain shift of spontaneous activity in MUT cerebellar slices. A Representative plots of spontaneous network activity in WT (left panel) and MUT (right panel) cerebellar slices recorded from 61 locations (X axis = time, Y axis = number of events, Z axis = recording electrode #). WT and KI slices show very low activity (upper panels) or high activity (lower panels). Inset shows a picture of the cerebellar slice on the hexagonal multi-electrode array. B No difference in the average frequency of spontaneous network activity in the cerebellum of WT (n=7 slices from 5 rats) and MUT (n=8 slices from 6 rats). Note an increased average frequency of positive spikes and reduced frequency of negative spikes in the MUT cerebellum (shown as mean ± SEM, analysis by Mann-Whitney test. *p<0.05). C Representative traces from spontaneous events of WT (left panel) and MUT (right panel). Note higher amplitude in positive spikes (lower panel) and negative spikes (upper panel) of the WT traces. An extremely large positive spike in WT and a complex spike in MUT are shown in middle panel traces. Scale bars y=20 μV, x=1 ms. D Analysis of spontaneous event amplitudes. Note the decreased amplitude of positive spikes (left panel, *p<0.05, Wilcoxon signed rank test) and decreased amplitude of negative spikes (right panel, ***p<0.001, Wilcoxon signed rank test) in the MUT compared with WT cerebellum. E Resting spontaneous activity in WT and MUT slices. Distribution of interspike interval (ISI) in 5 ms bins from WT (upper; n=7) and MUT (lower; n=8) slices during 10-min recordings. F Cumulative histogram of ISI analysis of events with 2 s or less time between them. Data shown in E re-analyzed using 1-ms bins. MUT slices show-reduced activity at high frequencies (above 20 Hz) compared with WT slices (p<0.0001, Mann-Whitney test). G Cumulative histogram of ISI of spontaneous activity in PF–PC connections in WT (n=4) and MUT slices (n=5) analyzed using 1-ms bins (p<0.0001, Mann-Whitney test)
The W246G ELOVL4 Mutation Severely Impairs Synaptic Plasticity at PF and CF Inputs to Purkinje Cells
Differences in neuronal network activity between WT and MUT cerebellum as well as the motor learning deficits of HET and MUT rats in the absence of neurodegeneration are consistent with the notion that synaptic function is impaired by the W246G ELOVL4 mutation. Therefore, we tested whether the W246G ELOVL4 mutation affected neurotransmission and long-term synaptic plasticity in the cerebellum. Long-term potentiation (LTP) and long-term depression (LTD) are hours-long increases and decreases, respectively, in synaptic strength in response to strong synaptic stimulation, and are important to motor learning. ELOVL4 is highly expressed in cerebellar granule cells (Fig. 4) and cells in the inferior olivary complex [37], and its VLC-SFA products that are incorporated into sphingolipids are known to modulate synaptic release in hippocampus [3]. To assess the functional effects of the W246G ELOVL4 mutation on long-term synaptic plasticity, we tested the function of PF and CF inputs to Purkinje cells in cerebellar slices prepared from 3- to 4-month-old WT and MUT rats.
The most numerous excitatory synaptic inputs to Purkinje cells are from PFs, which drive simple spikes in Purkinje cells [50]. To assess the function of PF-Purkinje cell synapses, acute ex vivo cerebellar slices from WT or MUT rats were stimulated using one electrode located in the molecular layer to stimulate PFs. The field potentials of excitatory post-synaptic potential (fEPSP) responses in the molecular layer were recorded every 30 s using a MED64 multi-electrode array system (MEA). A stimulation paradigm of 1 Hz for 10 min was used to induce PF LTP (modified from [51,52,53,54]). This stimulation paradigm does not require CF co-stimulation and indeed induced robust LTP in slices from WT, but not MUT, rats (Fig. 8). As expected, WT slices showed a significant increase in the slope of PF fEPSP 1 h after stimulation. However, no increase in the PF fEPSP slope was present in slices from MUT rats, indicating severe impairment of LTP at PF-Purkinje cell synapses (Fig. 8A, upper panel, WT = 145.4 ± 9.2% normalized slope, n=14 slices from 6 rats; MUT = 96.2 ± 3.8%, n=16 slices from 6 rats; mean ± SEM; ***p < 0.001, two-way repeated measures ANOVA with Bonferroni post-hoc test [RM ANOVA]). We also measured the basic physiological properties of the LTP at PF-Purkinje cell synapses by recording the characteristics of synaptic responses to a range of increasing stimuli (5–100 μA) before and after recording LTP. This “Input–Output” test allowed us to assess general physiological properties of PF-Purkinje cell synapses and to evaluate how LTP affects them. We found that, at stimulus intensity greater than 60 μA, MUT slices showed a significantly smaller fEPSP amplitude than WT slices before LTP (Fig. 8B; lower left panel, *p < 0.05, two-way RM ANOVA). Furthermore, after LTP induction, while the response for WT slices increased significantly (Fig. 8B; upper left panel, ***p < 0.001, two-way RM ANOVA), MUT slices showed no change in response (Fig. 8B; upper right panel, ns, two-way RM ANOVA), indicating that MUT animals had severely impaired synaptic plasticity. Accordingly, the difference between WT and MUT slices in synaptic response to stepwise stimuli became even more pronounced after LTP was induced (Fig. 8B; lower right panel, **p < 0.01, two-way RM ANOVA).

LTP at PF-Purkinje cell synapse is impaired in MUT rats. A LTP induced in WT rats by stimulation of the PFs in the molecular layer at 1 Hz for 10 min induces LTP in WT rat cerebellar slices (green circles, n = 14 slices from 6 rats), but not in cerebellar slices from MUT rats (red triangles, n = 16 slices from 6 rats). Each data point represents the average of two successive test responses. The vertical gray bar indicates the period of 1-Hz stimulation. Typical traces show EPSPs before (black line) and after (green line for WT, red line for MUT) 1-Hz stimulation in slices from WT (left) and MUT (right) rats (data are shown as mean ± SEM; ***p < 0.001, two-way repeated measures ANOVA with Bonferroni post hoc test [RM ANOVA]). B Baseline and potentiated synaptic strength in PF-PC synapses. Graph depicts amplitude of fEPSP evoked from WT slices (green, n=14 from 6 rats) and MUT slices (red, n=13 from 6 rats) by stepwise increase in the stimulus from 5 to 100 μA. WT slices show increased amplitudes after 1 h LTP (upper left panel, ***p < 0.001, two-way RM ANOVA). MUT slices show no increase in amplitudes after 1-h LTP (upper right panel, ns). Comparison of amplitudes between WT and MUT slices before LTP (lower left panel, *p < 0.05, two-way RM ANOVA) and after LTP (lower right panel, **p < 0.01, two-way RM ANOVA). C Changes in the synaptic responses during the 1-Hz stimulation trains. Normalized fEPSP amplitude in WT slices (green circles, n=14 slices from 6 rats) increased significantly compared with MUT slices (red triangles, n=14 slices from 6 rats). Each data point represents one fEPSP response as recorded every second and normalized to the amplitude of first response (data are shown as mean ± SEM, **p < 0.01, two-way ANOVA, last 100 s). D fEPSP average amplitude of the first response in WT and MUT slices (same data set as panel C; shown as mean ± SEM, ns, Mann-Whitney test). E Ratio of average amplitude of the last five responses (#596–600) divided by the average of the first five responses (#1–5) in WT and MUT slices (data are shown as mean ± SEM, ***p < 0.001, Mann-Whitney test)
As the baseline synaptic strength differed between WT and MUT PF fiber recordings, we next scrutinized the synaptic responses during the 1-Hz stimulus trains. We found that the applied low-intensity stimuli evoked similar responses from both genotypes (Fig. 8D), as the average amplitude of the first response was 395 ± 81.6 μV for WT and 344.3 ± 100 μV for MUT (p=0.66, Mann-Whitney test). During the stimulus train, WT responses increased significantly, but MUT responses remained stable (Fig. 8C, **p < 0.01, two-way ANOVA, last 100 s). This also was reflected in the ratio of average amplitude of the last five responses divided by the average of the first five responses (Fig. 8E), which increased during the 1-Hz stimulation only in WT slices (***p < 0.001, Mann-Whitney test). Taken together, these observations support the conclusion that PF synapses with the W246G ELOVL4 mutation are selectively defective in LTP, although they respond perfectly well to low-intensity single stimulation. Importantly, we tested for any sex-related differences in parallel fiber synaptic plasticity and found that both males and female MUT rats had similar LTP impairment (Suppl. Fig. 16). Thus, we have combined the data for both sexes in Fig. 8.
