
Abstract
The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the causative agent of human COVID-19, not only causes flu-like symptoms and gut microbiome complications but a large number of infected individuals also experience a host of neurological symptoms including loss of smell and taste, seizures, difficulty concentrating, decreased alertness, and brain inflammation. Although SARS-CoV-2 infections are not more prevalent in Parkinson’s disease patients, a higher mortality rate has been reported not only associated with older age and longer disease duration, but also through several mechanisms, such as interactions with the brain dopaminergic system and through systemic inflammatory responses. Indeed, a number of the neurological symptoms seen in COVID-19 patients, as well as the alterations in the gut microbiome, are also prevalent in patients with Parkinson’s disease. Furthermore, biochemical pathways such as oxidative stress, inflammation, and protein aggregation have shared commonalities between Parkinson’s disease and COVID-19 disease progression. In this review, we describe and compare the numerous similarities and intersections between neurodegeneration in Parkinson’s disease and RNA viral infections, emphasizing the current SARS-CoV-2 global health crisis.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder that primarily affects dopaminergic neurons in the substantia nigra [1]. Although this disorder mainly pertains to motor complication, PD patients also experience many non-motor symptoms such as neuropsychiatric (depression, dementia, apathy, anxiety), autonomic (constipation, urinary incontinence, excessive sweating), sleep disorders, and sensory abnormalities (loss of smell, pain, and paresthesia) [1]. The exact causes of sporadic PD are largely unknown, but it is clear that both environmental and genetic factors play a role. Indeed, the Braak hypothesis suggests that sporadic PD originates from an external pathogen that enters the body through the nasal cavity, which then migrates via the vagus nerve to the gut, causing complications such as changes in the gut microbiome and the advancement of Lewy Body (LB) pathology in the gut and in the nose similar to that of a viral infection [2, 3].
The novel SARS-CoV-2 coronavirus (COVID-19) pandemic was a result of a virus outbreak originating in Wuhan, China which quickly spread throughout the world. While flu-like symptoms such as fever, cough, and difficulty breathing appeared to be the predominant early warning signs of a COVID-19 infection, a large number of patients admitted to hospitals experienced a host of neurological symptoms including dizziness, loss of smell and taste, seizures, difficulty concentrating, decreased alertness, and brain inflammation [4]. In addition, complications in the gut microbiomes have also been noted as a result of COVID-19 [4].
Strikingly, a number of the neurological symptoms seen in COVID-19 patients, as well as the alterations in the gut microbiome, are also prevalent in patients with PD. Moreover, several biochemical pathways, including oxidative stress, inflammation, and protein aggregation, show similarities between PD and COVID-19. In this review, we describe the many intersections between neurodegeneration in PD and viral infections, with an emphasis on the novel SARS-CoV-2.
PD, Alpha-Synuclein, and RNA Viruses
The pathological hallmark of PD is the presence of LBs, neuronal inclusions constituted of aggregate protein [5]. The formation of LBs is seeded by the aggregation of its major component, the protein alpha-synuclein (a-syn). A-syn is an intrinsically disordered protein mainly found in neurons at the presynaptic terminals, but a-syn is also found in other tissues such as kidney, heart, and muscle cells [6]. While the physiological function of a-syn is not fully understood, a-syn has been implicated as a pre-synaptic protein that mediates neurotransmitter release [7, 8]. Duplications, triplications, and mutations of the a-syn encoding gene SNCA are linked to familial PD suggesting that an increase in a-syn and/or the expression of mutational variants contributes to neurodegeneration (Fig. 1) [9]. Many extrinsic factors such as post-translational modifications, oxidative stress, and metal binding have also been shown to affect multimerization and aggregation of a-syn, and due to its central effect on LB pathology, factors which affect the behavior or prevalence of a-syn are generally thought to be associated with PD [10, 11].

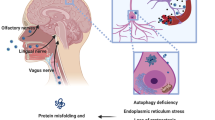
Parkinson’s disease contributing factors including heavy metals, inflammation, reactive oxygen species, and alpha-synuclein are elevated in response to RNA virus infection illustrating commonalities between neurodegeneration and viral infections
RNA viruses are linked to PD by their effect on a-syn (Table 1). For example, a-syn expression is upregulated in response to neurons infected by RNA viruses (Fig. 1) [12, 13]. It has been shown that a-syn supports expression of anti-viral interferon-stimulated genes [12]. In fact, a-syn restricts RNA viral replication, protecting the central nervous system (CNS) in infected mice [14]. Interestingly, a-syn expression also increases in enteric neurons of the gastrointestinal tract in response to infections with the single-stranded RNA norovirus followed by an inflammatory immune response [15, 16]. While the prevention of RNA virus progression is of immediate and primary concern to an individual’s health, elevated a-syn levels and prolonged inflammation are both linked to LB pathology and increased risk for PD [13].