A second key excitatory synaptic input to the Purkinje cells comes from the CFs, which drive complex spikes in Purkinje cells and are critical to motor learning [50]. The CF-Purkinje cell synapse has a high release probability [55] and shows pronounced LTD in response to tetanic stimulation. To assess the function of CF-Purkinje cell synapses, slices from WT or MUT rats were given tetanic stimulation to activate CFs at low frequency (1 Hz for 10 min) and CF fEPSP responses were recorded at the boundary of Purkinje cell and molecular layers every 30 s using the MEA system (modified from [51]). This stimulation paradigm induced strong LTD in slices from WT rats as expected, but did not induce LTD in slices from MUT rats (Fig. 9A). The level of depression was significantly higher in slices from WT rats than in slices from MUT rats 1 h after induction, indicating severe impairment in LTD in the presence of the ELOVL4 W246G mutation (WT = 60.5 ± 4.6% normalized slope, n=10 recordings from 5 rats; MUT = 95.98 ± 3.4%, n=14 recordings from 6 rats; mean ± SEM; ***p < 0.001, two-way RM ANOVA). We also recorded the Input–Output characteristics of synaptic responses to increasing stimuli (5–100 μA) before and after recording LTD to measure physiological properties of CF to PC synapses. We found that, at stimulus intensity greater than 50 μA, MUT slices showed a significantly smaller fEPSP amplitude than WT slices before LTD (Fig. 9B; lower left panel, ***p < 0.001, two-way RM ANOVA). However, after LTD induction, while the response for WT slices decreased significantly (Fig. 9B; upper left panel, *p < 0.05, two-way RM ANOVA), MUT slices showed no change in response (Fig. 9B; upper right panel, ns, p > 0.05, two-way RM ANOVA), indicating that MUT animals had impaired LTD. Moreover, after LTD induction, we found that WT and MUT slices still responded differently to strong stimuli (>50 μA), with MUT slices showing a significantly smaller amplitude than WT slices (Fig. 9B; lower right panel, *p < 0.05, two-way RM ANOVA). Finally, we analyzed the synaptic responses from CF during the 1-Hz stimulation (Fig. 9C–E), as described above for PF recordings. No significant difference was present between WT and MUT slices during the 600 pulses at 1 Hz.

LTD at CF-PC synapses is impaired in MUT rats. A Stimulating the CFs in the granule cell layer at 1 Hz for 10 min induces LTD in WT cerebellar slices (green circles, n = 10 slices from 5 rats), but not in cerebellar slices from MUT rats (red triangles, n = 14 slices from 6 rats). Each data point represents the average of two successive test responses. The vertical gray bar indicates the period of 1-Hz stimulation. Representative traces depict fEPSPs before (black line) and after (green line for WT and red line for MUT) application of the 1-Hz LTD protocol in WT and MUT slices (left and right inset, respectively) (data are shown as mean ± SEM; **p < 0.01, ***p < 0.001, two-way repeated measures ANOVA with Bonferroni post hoc test [RM ANOVA]). B Baseline and depressed synaptic strength in CF-PC synapses. Graph depicts amplitude of fEPSP evoked from WT slices (green, n=8 from 5 rats) and MUT slices (red, n=12 from 6 rats) by stepwise increase in the stimulus from 5 to 100 μA. Note the decreased baseline CF synaptic strength and lack of LTD in MUT slices after 1-Hz stimulus. WT slices show decreased amplitudes after 1-h LTD (upper left panel, *p < 0.05, two-way RM ANOVA). MUT slices show no changes in amplitudes after 1-h LTD (upper right panel, ns). Comparison of amplitudes between WT and MUT slices before LTD (lower left panel, ***p < 0.001, two-way RM ANOVA) and after LTD (lower left panel, *p<0.05, two-way RM ANOVA). C Changes in the synaptic responses during the 1-Hz stimulation trains. fEPSP amplitude in WT slices (green circles, n=10 slices from 5 rats) did not change significantly compared with MUT slices (red triangles, n=12 slices from 6 rats). Each data point represents one fEPSP response as recorded every second and normalized to the amplitude of first response (data are shown as mean ± SEM, ns, two-way ANOVA, last 100 s). D Average amplitude of the first fEPSP response in WT and MUT slices (data are shown as mean ± SEM, ns, Mann-Whitney test). E Ratio of average amplitude of the last five fEPSP responses (#596–600) divided by the average of the first five responses (#1–5) in WT and MUT slices (data are shown as mean ± SEM, ns, Mann-Whitney test)
Importantly, we tested for any sex-related differences in synaptic plasticity of climbing fibers and found that both male and female MUT rats had similar LTD impairment (Suppl. Fig. 16). Thus, we have combined the data for both sexes in Fig. 9. Sex was not a differentiating factor in WT rat synaptic physiology either, as presented in Suppl. Fig. 17.
These experiments together indicate that the W246G ELOVL4 mutation affected both LTP at PF-Purkinje cell synapses and LTD at CF-Purkinje cell synapses, which are thought to play a central role in cerebellar learning [45, 48, 53]. In view of no changes in the major synaptic signal transduction pathways in this rat model of SCA-34 analyzed so far, further studies should address the specific role of ELOVL4 in cerebellar synaptic plasticity and the cause of ataxia.
Discussion
These studies establish the W246G ELOVL4 knock-in rat as a model of ELOVL4 mutations that cause autosomal dominant SCA34 in humans. Importantly, heterozygous inheritance of the W246G ELOVL4 mutation results in motor deficits, mimicking the autosomal dominant inheritance pattern of human SCA34. These studies also demonstrate that homozygous inheritance of SCA34-causing mutations in ELOVL4 leads to motor impairment similar to that in heterozygotes, rather than the much more severe neuro-ichthyosis that arises from homozygous inheritance of STGD3-causing mutations in exon 6 [3] or recessive ELOVL4 mutations that cause early truncation of the protein [18, 19]. Our electrophysiological studies reveal severe impairment of long-term synaptic plasticity at PF and CF inputs to Purkinje cells, in agreement with our previous findings that deletion of ELOVL4 alters synaptic transmission in hippocampal neurons [3]. Neuroanatomical studies indicate that motor and synaptic impairments arise prior to any significant neurodegeneration, suggesting that SCA34 originates from synaptic dysfunction rather than neurodegeneration.
Motor Impairment Does Not Arise from Disrupted Cerebellar Cytoarchitecture or Early Neurodegeneration
Motor impairment is the hallmark of SCA34. Motor learning deficits were evident in HET and MUT rats on the rotarod test, consistent with ataxia observed in human SCA34 patients. However, the appearance of these deficits by 2 months of age was unexpected as human patients with the W246G ELOVL4 mutation have an average age of disease onset ranging from the third to sixth decade [16], similar to the SCA34-causing L168F and I171T mutations in ELOVL4 [14, 17]. However, other SCA34-causing extremely rare mutations (Q180P, T233M) lead to earlier disease onset in the mid-teens or early twenties [12, 13].
Morphometric measurements in the cerebellar cortex showed no evidence of atrophy by six months of age, well after the appearance of motor deficits in these animals. Similarly, extensive morphological and immunolabeling analyses revealed no gross disruptions in the layering, cellular organization, or synaptic organization of the cerebellum in HET or MUT rats out to at least 6 months of age. These studies provide strong evidence that the motor and synaptic impairments observed in HET and MUT rats precede any substantial neurodegeneration. Early functional impairment prior to neurodegeneration has been reported in a mouse model of SCA38 with homozygous knockout of ELOVL5, with motor impairment appearing by 3 months of age, followed by thinning of the cerebellar cortex by 6 months of age, without Purkinje cell loss out to at least 18 months of age [56]. Early appearance of motor impairment prior to neurodegeneration has also been reported in animal models of SCA6 [57, 58].
Relationship of ELOVL4 Function to Functional Synaptic Impairment
The different diseases caused by the various Elovl4 mutations are likely to arise from aberrant synthesis of its VLC-SFA and VLC-PUFA products. Previous studies of the STGD3-causing 5 base-pair deletion of Elovl4 [9, 59] showed that this mutation impairs synthesis of both VLC-SFA in skin [30,31,32,33] and VLC-PUFA in retina [32, 60, 61]. Our previous lipid analyses of baboon hippocampus showed that the 28:0 and 30:0 VLC-SFA products of ELOVL4 are incorporated into complex sphingolipids and are enriched selectively in synaptic vesicles [3]. No VLC-PUFA were detected in these studies. Studies performed using S+Elovl4mut/mut mice (5 bp deletion that causes STGD3) that express WT Elovl4 in the skin to prevent neonatal lethality showed that 28:0 and 30:0 VLC-SFA are key modulators of hippocampal synaptic function [3]. Homozygous S+Elovl4mut/mut mice developed intractable seizures by postnatal day 18 and died by postnatal day 21 [3], suggesting abnormal neuronal activity. This phenotype is strikingly similar to human patients homozygous for recessive ELOVL4 mutations that cause ELOVL4-neuroichthyosis [18, 19]. Electrophysiological recording of hippocampal slices from S+Elovl4mut/mut mice showed bursting, epileptiform spontaneous activity [3]. Hippocampal neurons cultured from S+Elovl4mut/mut mice showed accelerated presynaptic release kinetics that could be restored to WT kinetics by supplementation of the culture medium with VLC-SFA (28:0 + 30:0) [3]. Together, these studies establish that the VLC-SFA products of ELOVL4 can modulate synaptic function.
In contrast, our recent study of the W246G ELOVL4 mutation showed that this SCA34-causing mutation selectively impaired synthesis of VLC-SFA, but not VLC-PUFA [34]. Specifically, the W246G ELOVL4 mutation reduced, but did not eliminate, synthesis of the major VLC-SFA species (28:0 and 30:0) in the skin of MUT rats compared with WT and HET rats, consistent with EKV in MUT rats. In contrast, the W246G ELOVL4 mutation did not affect VLC-PUFA levels in the retina, consistent with the absence of retinal degeneration in HET and MUT rats. Importantly, we have shown here that maximal synaptic strengths of both PF and CF are reduced in SCA34 MUT cerebellum compared with WT synaptic connections (Figs. 8 and 9, before induction input/output curves), which mirrors the change in photopic b-wave in the retina of MUT rats [34]. These findings suggest that selective decrease of VLC-SFA synthesis by the W246G ELOVL4 mutation may contribute to SCA34. Homozygous inheritance of recessive ELOVL4 alleles that would lead to a severely truncated and presumably inactive ELOVL4 protein results in a much more severe neuro-ichthyotic disease characterized by seizures, spasticity, intellectual disability, ichthyosis, and early death [18, 19]. Similarly, homozygous inheritance of the 5 base-pair deletion, which would severely impair synthesis of both VLC-SFA and VLC-PUFA, leads to severe seizures [3] similar to the ELOVL4 neuro-ichthyotic syndrome.
The W246G ELOVL4 Mutation Disrupts Synaptic Plasticity at Excitatory Synapses onto Purkinje Cells
The finding that the W246G ELOVL4 mutation did not lead to gross disruption of cerebellar architecture or neurodegeneration suggests that the causal insult in SCA34 is likely to be a functional synaptopathy. An early functional synaptopathy also would be consistent with our early-onset motor deficits in the absence of structural disruption of cerebellar architecture or evidence of neurodegeneration. Cerebellar slice recordings from MUT rats showed that the W246G ELOVL4 mutation decreased transmission from PFs and CFs to Purkinje cells only moderately, but severely altered plasticity of these synapses in the cerebellum. Proper synaptic strength regulation is strikingly diminished in MUT cerebellar slices; neither PF LTP nor CF LTD could be evoked there. Moreover, disruption of cerebellar cortical synchronization, especially at higher frequencies, was combined with run-away focal PF activity only in MUT slices (Fig. 7). These changes were accompanied by a downregulation of delta band activity in the PF pathway and diminished gamma activity above 20 Hz in cerebellar circuits in the MUT rats. It is noteworthy that cerebellar delta band activity can modulate prefrontal cortical execution of motor task planning [62]. The output from Purkinje cells (especially in Crus 1) is also known to represent the phase difference of prefrontal cortical and hippocampal oscillations [63]. Interestingly, theta band activity, known to increase precise millisecond timing of PC firing independently of inhibitory inputs [64, 65], was spared in the MUT cerebellum.
Based on these findings, we suggest that the W246G ELOVL4 mutation is likely to cause synaptic dysfunction that leads to the development of the ataxia and other CNS symptoms characteristic of SCA34. Aberrant presynaptic release from PFs and CFs could disrupt the critical synchrony of signaling between these two synaptic populations, impairing motor learning and output. Alternatively, if the inhibitory input to Purkinje cells is altered relative to PF synaptic strength, it could then cause the observed pattern of cortical desynchronization, similarly to the serious breakdown of interfocal neighborhood interactions as originally predicted by Pellionisz and Szentagothai [66].
Plasticity at CF-Purkinje cell synapses is well characterized and the changes to CF-evoked complex spikes provide critical components of cerebellar learning [67]. Our results showed that CF stimulation at 1 Hz produced robust LTD in cerebellar slices from WT rats, but no induction of LTD in slices from MUT rats. Climbing fiber stimulation alone can induce postsynaptic LTD, which requires post-synaptic activation of mGluR1, protein kinase Cɣ (PKCɣ), and Ca2+ influx [55, 67, 68], and in turn leads to reduced numbers of GluA2-containing AMPA receptors in the postsynaptic membrane [68] and decreased amplitude of the synaptic response. The elimination of LTD by the W246G ELOVL4 mutation is strikingly similar to the elimination of LTD reported in a number of mouse strains with knockout or loss-of-function mutations in postsynaptic mGluR1, its downstream signaling molecules PKCɣ, phospholipase C-β4, PICK1, or the GluA2-containing AMPA receptor [45, 69,70,71,72,73]. Furthermore, mutations in PKCɣ and mGluR1 are known to cause human SCA14 [74, 75] and SCA44 [76]. As we have not detected changes in mGluR1 expression pattern (Fig. 5), downstream disruption in the mGluR1 signaling pathway is a leading candidate to cause the absence of CF LTD in MUT rat cerebellum. One obvious signal pathway involved in this effect is the PLC-DAG-PKC pathway. Interestingly, there were no differences in expression levels of the PKC gamma in WT, HET, and MUT rats (Suppl. Fig. 14). The other branch of this signaling pathway causes the release of calcium from internal calcium stores via inositol-(1,4,5)-tris phosphate (IP3), but there was no difference in the cerebellar expression pattern or levels of the IP3 receptors in WT, HET, and MUT rats (Suppl. Fig. 15). Other SCA models show similar CF deficits and disruption of glutamate transmission, leading to PC dysfunction [77]. Because CF LTD induction depends on an increase of postsynaptic Ca2+, a reduced pool of available Ca2+ in MUT cerebellum also could result in failure of LTD induction. Alterations in Ca2+ transients could have important functional roles [55, 78], as CF LTD decreases CF-evoked Ca2+ transients in Purkinje cells [78]. Thus, reduced Ca2+ transients in MUT rats could also play a role in the failure of LTD induction. Similar threshold mechanisms for Ca2+ have been widely reported in hippocampus and neocortex, where increased Ca2+ influx leads to LTP and reduced Ca2+ levels induce LTD. Interestingly, haploinsufficiency in CAMTA1, a member of the calmodulin-binding transcription activators, results in early-onset ataxia in humans [79,80,81], and nervous system deletion of CAMTA1 in mice causes severe ataxia with Purkinje cell degeneration and cerebellar atrophy [82].
We focused our studies on LTP of PF, as previously it was reported that blockade of PF LTD by T-588, a calcium release blocker [83], did not impair motor learning on rotarod [84]. Repetitive stimulation of PFs at low frequency similar to the stimuli used in this study evokes postsynaptic LTP at PF-Purkinje cell synapses by increasing postsynaptic AMPA receptor numbers via inhibition of AMPA receptor-PICK1 interaction to potentiate the postsynaptic response [85]. Induction of postsynaptic LTP at PF-Purkinje cell synapses requires low levels of postsynaptic Ca2+and protein phosphatase activation [86, 87]. However, it was reported that Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning, but induction of PF LTD was not affected [87]. It is suggested that motor learning results from the interaction of both LTP and LTD, and they originate from different cellular layers [88]. We see in our studies that the MUT rats show motor deficits and absence of LTP or LTD along with smaller evoked responses.
Further studies are needed to address effect of ELOVL4 mutations on AMPA receptor trafficking in Purkinje cells, and other forms of cerebellar synaptic plasticity as pure presynaptic LTP of PF [89] or mGLuR1-dependent PF plasticity of long-latency patches organized in parasagittal bands [90]. Remarkably, a mouse model of SCA type 2 caused by polyglutamine expansion in ataxin-2 had deficiency of C24/26-sphingomyelins contrasted with excess C18/20-sphingomyelin [91]. These changes in sphingomyelins were caused mostly by decreased levels of Elovl4 expression in the cerebellum [91]. This suggests that impaired VLC-SFA synthesis may be a significant factor in other forms of SCAs, even when there is no ELOVL4 mutation detected.
In summary, we have generated a rat model of SCA34 that recapitulates the phenotype found in humans. The animals develop ataxia and show impaired synaptic function in CF and PF inputs into Purkinje cells. In turn, the impaired synaptic plasticity at these two major excitatory inputs to Purkinje cells degrades the operation of cerebellar cortex as an effective forward controller of movements [88]. These synaptic functional changes occur in the absence of overt anatomical changes or of detectable changes in major synaptic protein expression levels in the cerebellum and prior to neurodegeneration, suggesting that the phenotype originates as a synaptic plasticity deficit that likely also causes ataxia as observed in SCA34 patients.