The protective function of a-syn against RNA viruses has obvious implications to the novel coronavirus SARS-CoV-2, especially in individuals with PD. Elevated a-syn expression may indeed serve as a protective mechanism against this RNA-virus; however, it is unlikely that aggregated a-syn contained within LBs will be affective in restricting RNA viral replication. In addition to viral replication, a-syn can also inhibit RNA virus transmission from the peripheral nervous system (PNS) to the CNS [17]. When peripherally injected with a non-neuroinvasive RNA virus, the brains of a-syn knockout mice showed a much higher viral load than the brains of heterozygous a-syn expressing mice [14, 17]. In addition, the same difference was not observed when inoculation was done intracerebrally or done on brain slice cultures [14, 17]. Since COVID-19 can manifest itself in both the CNS and PNS, its replication and its spread may be inhibited by elevated a-syn in individuals living with PD or possibly other synucleinopathies [18].
Viral Infections and Neurotransmitters
With regards to viral infections, it has been noted that there is a link between viruses, neurotransmitters, and neurotransmitter protein pathways. The acetylcholine (ACh) and dopaminergic pathways are obstructed during PD, as a result of degeneration of the substantia nigra and motor neurons [19]. However, in the case of viral infections, it has been shown that acetylcholine levels are affected throughout the progression of the virus. In the early stages of infection, acetylcholine levels seem to remain constant. However, ACh levels seem to rise around the peak point of the immune response. In addition, cholinergic lymphocytes, which make direct contact with macrophages in the lungs, appear as ACh levels increase which may be present as a result of the SARS-CoV-2 virus [20]. These lymphocytes, however, are also known to regulate pulmonary inflammation, a common symptom of COVID-19 [21]. In addition, it has also been shown that viruses lead to production of ACh via choline acetyltransferase (CHAT) enzyme activation [22]. With regards to recovery, however, the role of ACh is still unknown [21]. In addition to ACh, it has also been shown that viral infections play a role in the dopaminergic pathway. Theiler’s virus, a type of encephalomyelitis RNA virus, has been known to destroy the substantia nigra, which is the site of dopamine production [23]. In an additional study involving mice, the presence of viral RNA was observed, and the viral RNA was present in the substantia nigra within 3 days of infection and spread further throughout the brain within 10 days. However, 3 weeks later, the viral RNA was no longer detected [23]. As a result, it could be argued that these interactions between the substantia nigra and viral pathogens may support the PD Braak hypothesis. Furthermore, the interactions between neurotransmitter pathways and viral infections may lead to further research regarding the intersection of viral infections such as COVID-19 and PD.
Oxidative Stress
Oxidative stress plays an important role PD and reactive oxygen species (ROS) and their extensive production in the brain play an important role in dopaminergic neuronal cell loss death involving dopamine metabolism and high levels of iron and calcium (Fig. 1) [24]. ROS are naturally occurring in cells and are a necessary component of cellular homeostasis. Despite the importance of ROS in normal physiology, failure in ROS regulation by antioxidant proteins, such as superoxide dismutase (SOD1) and glutathione (GSH), can lead to oxidative stress which can have detrimental effects on cellular functions (Table 1) [25]. Mitochondria are key sites of ROS production and targets of ROS-induced damage by inhibition of the mitochondrial election transport chain (ETC) [26]. PINK1 and Parkin are PD-associated proteins important in mitochondrial homeostasis as well as ROS homeostasis [26].
Many viruses, such as hepatitis C (HCV), are known to cause oxidative stress by changing the antioxidant balance within cells (Fig. 1) [27]. HCV belongs to a family of RNA viruses that cause damage and cirrhosis of the liver and patients with chronic hepatitis C have increased ROS levels triggering immune responses and increased inflammation [28]. Another virus, Zika virus (ZIKV), which has been extensively studied due to its link with congenital malformations, has revealed that astrocytes are the targets of ZIKV [28]. In a recent study, it was shown that ROS imbalance, coupled with mitochondrial defects, trigger DNA damage in induced pluripotent stem cell (iPSC)–derived astrocytes, causing neuronal loss and motor defects [29]. Interestingly, ZIKV belongs to the Flaviviridae family of viruses, as does HCV, and it replicates within the endoplasmic reticulum (ER) causing an increase in ROS in both the ER and mitochondria [30, 31]. This increase in ROS causes breaks in DNA, activating the DNA damage response ultimately causing apoptosis [30, 31]. Reactive gliosis is a condition whereby neuroinflammatory conditions caused by bacterial or viral infections cause an inflammatory environment culminating in astrocyte reactivity [32]. These infected astrocytes correlate to an increase in pro-inflammatory chemokines and cytokines, and it has been speculated that surviving children infected by ZIKV might show an increased rate of neurological disorders such as PD or Alzheimer’s disease (AD) later on in life [30, 31]. Although a definite connection is yet to be made between viral infections, such as ZIKV and hepatitis C, with respect to increased risk of sporadic PD it is noteworthy that increased ROS and mitochondrial dysfunction are found in both viral infections and in PD (Table 1).