Conclusion
Different ELOVL4 mutations cause different tissue-specific disorders in humans. Here we report the first animal model of SCA34 caused by W246G ELOVL4 mutation that led to age-related impaired synaptic and motor function, prior to the onset of overt neurodegeneration and cerebellar atrophy. Our results suggest that in SCA34 patients, neuronal deficits likely arise from the impaired ability of W246G ELOVL4 to synthesize VLC-SFA, which we have previously shown to be enriched in synaptic vesicles and are necessary for modulating presynaptic release kinetics. Taken together, our findings suggest that onset of ataxia in SCA34 is associated with a synaptopathy present from early ages that over time leads to cerebellar degeneration and the severe motor impairments that characterize the disease. These findings support the essential role of ELOVL4 and its VLC-FA products in physiological neuronal function.
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files). Any other information needed on the datasets used and/or analyzed during the current study is available from the corresponding author on reasonable request.
Abbreviations
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CF:
-
climbing fibers
- EKV:
-
erythrokeratodermia variabilis
- ELOVL4:
-
fatty acid elongase-4 or elongation of very long chain fatty acids-4
- fEPSP:
-
field excitatory postsynaptic potential
- GABA :
-
γ-aminobutyric acid
- GAD65/67 :
-
glutamate decarboxylase-65/67
- GFAP :
-
glial fibrillary acidic protein
- IP3:
-
inositol-(1,4,5)-tris phosphate
- KI:
-
knock-in
- LTD:
-
long-term depression
- LTP:
-
long-term potentiation
- MEA:
-
multi-electrode array
- mGluR1:
-
metabotropic glutamate receptor subtype 1
- PF:
-
parallel fibers
- PSD-95:
-
postsynaptic density protein-95
- SCA34:
-
autosomal dominant spinocerebellar ataxia-34
- STGD3:
-
autosomal dominant Stargardt-like macular dystrophy
- SYT2:
-
synaptotagmin 2
- VGluT1/2 :
-
vesicular glutamate transporter (VGLUT)/(VGLUT2)
- VLC-FA:
-
very long chain fatty acids
- VLC-PUFA:
-
very long chain polyunsaturated fatty acids
- VLC-SFA:
-
very long chain saturated fatty acids
References
Agbaga MP, Brush RS, Mandal MN, Henry K, Elliott MH, Anderson RE (2008) Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc Natl Acad Sci U S A 105(35):12843–12848. https://doi.org/10.1073/pnas.0802607105
Agbaga MP, Mandal MN, Anderson RE (2010) Retinal very long-chain PUFAs: new insights from studies on ELOVL4 protein. J Lipid Res 51(7):1624–1642. https://doi.org/10.1194/jlr.R005025
Hopiavuori BR, Deak F, Wilkerson JL, Brush RS, Rocha-Hopiavuori NA, Hopiavuori AR, Ozan KG, Sullivan MT et al (2018) Homozygous expression of mutant ELOVL4 leads to seizures and death in a novel animal model of very long-chain fatty acid deficiency. Mol Neurobiol 55(2):1795–1813. https://doi.org/10.1007/s12035-017-0824-8
Mandal MN, Ambasudhan R, Wong PW, Gage PJ, Sieving PA, Ayyagari R (2004) Characterization of mouse orthologue of ELOVL4: genomic organization and spatial and temporal expression. Genomics 83(4):626–635. https://doi.org/10.1016/j.ygeno.2003.09.020
McMahon A, Lu H, Butovich IA (2014) A role for ELOVL4 in the mouse meibomian gland and sebocyte cell biology. Invest Ophthalmol Vis Sci 55(5):2832–2840. https://doi.org/10.1167/iovs.13-13335
Poulos A, Johnson DW, Beckman K, White IG, Easton C (1987) Occurrence of unusual molecular species of sphingomyelin containing 28-34-carbon polyenoic fatty acids in ram spermatozoa. Biochem J 248(3):961–964. https://doi.org/10.1042/bj2480961
Deák F, Anderson RE, Fessler JL, Sherry DM (2019) Novel cellular functions of very long chain-fatty acids: insight from ELOVL4 mutations. Front Cell Neurosci 13:428. https://doi.org/10.3389/fncel.2019.00428
Bardak H, Gunay M, Ercalik Y, Bardak Y, Ozbas H, Bagci O, Ayata A, Sonmez M et al (2016) Analysis of ELOVL4 and PRPH2 genes in Turkish Stargardt disease patients. Genet Mol Res 15(4). https://doi.org/10.4238/gmr15048774
Bernstein PS, Tammur J, Singh N, Hutchinson A, Dixon M, Pappas CM, Zabriskie NA, Zhang K et al (2001) Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest Ophthalmol Vis Sci 42(13):3331–3336
Edwards AO, Donoso LA, Ritter R 3rd (2001) A novel gene for autosomal dominant Stargardt-like macular dystrophy with homology to the SUR4 protein family. Invest Ophthalmol Vis Sci 42(11):2652–2663
Maugeri A, Meire F, Hoyng CB, Vink C, Van Regemorter N, Karan G, Yang Z, Cremers FP et al (2004) A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Invest Ophthalmol Vis Sci 45(12):4263–4267. https://doi.org/10.1167/iovs.04-0078
Bourassa CV, Raskin S, Serafini S, Teive HA, Dion PA, Rouleau GA (2015) A new ELOVL4 mutation in a case of spinocerebellar ataxia with erythrokeratodermia. JAMA Neurol 72(8):942–943. https://doi.org/10.1001/jamaneurol.2015.0888
Bourque PR, Warman-Chardon J, Lelli DA, LaBerge L, Kirshen C, Bradshaw SH, Hartley T, Boycott KM (2018) Novel ELOVL4 mutation associated with erythrokeratodermia and spinocerebellar ataxia (SCA 34). Neurol Genet 4(4):e263. https://doi.org/10.1212/NXG.0000000000000263
Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, Martin C, Meloche C, Gravel M, Drouin CA, Rouleau GA et al (2014) Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol 71(4):470–475. https://doi.org/10.1001/jamaneurol.2013.6337
Ozaki K, Ansai A, Nobuhara K, Araki T, Kubodera T, Ishii T, Higashi M, Sato N et al (2019) Prevalence and clinicoradiological features of spinocerebellar ataxia type 34 in a Japanese ataxia cohort. Parkinsonism Relat Disord 65:238–242. https://doi.org/10.1016/j.parkreldis.2019.05.019
Ozaki K, Doi H, Mitsui J, Sato N, Iikuni Y, Majima T, Yamane K, Irioka T et al (2015) A novel mutation in ELOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a broadened spectrum of SCA34. JAMA Neurol 72(7):797–805. https://doi.org/10.1001/jamaneurol.2015.0610
Xiao C, MB E, Rexach J, Knight-Johnson A, Khemani P, Fogel BL, Das S, MS E et al (2019) A family with spinocerebellar ataxia and retinitis pigmentosa attributed to an ELOVL4 mutation. Neurol Genet 5(5):e357. https://doi.org/10.1212/NXG.0000000000000357
Aldahmesh MA, Mohamed JY, Alkuraya HS, Verma IC, Puri RD, Alaiya AA, Rizzo WB, Alkuraya FS (2011) Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am J Hum Genet 89(6):745–750. https://doi.org/10.1016/j.ajhg.2011.10.011
Mir H, Raza SI, Touseef M, Memon MM, Khan MN, Jaffar S, Ahmad W (2014) A novel recessive mutation in the gene ELOVL4 causes a neuro-ichthyotic disorder with variable expressivity. BMC Med Genet 15:25. https://doi.org/10.1186/1471-2350-15-25
Brush RS, Tran JT, Henry KR, McClellan ME, Elliott MH, Mandal MN (2010) Retinal sphingolipids and their very-long-chain fatty acid-containing species. Invest Ophthalmol Vis Sci 51(9):4422–4431. https://doi.org/10.1167/iovs.09-5134
Poulos A, Sharp P, Johnson D, Easton C (1988) The occurrence of polyenoic very long chain fatty acids with greater than 32 carbon atoms in molecular species of phosphatidylcholine in normal and peroxisome-deficient (Zellweger's syndrome) brain. Biochem J 253(3):645–650. https://doi.org/10.1042/bj2530645
Robinson BS, Johnson DW, Poulos A (1990) Unique molecular species of phosphatidylcholine containing very-long-chain (C24-C38) polyenoic fatty acids in rat brain. Biochem J 265(3):763–767. https://doi.org/10.1042/bj2650763
Agbaga MP, Merriman DK, Brush RS, Lydic TA, Conley SM, Naash MI, Jackson S, Woods AS et al (2018) Differential composition of DHA and very-long-chain PUFAs in rod and cone photoreceptors. J Lipid Res 59(9):1586–1596. https://doi.org/10.1194/jlr.M082495
Aveldano MI (1987) A novel group of very long chain polyenoic fatty acids in dipolyunsaturated phosphatidylcholines from vertebrate retina. J Biol Chem 262(3):1172–1179
Aveldano MI, Sprecher H (1987) Very long chain (C24 to C36) polyenoic fatty acids of the n-3 and n-6 series in dipolyunsaturated phosphatidylcholines from bovine retina. J Biol Chem 262(3):1180–1186
Rotstein NP, Pennacchiotti GL, Sprecher H, Aveldano MI (1996) Active synthesis of C24:5, n-3 fatty acid in retina. Biochem J 316(Pt 3):859–864. https://doi.org/10.1042/bj3160859
Rabionet M, Bayerle A, Jennemann R, Heid H, Fuchser J, Marsching C, Porubsky S, Bolenz C et al (2015) Male meiotic cytokinesis requires ceramide synthase 3-dependent sphingolipids with unique membrane anchors. Hum Mol Genet 24(17):4792–4808. https://doi.org/10.1093/hmg/ddv204
Rabionet M, van der Spoel AC, Chuang CC, von Tumpling-Radosta B, Litjens M, Bouwmeester D, Hellbusch CC, Korner C et al (2008) Male germ cells require polyenoic sphingolipids with complex glycosylation for completion of meiosis: a link to ceramide synthase-3. J Biol Chem 283(19):13357–13369. https://doi.org/10.1074/jbc.M800870200
Santiago Valtierra FX, Penalva DA, Luquez JM, Furland NE, Vasquez C, Reyes JG, Aveldano MI, Oresti GM (2018) Elovl4 and Fa2h expression during rat spermatogenesis: a link to the very-long-chain PUFAs typical of germ cell sphingolipids. J Lipid Res 59(7):1175–1189. https://doi.org/10.1194/jlr.M081885
Cameron DJ, Tong Z, Yang Z, Kaminoh J, Kamiyah S, Chen H, Zeng J, Chen Y et al (2007) Essential role of Elovl4 in very long chain fatty acid synthesis, skin permeability barrier function, and neonatal survival. Int J Biol Sci 3(2):111–119. https://doi.org/10.7150/ijbs.3.111
Li W, Sandhoff R, Kono M, Zerfas P, Hoffmann V, Ding BC, Proia RL, Deng CX (2007) Depletion of ceramides with very long chain fatty acids causes defective skin permeability barrier function, and neonatal lethality in ELOVL4 deficient mice. Int J Biol Sci 3(2):120–128. https://doi.org/10.7150/ijbs.3.120
McMahon A, Butovich IA, Mata NL, Klein M, Ritter R 3rd, Richardson J, Birch DG, Edwards AO et al (2007) Retinal pathology and skin barrier defect in mice carrying a Stargardt disease-3 mutation in elongase of very long chain fatty acids-4. Mol Vis 13:258–272
Vasireddy V, Uchida Y, Salem N Jr, Kim SY, Mandal MN, Reddy GB, Bodepudi R, Alderson NL et al (2007) Loss of functional ELOVL4 depletes very long-chain fatty acids (> or =C28) and the unique omega-O-acylceramides in skin leading to neonatal death. Hum Mol Genet 16(5):471–482. https://doi.org/10.1093/hmg/ddl480
Agbaga MP, Stiles MA, Brush RS, Sullivan MT, Machalinski A, Jones KL, Anderson RE, Sherry DM (2020) The Elovl4 spinocerebellar ataxia-34 mutation 736T>G (p.W246G) impairs retinal function in the absence of photoreceptor degeneration. Mol Neurobiol 57:4735–4753. https://doi.org/10.1007/s12035-020-02052-8
Huang M, Verbeek DS (2019) Why do so many genetic insults lead to Purkinje Cell degeneration and spinocerebellar ataxia? Neurosci Lett 688:49–57. https://doi.org/10.1016/j.neulet.2018.02.004
Paulson HL, Shakkottai VG, Clark HB, Orr HT (2017) Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat Rev Neurosci 18(10):613–626. https://doi.org/10.1038/nrn.2017.92
Sherry DM, Hopiavuori BR, Stiles MA, Rahman NS, Ozan KG, Deak F, Agbaga MP, Anderson RE (2017) Distribution of ELOVL4 in the developing and adult mouse brain. Front Neuroanat 11:38. https://doi.org/10.3389/fnana.2017.00038
Kuny S, Cho WJ, Dimopoulos IS, Sauve Y (2015) Early onset ultrastructural and functional defects in RPE and photoreceptors of a Stargardt-like macular dystrophy (STGD3) transgenic mouse model. Invest Ophthalmol Vis Sci 56(12):7109–7121. https://doi.org/10.1167/iovs.15-17567
Schori C, Agbaga MP, Brush RS, Ayyagari R, Grimm C, Samardzija M (2018) Elovl4 5-bp deletion does not accelerate cone photoreceptor degeneration in an all-cone mouse. PLoS One 13(1):e0190514. https://doi.org/10.1371/journal.pone.0190514
Luong TN, Carlisle HJ, Southwell A, Patterson PH (2011) Assessment of motor balance and coordination in mice using the balance beam. J Vis Exp 49. https://doi.org/10.3791/2376
Shiotsuki H, Yoshimi K, Shimo Y, Funayama M, Takamatsu Y, Ikeda K, Takahashi R, Kitazawa S et al (2010) A rotarod test for evaluation of motor skill learning. J Neurosci Methods 189(2):180–185. https://doi.org/10.1016/j.jneumeth.2010.03.026
Brooks SP, Dunnett SB (2009) Tests to assess motor phenotype in mice: a user's guide. Nat Rev Neurosci 10(7):519–529. https://doi.org/10.1038/nrn2652
Robinson KJ, Watchon M, Laird AS (2020) Aberrant cerebellar circuitry in the spinocerebellar ataxias. Front Neurosci 14:707. https://doi.org/10.3389/fnins.2020.00707
Nusser Z, Mulvihill E, Streit P, Somogyi P (1994) Subsynaptic segregation of metabotropic and ionotropic glutamate receptors as revealed by immunogold localization. Neuroscience 61(3):421–427. https://doi.org/10.1016/0306-4522(94)90421-9
Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S (1994) Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 79(2):377–388
Ohtani Y, Miyata M, Hashimoto K, Tabata T, Kishimoto Y, Fukaya M, Kase D, Kassai H et al (2014) The synaptic targeting of mGluR1 by its carboxyl-terminal domain is crucial for cerebellar function. J Neurosci 34(7):2702–2712. https://doi.org/10.1523/jneurosci.3542-13.2014
Feil R, Hartmann J, Luo C, Wolfsgruber W, Schilling K, Feil S, Barski JJ, Meyer M et al (2003) Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. J Cell Biol 163(2):295–302. https://doi.org/10.1083/jcb.200306148
Ito M (2006) Cerebellar circuitry as a neuronal machine. Prog Neurobiol 78(3-5):272–303. https://doi.org/10.1016/j.pneurobio.2006.02.006
Kessler M, Kiliman B, Humes C, Arai AC (2008) Spontaneous activity in Purkinje cells: multi-electrode recording from organotypic cerebellar slice cultures. Brain Res 1218:54–69. https://doi.org/10.1016/j.brainres.2008.04.063
Hansel C, Linden DJ, D’Angelo E (2001) Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci 4(5):467–475. https://doi.org/10.1038/87419
Chen C, Thompson RF (1995) Temporal specificity of long-term depression in parallel fiber--Purkinje synapses in rat cerebellar slice. Learning & memory (Cold Spring Harbor, NY) 2(3-4):185–198. https://doi.org/10.1101/lm.2.3-4.185
Lev-Ram V, Wong ST, Storm DR, Tsien RY (2002) A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci U S A 99(12):8389–8393. https://doi.org/10.1073/pnas.122206399
Ito M (1982) Experimental verification of Marr-Albus plasticity assumption for the cerebellum. Acta Biol Acad Sci Hung 33(2-3):189–199
van Woerden GM, Hoebeek FE, Gao Z, Nagaraja RY, Hoogenraad CC, Kushner SA, Hansel C, De Zeeuw CI et al (2009) betaCaMKII controls the direction of plasticity at parallel fiber-Purkinje cell synapses. Nat Neurosci 12(7):823–825. https://doi.org/10.1038/nn.2329
Hansel C, Linden DJ (2000) Long-term depression of the cerebellar climbing fiber--Purkinje neuron synapse. Neuron 26(2):473–482
Hoxha E, Gabriele RMC, Balbo I, Ravera F, Masante L, Zambelli V, Albergo C, Mitro N et al (2017) Motor deficits and cerebellar atrophy in Elovl5 knock out mice. Front Cell Neurosci 11:343. https://doi.org/10.3389/fncel.2017.00343
Jayabal S, Ljungberg L, Erwes T, Cormier A, Quilez S, El Jaouhari S, Watt AJ (2015) Rapid onset of motor deficits in a mouse model of spinocerebellar ataxia type 6 precedes late cerebellar degeneration. eNeuro 2(6):ENEURO.0094–ENEU15.2015. https://doi.org/10.1523/ENEURO.0094-15.2015
Unno T, Wakamori M, Koike M, Uchiyama Y, Ishikawa K, Kubota H, Yoshida T, Sasakawa H et al (2012) Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc Natl Acad Sci U S A 109(43):17693–17698. https://doi.org/10.1073/pnas.1212786109
Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML et al (2001) A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet 27(1):89–93. https://doi.org/10.1038/83817
Mandal NA, Tran JT, Zheng L, Wilkerson JL, Brush RS, McRae J, Agbaga MP, Zhang K et al (2014) In vivo effect of mutant ELOVL4 on the expression and function of wild-type ELOVL4. Invest Ophthalmol Vis Sci 55(4):2705–2713. https://doi.org/10.1167/iovs.13-13198
Logan S, Agbaga MP, Chan MD, Kabir N, Mandal NA, Brush RS, Anderson RE (2013) Deciphering mutant ELOVL4 activity in autosomal-dominant Stargardt macular dystrophy. Proc Natl Acad Sci U S A 110(14):5446–5451. https://doi.org/10.1073/pnas.1217251110