Inflammation
Brain inflammation has long been implicated as a risk factor (Fig. 1) [33, 34], as well as a pathological effect of PD (Table 1) [35,36,37,38]. The primary facilitators of the neuroinflammatory response are microglia. Microglia release immune factors when activated in response to trauma, viral infection, and aggregated proteins such as a-syn [13, 39]. In addition, astrocytes can contribute to the inflammatory response when activated by RNA viral infection (Fig. 1) [40]. Neuroinflammation has been described as a double-edged sword in regards to PD as on one hand it is neuroprotective in the short term but acts in a neurotoxic manner when chronically sustained [39, 41]. Inflammatory cytokines, reported as increased in PD patients [42], have been shown to exacerbate cognition, depression, anxiety, and sleep disturbances [43, 44]. Similarly, COVID-19 patients have shown a wide range of neurological disorders including psychosis/delirium, inflammation of the brain, ischemic stroke, and multisystem inflammatory syndrome (Table 1) [45, 46]. Interestingly, pro-inflammatory cytokines tumor necrosis factor alpha (TNFα), interleukin (IL)-2, and IL-6 are found at higher levels in the brains of PD patients and the cerebrospinal fluid of COVID-19 patients [47, 48]. In addition, many COVID-19 cases result in a cytokine storm, a massive immune response that upregulates pro-inflammatory cytokines [49]. Increased inflammation, due to COVID-19 in PD patients, may exacerbate non-motor symptoms, and it has been suggested that PD patients recovering from COVID-19 show an extended period before reaching baseline. However, it is unclear if the exact mechanisms of inflammation in PD, MSA, and COVID-19 are the same.
Metals, Viral Infections, and PD
It has been noted that exposure to heavy metals such as manganese and iron has played a role in the progression of PD and other neurological diseases (Fig. 1) [50]. The most common form of exposure to these metals is a result of the environment and abnormal accumulations in the body. The presence of these metals results in multiple oxidative stress pathways that can lead to the oxidation of dopamine and production of free radicals [50]. Similarly, it has been noted that heavy metals also play a role regarding some viral infections. Because many viruses use iron to replicate themselves, large iron buildups may form leading to potential neurodegeneration (Fig. 1) [51]. Like many RNA viruses including most coronaviruses, the replication of the SARS-CoV-2 virus is dependent on manganese and iron, so it is possible that contracting COVID-19 may lead to the future onset of PD [52]. In addition, it has been found that heavy metals are found at higher concentrations in hepatitis C–infected individuals [53]. Therefore, people that contract RNA viral infections that are dependent on heavy metals should be aware of the potential risk of developing PD in the future.
COVID-19 and the Braak Hypothesis
The Braak hypothesis states that sporadic PD originates in the gut and the nasal cavity where a pathogen travels along the vagus nerve and olfactory tract toward the brain [54, 55]. The Braak hypothesis is supported by longitudinal clinical data which demonstrate that loss of smell and gastrointestinal dysfunction represent early PD symptoms, often preceding a definite PD diagnosis once motor symptoms present themselves [3, 56,57,58,59]. Further, due to evidence of LB formation in the olfactory epithelium [60, 61] and the enteric nervous system [62, 63], a-syn aggregation has been implicated as the pathogen referenced in the Braak hypothesis. That the staging presented by the Braak hypothesis resembles in many cases that of COVID-19 symptom progression is cause for concern. In essence, the Braak hypothesis describes a mode of pathogen transmission, likely a-syn aggregates, which can bypass the blood–brain barrier and originate in two areas that are indeed affected by COVID-19.