Parker KL, Kim YC, Kelley RM, Nessler AJ, Chen KH, Muller-Ewald VA, Andreasen NC, Narayanan NS (2017) Delta-frequency stimulation of cerebellar projections can compensate for schizophrenia-related medial frontal dysfunction. Mol Psychiatry 22(5):647–655. https://doi.org/10.1038/mp.2017.50
McAfee SS, Liu Y, Sillitoe RV, Heck DH (2019) Cerebellar lobulus simplex and Crus I differentially represent phase and phase difference of prefrontal cortical and hippocampal oscillations. Cell Rep 27(8):2328–2334.e2323. https://doi.org/10.1016/j.celrep.2019.04.085
De Zeeuw CI, Hoebeek FE, Schonewille M (2008) Causes and consequences of oscillations in the cerebellar cortex. Neuron 58(5):655–658. https://doi.org/10.1016/j.neuron.2008.05.019
Gandolfi D, Lombardo P, Mapelli J, Solinas S, D'Angelo E (2013) θ-Frequency resonance at the cerebellum input stage improves spike timing on the millisecond time-scale. Front Neural Circuits 7:64. https://doi.org/10.3389/fncir.2013.00064
Pellionisz A, Szentàgothai J (1974) Dynamic single unit simulation of a realistic cerebellar network model. II Purkinje cell activity within the basic circuit and modified by inhibitory systems. Brain Res 68(1):19–40. https://doi.org/10.1016/0006-8993(74)90531-9
Ohtsuki G, Piochon C, Hansel C (2009) Climbing fiber signaling and cerebellar gain control. Front Cell Neurosci 3:4. https://doi.org/10.3389/neuro.03.004.2009
Shen Y, Hansel C, Linden DJ (2002) Glutamate release during LTD at cerebellar climbing fiber-Purkinje cell synapses. Nat Neurosci 5(8):725–726. https://doi.org/10.1038/nn895
Hashimoto K, Miyata M, Watanabe M, Kano M (2001) Roles of phospholipase Cbeta4 in synapse elimination and plasticity in developing and mature cerebellum. Mol Neurobiol 23(1):69–82. https://doi.org/10.1385/MN:23:1:69
Ichise T, Kano M, Hashimoto K, Yanagihara D, Nakao K, Shigemoto R, Katsuki M, Aiba A (2000) mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science 288(5472):1832–1835. https://doi.org/10.1126/science.288.5472.1832
Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S (1997) Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron 18(1):71–79. https://doi.org/10.1016/s0896-6273(01)80047-7
Shuvaev AN, Horiuchi H, Seki T, Goenawan H, Irie T, Iizuka A, Sakai N, Hirai H (2011) Mutant PKCgamma in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J Neurosci 31(40):14324–14334. https://doi.org/10.1523/JNEUROSCI.5530-10.2011
Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM et al (2006) Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron 49(6):845–860. https://doi.org/10.1016/j.neuron.2006.02.025
Chen DH, Brkanac Z, Verlinde CL, Tan XJ, Bylenok L, Nochlin D, Matsushita M, Lipe H et al (2003) Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet 72(4):839–849. https://doi.org/10.1086/373883
van de Warrenburg BP, Verbeek DS, Piersma SJ, Hennekam FA, Pearson PL, Knoers NV, Kremer HP, Sinke RJ (2003) Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurology 61(12):1760–1765. https://doi.org/10.1212/01.wnl.0000098883.79421.73
Watson LM, Bamber E, Schnekenberg RP, Williams J, Bettencourt C, Lickiss J, Jayawant S, Fawcett K et al (2017) Dominant mutations in GRM1 cause spinocerebellar ataxia type 44. Am J Hum Genet 101(5):866. https://doi.org/10.1016/j.ajhg.2017.10.008
Smeets CJ, Verbeek DS (2016) Climbing fibers in spinocerebellar ataxia: a mechanism for the loss of motor control. Neurobiol Dis 88:96–106. https://doi.org/10.1016/j.nbd.2016.01.009
Weber JT, De Zeeuw CI, Linden DJ, Hansel C (2003) Long-term depression of climbing fiber-evoked calcium transients in Purkinje cell dendrites. Proc Natl Acad Sci U S A 100(5):2878–2883. https://doi.org/10.1073/pnas.0536420100
Huentelman MJ, Papassotiropoulos A, Craig DW, Hoerndli FJ, Pearson JV, Huynh KD, Corneveaux J, Hänggi J et al (2007) Calmodulin-binding transcription activator 1 (CAMTA1) alleles predispose human episodic memory performance. Hum Mol Genet 16(12):1469–1477. https://doi.org/10.1093/hmg/ddm097
Thevenon J, Lopez E, Keren B, Heron D, Mignot C, Altuzarra C, Béri-Dexheimer M, Bonnet C et al (2012) Intragenic CAMTA1 rearrangements cause non-progressive congenital ataxia with or without intellectual disability. J Med Genet 49(6):400–408. https://doi.org/10.1136/jmedgenet-2012-100856
Magnin E, Blagosklonov O, Sylvestre G, Minot D, Thevenon J, Faivre L, Boulahdour H, Thauvin-Robinet C et al (2014) Neuropsychological and neuroimaging phenotype induced by a CAMTA1 mutation. Brain Dev 36(8):711–715. https://doi.org/10.1016/j.braindev.2013.09.008
Long C, Grueter CE, Song K, Qin S, Qi X, Kong YM, Shelton JM, Richardson JA et al (2014) Ataxia and Purkinje cell degeneration in mice lacking the CAMTA1 transcription factor. Proc Natl Acad Sci U S A 111(31):11521–11526. https://doi.org/10.1073/pnas.1411251111
Kimura T, Sugimori M, Llinás RR (2005) Purkinje cell long-term depression is prevented by T-588, a neuroprotective compound that reduces cytosolic calcium release from intracellular stores. Proc Natl Acad Sci U S A 102(47):17160–17165. https://doi.org/10.1073/pnas.0508190102
Welsh JP, Yamaguchi H, Zeng XH, Kojo M, Nakada Y, Takagi A, Sugimori M, Llinás RR (2005) Normal motor learning during pharmacological prevention of Purkinje cell long-term depression. Proc Natl Acad Sci U S A 102(47):17166–17171. https://doi.org/10.1073/pnas.0508191102
Anggono V, Huganir RL (2012) Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol 22(3):461–469. https://doi.org/10.1016/j.conb.2011.12.006
Belmeguenai A, Hosy E, Bengtsson F, Pedroarena CM, Piochon C, Teuling E, He Q, Ohtsuki G et al (2010) Intrinsic plasticity complements long-term potentiation in parallel fiber input gain control in cerebellar Purkinje cells. J Neurosci 30(41):13630–13643. https://doi.org/10.1523/JNEUROSCI.3226-10.2010
Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, Gao Z, Badura A et al (2010) Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron 67(4):618–628. https://doi.org/10.1016/j.neuron.2010.07.009
Hoxha E, Tempia F, Lippiello P, Miniaci MC (2016) Modulation, plasticity and pathophysiology of the parallel fiber-Purkinje cell synapse. Frontiers in synaptic neuroscience 8:35. https://doi.org/10.3389/fnsyn.2016.00035
van Beugen BJ, Nagaraja RY, Hansel C (2006) Climbing fiber-evoked endocannabinoid signaling heterosynaptically suppresses presynaptic cerebellar long-term potentiation. J Neurosci 26(32):8289–8294. https://doi.org/10.1523/jneurosci.0805-06.2006
Wang X, Chen G, Gao W, Ebner TJ (2011) Parasagittally aligned, mGluR1-dependent patches are evoked at long latencies by parallel fiber stimulation in the mouse cerebellar cortex in vivo. J Neurophysiol 105(4):1732–1746. https://doi.org/10.1152/jn.00717.2010
Sen NE, Arsovic A, Meierhofer D, Brodesser S, Oberschmidt C, Canet-Pons J, Kaya ZE, Halbach MV et al (2019) In human and mouse spino-cerebellar tissue, ataxin-2 expansion affects ceramide-sphingomyelin metabolism. Int J Mol Sci 20(23). https://doi.org/10.3390/ijms20235854
Trevarrow B, Marks DL, Kimmel CB (1990) Organization of hindbrain segments in the zebrafish embryo. Neuron 4(5):669–679. https://doi.org/10.1016/0896-6273(90)90194-k
Acknowledgements
The monoclonal antibodies against VGluT1 (clone N28/9), GluA1 (clone N355/1), GluA2 (clone L21/32), and PSD-95 (clone 75-028) were developed by and/or obtained from the UC Davis/NIH NeuroMab Facility, supported by NIH grant U24NS050606 and maintained by the Department of Neurobiology, Physiology and Behavior, College of Biological Sciences, University of California, Davis, CA 95616. The synaptotagmin 2 monoclonal antibody generated by Trevarrow et al. [92] was obtained from the Zebrafish International Resource Center, which is supported by grant P40RR012546 from the NIH-NCRR. We thank Drs. Anna Csiszar, Tamas Kiss, and Zoltan Ungvari and the Oklahoma Nathan Shock Center for advice on the behavioral assessment. We are grateful to Dr. Blake Hopiavuori and Nicole A. Rocha-Hopiavuori for the programming help with data extraction and analysis of the spontaneous cerebellar network activity.
Funding
Supported by a Multi-PI Team Science grant from the Presbyterian Health Foundation (DMS, REA, and MPA). NIH grants R01EY04149, P30EY021725, and R21NS090117 (REA) and unrestricted grant support from Research to Prevent Blindness to Dr. Gregory Skuta at the Dean McGee Eye Institute. NIH grants R01 EY030513 and R21 AR076035 and the Oklahoma Center for Advancement of Science and Technology (OCAST) (MPA). Research reported in this publication was supported in part by 1P20GM125528 “Cellular and Molecular GeroScience CoBRE” from the National Institute of General Medical Sciences, 3P30AG050911-04S1, NIH grant R01 AG062655 (FD) and a supplement grant to Oklahoma Nathan Shock Center (FD), and by an Oklahoma Center for Adult Stem Cell Research shared equipment grant (DMS).
Author information
Authors and Affiliations
Contributions
RYN, AO, DMS, REA, M-PA, and FD designed the research; RYN, AO, DMS, JLF, MAS, FL, KM, MA, and RSB performed the research and analyzed the data; RYN, DMS, REA, M-PA, and FD wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Additional information
The human ELOVL4 and rodent Elovl4 genes are in written in italics, respectively, while the protein for all species is represented as ELOVL4.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Raghavendra Y. Nagaraja and David M. Sherry are co-first authors.
Supplementary Information
Supplemental Fig. 1.
Generation and characterization of c.736 T>G, p.W246G knock-in Long Evans rat using the CRISPR/Cas9 system. a. Schematic depiction of targeting strategy. The genomic region of rat Elovl4 locus is diagrammed (gene is oriented from left to right; total size is 28.21 kb). Solid bars represent open reading frame (exons); open bars represent untranslated regions. The sgRNA cut site and the single strand oligonucleotide donor sequencing with homology arms and the mutation site is shown by the red arrow. b. Genotyping of WT, HET, and MUT SCA34-KI rats by StyI restriction analysis. 704 bp PCR products were generated from WT, HET, and MUT rats using PCR primers. The amplicons were run undigested or purified and digested with StyI restriction enzyme. Left panel: Undigested amplicons ran at 704 bp for WT, HET, and MUT rats, as expected. Right panel: Digestion with StyI restriction enzyme showed the WT PCR product is resistant to Sty I digestion. In contrast, HET rats show two bands, one corresponding to the WT PCR product and a smaller, digested fragment arising from the mutant PCR product containing the StyI digestion site. MUT rats show only a single band corresponding to the digested, mutant PCR product with no WT PCR product, as expected. (From: Agbaga et al., 2020[34]). (PNG 1092 kb)
Supplemental Fig. 2.
Sanger sequencing confirms appropriate gene editing. Sequencing from the 5’-3 primer direction (left to right on the figure), Sanger DNA sequencing of WT, HET, and MUT rat DNA sequences confirms the single point mutation c.736 T>G in the rat Elovl4 genome. Box and arrows indicate site of gene editing. a. MUT. b HET. c. WT. (From: Agbaga et al., 2020[34]). (PNG 1558 kb)
Supplemental Fig. 3.
Whole genome sequence analysis. Reading from the 3’-5 primer direction (right to left on the figure), whole genome sequencing of WT and MUT rats confirms knockin of the 736 T>G, p.W246G mutant Elovl4 without any major off target effects in MUT rats (MUT). The box highlights the position of the 736 T>G mutation. Each gray bar represents a NextGen sequence. Colored bases differ from the WT sequence. Bases matching the WT are shown in gray to highlight only mutant bases. Examples of whole genome sequencing from two WT (WT 1 and WT2) and two MUT (MUT1 and MUT2) rats are shown. (From: Agbaga et al., 2020[34]). (PNG 385 kb)
Supplemental Fig. 4.
Gross physical phenotype of SCA34-KI rats. a. Gross appearance of WT and HET SCA34-KI rats is similar. MUT SCA34-KI rats show hair loss and erythrokeratodermia variabilis (EKV). b. Comparison of the underside of WT and MUT SCA34-KI rats showing marked hair loss and EKV. c. Hair loss around the eyes nose and ears (arrows) on a MUT SCA34-KI rat. d. WT rat pups show normal eyelids with full opening (P45 shown). e. MUT SCA34-KI rats show stiff, swollen eyelids at early ages that open incompletely (P45 shown), which resolves by about P60. Hair loss around the eye is also common in MUT rats. f. Adult MUT rats show complete opening of the lids. (From: Agbaga et al., 2020[34]). (PNG 3491 kb)
Supplemental Fig. 5.
The W246G mutation in ELOVL4 impairs VLC-SFA synthesis. a. Analysis of VLC-SFA in skin. Levels of VLC-SFA (28:0 and 30:0) and total VLC-SFA (28:0+30:0) were significantly reduced in the skin of MUT rats compared with WT and HET rats. b. Levels of 26:0, the direct precursor for VLC-SFA synthesis, did not differ significantly across genotypes. However, levels of 24:0 were significantly elevated in the skin of HET and MUT rats compared with WT rats. (Data are shown as mean +/- St. Dev. Analysis by 1-way ANOVA with Tukey's post-hoc test. *, p<0.05; **, p<0.01: ***, p<0.001). (From: Agbaga et al., 2020[34]). (PNG 64 kb)
Supplemental Fig. 6.
W246G ELOVL4 retains the ability to synthesize VLC-PUFA. Analysis of retinal glycerophospholipids from WT, HET, and MUT SCA34-KI rats shows that the W246G mutant form of ELOVL4 retains the capacity to synthesize VLC-PUFA at normal levels. No differences in total retinal levels of VLC-PUFA were present among WT, HET, and MUT rats. a: VLC-PUFA were detected specifically in the phosphatidylcholine fraction (PC), but total VLC-PUFA levels showed no differences among WT, HET, and MUT rat retina. However, significant differences were detected in non-VLC-FA (PC 34:01 and PC 40:06) among genotypes. b: No VLC-PUFA were detected in the phosphatidylethanolamine (PE) fraction of WT, HET, or MUT rat retina. However, statistically significant differences were detected in PE 40:06 and PC 44:12. c: No VLC-PUFA were detected in the phosphatidylserine (PS) fraction of WT, HET, or MUT rat retina. No significant differences were detected in any lipid species in the PS fraction. (Analysis by 1-way ANOVA with Tukey's post-hoc test. Data are shown as mean +/- St. Dev. *, MUT differs from WT; $, HET differs from WT; #, MUT differs from WT). (From: Agbaga et al., 2020[34]). (PNG 123 kb)
Supplemental Fig. 7.