Viral Infections, the Gut, and PD
The human body is inhabited by a diverse microflora which plays a role in many physiological and metabolic processes [64]. A healthy commensal microbiota plays a role in the breakdown of dietary substances that are too large to be digested and it has been shown to be essential for protecting the host against a variety of pathogenic infections [65]. Viral infections are among some of the most common invading pathogens that effect the host microbiota. In fact, our gut flora regulates viral expression yielding beneficial outcomes; however, it can also be regulated by viruses causing dysbiosis and a multitude of gastrointestinal (GI) issues [66]. The gut microbiota is decreased in older adults due to a number of factors that include diet, environmental factors, and genetics, and the rate and duration of viral infections are much higher in those with a compromised gut flora [67]. Patients with PD display an array of GI complications such as dysbiosis, constipation, and dysphasia (Table 1) [68], and these complications can occur up to 10 years before hallmark motor symptoms occur suggesting a possible link between the gut and the progression of PD (Fig. 2) [69]. Indeed, individuals with PD are at a higher risk of infection due to a compromised gut microbiota as well as severity of infections and emerging evidence has suggested that a rich and microbiota can play an essential role in modulating host immune response [70]. This occurs by stimulating the production of various pro-inflammatory cytokines during infection (Fig. 2).

The brain–gut microbiota axis: Schematic diagram highlighting the relationship between the brain and gut microbiota. A rich and diverse microflora allows for healthy immune and regulatory mediators, whereby a compromised gut microflora caused by viral infections, stress, antibiotics, and poor diet can cause a lack in immune response contributing to abnormal production of inflammatory cytokines which can lead to neuroinflammation
Commensal microbiota has also been shown to directly suppress viral infections especially in certain sites where viruses can gain entry into the host [66]. Supporting this notion is the fact that Enterococcus faecium can prevent infection by the influenza virus by direct absorption and trapping as well as producing various metabolites to prevent viral infection [71]. Studies have also shown that microbiota depletion, due to antibiotic treatment, can result in significantly higher viral shedding [72]. Similarly, a study found that fecal microbiota transplants in rhesus macaques infected with simian immunodeficiency virus (SIV) induced greater antiviral immunity [73]. The immune gut homeostasis is delicately orchestrated by the fine tuning of the regulatory balance of pro-inflammatory responses, such as Th17, versus inflammatory regulatory T cells (Tregs) (Fig. 2) [73]. For example, gut probiotics like Lactobacillus paracasei increase pro-inflammatory cytokines like IL-33, IL-β, IL-12, and INFγ during influenza virus infection [74]. Quite a few studies have also shown that viral infections such as HCV/HBV can cause a profound alteration in gut microbiota causing dysbiosis, and this reduced gut diversity caused an increase in severity of infections [75]. A healthy gut microbiome is therefore essential in maintaining an optimal immune system to fight off pathogenic infections including viral infections.
It has been noted that severe viral infections could possibly increase the risk of developing PD later in life [76]. Clearly, viral infections are not the primary cause of PD but may act as vital triggers as alluded to previously in this review [77]. For example, it is known that virions can pass the blood–brain barrier and elicit inflammatory responses in the brain such as those observed in encephalitis [77]. It has also been documented that individuals infected with hepatitis C are 30% more likely to develop PD than individuals who never had the virus [78]. Similarly, PD patients show a statistically higher antibody titer against the HSV-1 virus than healthy controls, and this autoimmunity has been further highlighted in mechanisms of a-syn molecular mimicry [79]. Indeed, the immunological cross-reactivity between HSV-1 and a-syn has been shown to cause destruction of dopaminergic neurons of the substantia nigra [80]. In many cases, SARS-CoV-2 primarily affects lung function through binding to ACE2 receptors present on the alveolar epithelial cells [81]. However, evidence has suggested that SARS-CoV-2 RNA can be detected in the stool of some patients with COVID-19 [82]. A recent study obtained blood and stool records from 100 patients with confirmed COVID-19, and 27 out of the 100 stool samples were collected 30 days after infection. The study discovered that the gut microbiome was significantly altered in patients with COVID-19 compared to control patients [83], showing a decrease in Faecalibacterium prausnitzii, Eubacterium rectale, and bifidobacteria. Furthermore, the study showed elevated levels of C reactive protein and lactate dehydrogenase, and it was suggested that altered gut microbiota is involved in COVID-19 severity and this dysbiosis is observed after the infection has passed.
These data suggest that there may be a link between the gut microbiota and COVID-19 severity, particularly as the higher COVID-19 mortality rate is seen within the elderly population that have a decreased gut microbiota. Interestingly, PD patients with chronic GI conditions suffer from a decrease in gut flora causing constant bacterial infections [84]. This may suggest an increased chance of COVID-19 mortality for a patient with PD.
The Olfactory Tract
Loss of smell is one of the earliest symptoms of PD and the olfactory epithelium (OE) is becoming a target tissue to study PD and brain aging in general (Table 1) [2, 3, 85,86,87]. Indeed, olfactory dysfunction is as common as the other cardinal motor symptoms in PD and more prevalent than resting tremor [88]. Idiopathic hyposmia has been associated with an increased risk for PD in first-degree relatives of PD individuals [85, 89], and it has been suggested that neurologists should screen high-risk patients with olfactory tests [90].