Male WT, HET, and MUT rats all show poor performance on the rotarod test (distance traveled variable shown). (A) Rotarod performance of male WT, HET, and MUT rats at 2 months of age. (B) Rotarod performance of WT, HET, and MUT male rats at 6 months of age. (n= 5 WT, 7 HET, 4 MUT). Analysis by 1-way ANOVA with Tukey’s post-hoc test showed no significant difference across genotypes. (PNG 114 kb)
Supplemental Fig. 8.
Purkinje cell organization in WT, HET, and MUT rat cerebellum is comparable from P45 to 6 months. Immunolabeling for calbindin shows that Purkinje cells form a monolayer in the Purkinje cell layer (PCL), as appropriate, and have comparable organization across age-matched WT, HET, and MUT rats. (A-C) Postnatal day 45 (P45). (D-F) 2 months of age. (G-I) 6 months of age. Panels G-I of this figure match panels A-C shown in Figure 5. ML, molecular layer; GCL, granule cell layer. Scale bars = 200 μm for each row. (PNG 1872 kb)
Supplemental Fig. 9.
Granule cells in WT, HET, and MUT rat cerebellum are comparable from P45 to 6 months. Immunolabeling for NeuN, a granule cell marker, shows that granule cells are distributed appropriately in the granule cell layer (GCL) in WT, HET, and MUT rat cerebellum from P45 to 6 months of age. A small population of NeuN-positive cells is also present in the molecular layer (ML), as appropriate. (A-B) P45. (D-F) 2 months. (G-I) 6 months. Panels G-I of this figure match panels G-I shown in Figure 5. PCL, Purkinje cell layer. Scale bars = 200 μm for each row. (PNG 1441 kb)
Supplemental Fig. 10.
Immunolabeling for Vesicular Glutamate Transporter 1 (VGluT1) in WT, HET, and MUT rat cerebellum is comparable from P45 to 6 months of age. Immunolabeling for VGluT1 shows that mossy fiber inputs project to the glomeruli of the granule cell layer (GCL) and that PFs are distributed throughout the entire molecular layer (ML) of WT, HET, and MUT cerebellum from P45 to 6 months of age, as appropriate. (A-C) P45. (D-F) 2 months. (G-I) 6 months. Panels G-I of this figure match panels J-L shown in Figure 5. PCL, Purkinje cell layer. Scale bars = 200 μm for each row. (PNG 1781 kb)
Supplemental Fig. 11.
Immunolabeling for synaptotagmin 2 (Syt2) in WT, HET, and MUT rat cerebellum is comparable from P45 to 6 months of age. Syt2 is present in the glomeruli of the granule cell layer (GCL) and throughout the molecular layer (ML), as appropriate. (A-C) P45. (D-F) 2 months. (G-I) 6 months. Panels G-I of this figure match panels M-O shown in Figure 5. PCL, Purkinje cell layer. Scale bars = 200 μm for each row. (PNG 2581 kb)
Supplemental Fig. 12.
(A-C) Distribution of Protein Kinase Cɣ (PKCɣ) immunolabeling is comparable in WT (A), HET (B), and MUT (C) cerebellum. Scale bar = 100 μm for all panels. (D) Western blotting revealed no significant difference in PKG levels in WT, HET, and MUT cerebellum. (PNG 997 kb)
Supplemental Fig. 13.
(A-C) Distribution of Inositol Phosphate 3 Receptor (IP3R) immunolabeling is comparable in WT (A), HET (B), and MUT (C) cerebellum. Scale bar = 100 μm for all panels. (D) Western blotting revealed no significant difference in IP3R levels in WT, HET, and MUT cerebellum. (PNG 933 kb)
Supplemental Fig. 14.
Immunolabeling for Glial Fibrillary Acidic Protein (GFAP) in WT, HET, and MUT rat cerebellum is comparable from P45 to 6 months of age. Immunolabeling for GFAP shows that astrocytes are distributed appropriately, with no evidence of gliosis, in the WT, HET, and MUT rat cerebellum. (A-C) P45. (D-F) 2 months. (G-I) 6 months. ML, molecular layer; PCL, Purkinje cell layer; GCL, granule cell layer; bv, blood vessel. Scale bars = 200 μm for each row. (PNG 1572 kb)
Supplemental Fig. 15.
Microglia show “quiescent” ramified morphology in WT, HET, and MUT rat cerebellum. Tomato lectin (TL) labels blood vessels (bv) and microglial cells (arrowheads). Microglia show ramified morphology with symmetrically distributed processes typical of quiescent glia performing surveillance functions in the cerebellum of WT, HET, and MUT rats. (A-C) WT. (D-F) HET. (G-I) MUT. Molecular layer (ML). All images shown at 6 months of age. Scale bar = 200 μm for all panels. (PNG 1519 kb)
Supplemental Fig. 16.
Impaired synaptic plasticity in both male and female mutant rats. (A) LTP induced by stimulation of the PFs in the molecular layer at 1 Hz for 10 min induces LTP in female WT rat cerebellar slices (upper panel, green circles, ♀WT = 155.17 ± 17.38% normalized slope, n = 6 slices from 3 rats), but not in cerebellar slices from female MUT rats (upper panel, red triangles, ♀MUT = 97.87 ± 7.42% normalized slope, n = 8 slices from 3 rats). Similarly, the stimulation of the PFs at 1 Hz induces LTP in male WT rat cerebellar slices (lower panel, green circles, ♂WT = 140.45 ± 8.18% normalized slope, n = 8 slices from 3 rats), but not in cerebellar slices from male MUT rats (lower panel, red triangles, ♂MUT = 93.12 ± 5.94% normalized slope, n = 8 slices from 3 rats). (B) Stimulating the CFs in the granule cell layer at 1 Hz for 10 min induces LTD in female WT cerebellar slices (upper panel, green circles, ♀WT = 59.72 ± 6.66% normalized slope, n = 6 slices from 3 rats), but not in cerebellar slices from female MUT rats (upper panel, red triangles, ♀MUT = 92.13 ± 4.54% normalized slope, n = 8 slices from 3 rats). Similarly, the stimulation of the CFs at 1 Hz induces LTD in male WT rat cerebellar slices (lower panel, green circles, ♂WT = 63.13 ± 4.85% normalized slope, n = 4 slices from 2 rats), but not in cerebellar slices from male MUT rats (lower panel, red triangles, ♂MUT = 92.12 ± 8.14% normalized slope, n = 6 slices from 3 rats). Each data point represents the average of two successive test responses. The vertical gray bar indicates the period of 1 Hz stimulation. Data are shown as mean ± SEM. * p < 0.05, ** p < 0.01, ***p < 0.001, **** p < 0.0001, two-way repeated measures ANOVA with Bonferroni post-hoc test. (PNG 110 kb)
Supplemental Fig. 17.
Synaptic plasticity responses do not differ in female and male rats. (A) LTP induced by stimulation of the PFs in the molecular layer at 1 Hz for 10 min does not induce LTP in female MUT rats (upper panel, red triangles, ♀MUT = 97.87 ± 7.42% normalized slope, n = 8 slices from 3 rats) nor in cerebellar slices from male MUT rats (upper panel, red and black triangles, ♂MUT = 93.12 ± 5.94% normalized slope, n = 8 slices from 3 rats). The stimulation of the PFs at 1 Hz induces LTP in female WT rat cerebellar slices (lower panel, green circles, ♀WT = 155.17 ± 17.38% normalized slope, n = 6 slices from 3 rats), and also in male WT rat cerebellar slices (lower panel, green and black circles, ♂WT = 140.45 ± 8.18% normalized slope, n = 8 slices from 3 rats). (B) Stimulating the CFs in the granule cell layer at 1 Hz for 10 min does not induce LTD in cerebellar slices from female MUT rats (upper panel, red triangles, ♀MUT = 92.13 ± 4.54% normalized slope, n = 8 slices from 3 rats) nor in cerebellar slices from male MUT rats (upper panel, red and black triangles, ♂MUT = 92.12 ± 8.14% normalized slope, n = 6 slices from 3 rats). The stimulation of the CFs at 1 Hz induces LTD in female WT cerebellar slices (lower panel, green circles, ♀WT = 59.72 ± 6.66% normalized slope, n = 6 slices from 3 rats), and also in male WT rat cerebellar slices (lower panel, green and black circles, ♂WT = 63.13 ± 4.85% normalized slope, n = 4 slices from 2 rats). Each data point represents the average of two successive test responses. The vertical gray bar indicates the period of 1 Hz stimulation. Data are shown as mean ± SEM. p>0.05 for all panels, two-way repeated measures ANOVA with Bonferroni post-hoc test. (PNG 99 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Nagaraja, R.Y., Sherry, D.M., Fessler, J.L. et al. W246G Mutant ELOVL4 Impairs Synaptic Plasticity in Parallel and Climbing Fibers and Causes Motor Defects in a Rat Model of SCA34. Mol Neurobiol 58, 4921–4943 (2021). https://doi.org/10.1007/s12035-021-02439-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02439-1