Like PD, olfactory dysfunction is one of the earliest symptoms of COVID-19 infection and is therefore a key diagnostic criteria (Table 1) [91, 92]. With inhalation of SARS-CoV-2, the primary mode of infection, the nasal cavity and olfactory epithelium represent important targets for the virus. ACE2, critical to SARS-CoV-2 entry into cells, is expressed in sustentacular cells and basal cells of the olfactory epithelium, but is absent in olfactory sensory neurons [93]. Sustentacular cells act as support cells which provide major physical, metabolic, secretory, and absorptive support functions for the olfactory sensory neurons (OSN) [94] while basal cells of the OE act as stem cells producing OSNs since their lifespan is only a few weeks [94]. There are two mechanisms that could be at play here. First, RNA viral infection has been previously shown to be capable of inducing a-syn seeding in neurons [95]. Due to the prion-like activity of a-syn aggregates, a seeding event in any cell type of the OE can cause the propagation of aggregation throughout the OE toward the brain. Another mechanism of PD pathogenesis could be that the sustentacular cells and basal cells cannot function normally after infection and cause alterations in the OSNs. In fact, glial cell loss-of-function or gain-of-toxic-function has been thought to participate in the pathogenesis of PD [96]. A variety of PD risk factors are associated with altered proteostasis and metabolism in neurons and a single seeding event, be it caused by viral infection or disrupted homeostasis, could contribute to the pathology established in the Braak hypothesis [97].
PD Non-Motor Symptoms and COVID-19
One of the most common non-motor symptoms of PD is depression and typically, 30–40% of PD patients experience some form of depression in their lifetime post diagnosis [98]. It has been hypothesized that increased stress and a loss of emotional control are a result of a lack of dopamine-dependent adaptation [98]. In times of crisis, such as during the COVID-19 pandemic, stress-induced depression and anxiety may increase as a result of medical, financial, and social factors. For PD patients who have contracted the SARS-CoV-2 virus, it has been found that there is an increase in non-motor symptoms including trouble sleeping, mood changes, cognitive changes, and autonomic problems which all have ties to depression [99]. However, not only PD patients have experienced an increase in depression as a result of the pandemic. Factors including a lack of knowledge of infectious diseases, the effects of quarantine, inadaptability, and the fear for one’s health all affect one’s mental health whether that person has been infected or not [100].
COVID-19 and PD Guidelines
During the COVID-19 pandemic, numerous guidelines have been published in order to keep the world’s populations safe and healthy. For many patients with PD, these guidelines are of course generally identical including the practice of social distancing and self-isolation if infected in order to prevent any further spread of the virus [101]. However, it has been noted that people with PD should remain active and engage in different forms of physical activity, particularly while spending time at home [102].
With regards to contagion, there does not seem to be a correlation between PD and contraction of the SARS-CoV-2 virus. However, if a PD patient does contract COVID-19, several complications may arise. It has been noted that PD patients are at larger risk of developing pneumonia and other respiratory infection, so clearly a SARS-CoV-2 infection may in many cases lead to a worsening of PD symptoms [101]. Indeed, urinary tract infections, pneumonia, or the flu can temporarily worsen PD symptoms, and it is therefore important for patients to have their PD medications readily available.
As with any underlying conditions, PD patients should consult with their physician in order to ensure that their PD medications are compatible with medications used to treat symptoms of COVID-19. It should be noted that certain cold and flu medications should not be administered together with, for example, MAO-B inhibitors, such as Azilect/rasagiline or Xadago/safinamide, frequently used to treat PD symptoms [101].
In the past few years, it has been noted that the number of PD cases has been on the rise. Although this rise in cases may be in part be attributed to an increasingly aging population, it is also believed that external factors such as pesticides, smoking, and viral infections may contribute to the increase [103].
Concluding Remarks
The global COVID-19 health crisis has challenged the way of life across the planet, affecting the economy, social interactions, and our health and safety. In this review, we have highlighted the intersections between PD, viral infections, and COVID-19 with an emphasis on the many similarities between RNA viral pathways and neurodegeneration in PD. Indeed, the onset and progression of PD, as detailed in the Braak hypothesis, as well as the pathogenic nature, molecular mechanisms, and symptom development of the disorder share many similarities with the SARS-CoV-2 virus and COVID-19. As further research is conducted, more evidence of a possible correlation between PD, viral infections, and the current SARS-CoV-2 virus will become available.
Data Availability
All data are available and can be shared.
Code Availability
Not applicable.
References
Elkouzi A. https://www.parkinson.org/understanding-parkinsons/what-is-parkinsons. Accessed September 14, 2020
Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318(1):121–134. https://doi.org/10.1007/s00441-004-0956-9
Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211. https://doi.org/10.1016/S0197-4580(02)00065-9
Garg D, Dhamija RK (2020) The challenge of managing Parkinson’s disease patients during the COVID-19 pandemic. Ann Indian Acad Neurol 23(Suppl 1):S24–S27. https://doi.org/10.4103/aian.AIAN_295_20
Tanaka K (1843) Matsuda N (2014) Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim Biophys Acta 1:197–204. https://doi.org/10.1016/j.bbamcr.2013.03.012
Burré J, Sharma M, Südhof TC (2018) Cell biology and pathophysiology of α-synuclein. Cold Spring Harb Perspect Med 8(3):a024091. https://doi.org/10.1101/cshperspect.a024091
Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123(3):383–396. https://doi.org/10.1016/j.cell.2005.09.028
Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH (2005) Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci 25(47):10913–10921. https://doi.org/10.1523/jneurosci.2922-05.2005
Abdullah R, Basak I, Patil KS, Alves G, Larsen JP, Møller SG (2015) Parkinson’s disease and age: the obvious but largely unexplored link. Exp Gerontol 68:33–38. https://doi.org/10.1016/j.exger.2014.09.014
Cavallarin N, Vicario M, Negro A (2010) The role of phosphorylation in synucleinopathies: focus on Parkinson’s disease. CNS Neurol Disord Drug Targets 9(4):471–481
Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281(40):29739–29752. https://doi.org/10.1074/jbc.M600933200
Massey AR, Monogue B, Chen Y, Lesteberg K, Johnson ME, Bergkvist L, Steiner JA, Ma J, Mahalingam R, Kleinschmidt-Demasters BK, Galvis ME, Brundin P, Kunath T, Beckham JD (2020) Alpha-synuclein expression supports interferon stimulated gene expression in neurons. bioRxiv:2020.2004.2025.061762. https://doi.org/10.1101/2020.04.25.061762
Bantle CM, Phillips AT, Smeyne RJ, Rocha SM, Olson KE, Tjalkens RB (2019) Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinson’s Dis 5(1):20
Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE, Beckham JD (2015) Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol 90(6):2767–2782. https://doi.org/10.1128/jvi.02949-15
Labrie V, Brundin P (2017) Alpha-synuclein to the rescue: immune cell recruitment by alpha-synuclein during gastrointestinal infection. J Innate Immun 9(5):437–440. https://doi.org/10.1159/000479653
Stolzenberg E, Berry D, Yang D, Lee EY, Kroemer A, Kaufman S, Wong GCL, Oppenheim JJ, Sen S, Fishbein T, Bax A, Harris B, Barbut D, Zasloff MA (2017) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9(5):456–463. https://doi.org/10.1159/000477990
Massey AR, Beckham JD (2016) Alpha-synuclein, a novel viral restriction factor hiding in plain sight. DNA Cell Biol 35(11):643–645. https://doi.org/10.1089/dna.2016.3488
Ait Wahmane S, Achbani A, Ouhaz Z, Elatiqi M, Belmouden A, Nejmeddine M (2020) The possible protective role of α-synuclein against severe acute respiratory syndrome coronavirus 2 infections in patients with Parkinson’s disease. Mov Disord 35(8):1293–1294. https://doi.org/10.1002/mds.28185
Rizzi G, Tan KR (2017) Dopamine and acetylcholine, a circuit point of view in Parkinson’s disease. Front Neural Circuits 11:110–110. https://doi.org/10.3389/fncir.2017.00110
Horkowitz AP, Schwartz AV, Alvarez CA, Herrera EB, Thoman ML, Chatfield DA, Osborn KG, Feuer R, George UZ, Phillips JA (2020) Acetylcholine regulates pulmonary pathology during viral infection and recovery. Immunotargets Ther 9:333–350. https://doi.org/10.2147/ITT.S279228
Phillips JA, Horkowitz A, Feuer R (2019) Acetylcholine and cholinergic lymphocytes in the immune response to influenza. The Journal of Immunology. 202(1 Supplement):66.20
Contoli M, Marcellini A, Casolari P, Caramori G, Papi A (2016) Role of the acetylcholine in the virus-induced bronchoconstriction. Eur Respir J 48(supp 60):PA4103. https://doi.org/10.1183/13993003.congress-2016.PA4103
Oliver KR, Brennan P, Fazakerley JK (1997) Specific infection and destruction of dopaminergic neurons in the substantia nigra by Theiler’s virus. J Virol 71(8):6179–6182. https://doi.org/10.1128/JVI.71.8.6179-6182.1997
Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3(4):461–491. https://doi.org/10.3233/JPD-130230
Hauser DN, Hastings TG (2013) Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol Dis 51:35–42. https://doi.org/10.1016/j.nbd.2012.10.011
Zhu J, Chu CT (2010) Mitochondrial dysfunction in Parkinson’s disease. J Alzheimers Dis 20(Suppl 2):S325-334. https://doi.org/10.3233/JAD-2010-100363
Muriel P (2009) Role of free radicals in liver diseases. Hepatol Int 3(4):526–536. https://doi.org/10.1007/s12072-009-9158-6
Paracha UZ, Fatima K, Alqahtani M, Chaudhary A, Abuzenadah A, Damanhouri G, Qadri I (2013) Oxidative stress and hepatitis C virus. Virol J 10:251. https://doi.org/10.1186/1743-422X-10-251
Ledur PF, Karmirian K, Pedrosa C, Souza LRQ, Assis-de-Lemos G, Martins TM, Ferreira J, de Azevedo Reis GF, Silva ES, Silva D, Salerno JA, Ornelas IM, Devalle S, Madeiro da Costa RF, Goto-Silva L, Higa LM, Melo A, Tanuri A, Chimelli L, Murata MM, Garcez PP, Filippi-Chiela EC, Galina A, Borges HL, Rehen SK (2020) Zika virus infection leads to mitochondrial failure, oxidative stress and DNA damage in human iPSC-derived astrocytes. Sci Rep 10(1):1218. https://doi.org/10.1038/s41598-020-57914-x
Li G, Poulsen M, Fenyvuesvolgyi C, Yashiroda Y, Yoshida M, Simard JM, Gallo RC, Zhao RY (2017) Characterization of cytopathic factors through genome-wide analysis of the Zika viral proteins in fission yeast. Proc Natl Acad Sci U S A 114(3):E376–E385. https://doi.org/10.1073/pnas.1619735114
Medvedev R, Ploen D, Hildt E (2016) HCV and oxidative stress: implications for HCV life cycle and HCV-associated pathogenesis. Oxid Med Cell Longev 2016:9012580. https://doi.org/10.1155/2016/9012580
Potokar M, Jorgacevski J, Zorec R (2019) Astrocytes in Flavivirus infections. Int J Mol Sci 20(3):691. https://doi.org/10.3390/ijms20030691
McGeer PL, Itagaki S, Boyes BE, McGeer E (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38(8):1285–1285
Racette BA, Gross A, Vouri SM, Camacho-Soto A, Willis AW, Searles Nielsen S (2018) Immunosuppressants and risk of Parkinson disease. Ann Clin Transl Neurol 5(7):870–875. https://doi.org/10.1002/acn3.580
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994) Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci Lett 180(2):147–150
Müller T, Blum-Degen D, Przuntek H, Kuhn W (1998) Short communication interleukin-6 levels in cerebrospinal fluid inversely correlate to severity of Parkinson’s disease. Acta Neurol Scand 98(2):142–144
Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Investig 119(1):182–192. https://doi.org/10.1172/jci36470
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21(2):404–412. https://doi.org/10.1016/j.nbd.2005.08.002
Tansey MG, McCoy MK, Frank-Cannon TC (2007) Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208(1):1–25. https://doi.org/10.1016/j.expneurol.2007.07.004
Klein RS, Garber C, Funk KE, Salimi H, Soung A, Kanmogne M, Manivasagam S, Agner S, Cain M (2019) Neuroinflammation during RNA viral infections. Annu Rev Immunol 37:73–95. https://doi.org/10.1146/annurev-immunol-042718-041417
Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease—a double-edged sword. Neuron 35(3):419–432. https://doi.org/10.1016/S0896-6273(02)00794-8
Koziorowski D, Tomasiuk R, Szlufik S, Friedman A (2012) Inflammatory cytokines and NT-proCNP in Parkinson’s disease patients. Cytokine 60(3):762–766. https://doi.org/10.1016/j.cyto.2012.07.030
Menza M, Dobkin RD, Marin H, Mark MH, Gara M, Bienfait K, Dicke A, Kusnekov A (2010) The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease. Psychosomatics 51(6):474–479. https://doi.org/10.1176/appi.psy.51.6.474
Lindqvist D, Kaufman E, Brundin L, Hall S, Surova Y, Hansson O (2012) Non-motor symptoms in patients with Parkinson’s disease – correlations with inflammatory cytokines in serum. PLoS ONE 7(10):e47387. https://doi.org/10.1371/journal.pone.0047387
Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, Jayaseelan DL, Kumar G et al (2020) The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. https://doi.org/10.1093/brain/awaa240
Feldstein LR, Rose EB, Horwitz SM, Collins JP, Newhams MM, Son MBF, Newburger JW, Kleinman LC et al (2020) Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med 383(4):334–346. https://doi.org/10.1056/NEJMoa2021680
Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Cytokines in Parkinson’s disease. J Neural Transm Suppl 58:143–151
Achar A, Ghosh C (2020) COVID-19-associated neurological disorders: the potential route of CNS invasion and blood–brain relevance. Cells 9(11):2360. https://doi.org/10.3390/cells9112360
Nile SH, Nile A, Qiu J, Li L, Jia X, Kai G (2020) COVID-19: pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev 53:66–70. https://doi.org/10.1016/j.cytogfr.2020.05.002
Montgomery EB Jr (1995) Heavy metals and the etiology of Parkinson’s disease and other movement disorders. Toxicology 97(1–3):3–9. https://doi.org/10.1016/0300-483x(94)02962-t
Drakesmith H, Prentice A (2008) Viral infection and iron metabolism. Nat Rev Microbiol 6(7):541–552. https://doi.org/10.1038/nrmicro1930
Ahn DG, Choi JK, Taylor DR, Oh JW (2012) Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Adv Virol 157(11):2095–2104. https://doi.org/10.1007/s00705-012-1404-x
Aslam N, Iqbal MS, Hussain SM, Rizwan M, Naseer QU, Afzal M, Muneer R, Batool F (2019) Effects of chelating agents on heavy metals in hepatitis C virus (HCV) patients. Math Biosci Eng 16(3):1138–1149. https://doi.org/10.3934/mbe.2019054
Hawkes CH, Del Tredici K, Braak H (2007) Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 33(6):599–614. https://doi.org/10.1111/j.1365-2990.2007.00874.x
Hawkes C, Del Tredici K, Braak H (2009) Parkinson’s disease: the dual hit theory revisited. Ann N Y Acad Sci 1170:615–622. https://doi.org/10.1111/j.1749-6632.2009.04365.x
Ross GW, Petrovitch H, Abbott RD, Tanner CM, Popper J, Masaki K, Launer L, White LR (2008) Association of olfactory dysfunction with risk for future Parkinson’s disease. Ann Neurol 63(2):167–173. https://doi.org/10.1002/ana.21291
Ross GW, Abbott RD, Petrovitch H, Tanner CM, Davis DG, Nelson J, Markesbery WR, Hardman J et al (2006) Association of olfactory dysfunction with incidental Lewy bodies. Mov Disord 21(12):2062–2067. https://doi.org/10.1002/mds.21076
Cersosimo MG, Raina GB, Pecci C, Pellene A, Calandra CR, Gutiérrez C, Micheli FE, Benarroch EE (2013) Gastrointestinal manifestations in Parkinson’s disease: prevalence and occurrence before motor symptoms. J Neurol 260(5):1332–1338. https://doi.org/10.1007/s00415-012-6801-2
Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, Kordower JH (2012) Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord 27(6):709–715. https://doi.org/10.1002/mds.23838
Saito Y, Shioya A, Sano T, Sumikura H, Murata M, Murayama S (2016) Lewy body pathology involves the olfactory cells in Parkinson’s disease and related disorders. Mov Disord 31(1):135–138. https://doi.org/10.1002/mds.26463
Funabe S, Takao M, Saito Y, Hatsuta H, Sugiyama M, Ito S, Kanemaru K, Sawabe M et al (2013) Neuropathologic analysis of Lewy-related α-synucleinopathy in olfactory mucosa. Neuropathology 33(1):47–58. https://doi.org/10.1111/j.1440-1789.2012.01329.x
Kupsky WJ, Grimes MM, Sweeting J, Bertsch R, Cote LJ (1987) Parkinson’s disease and megacolon: concentric hyaline inclusions (Lewy bodies) in enteric ganglion cells. Neurology 37(7):1253–1255. https://doi.org/10.1212/wnl.37.7.1253
Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F (1988) Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol 76(3):217–221. https://doi.org/10.1007/bf00687767
Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A et al (2018) Environment dominates over host genetics in shaping human gut microbiota. Nature 555(7695):210–215. https://doi.org/10.1038/nature25973
Piewngam P, Zheng Y, Nguyen TH, Dickey SW, Joo HS, Villaruz AE, Glose KA, Fisher EL et al (2018) Pathogen elimination by probiotic Bacillus via signalling interference. Nature 562(7728):532–537. https://doi.org/10.1038/s41586-018-0616-y
Karst SM (2016) The influence of commensal bacteria on infection with enteric viruses. Nat Rev Microbiol 14(4):197–204. https://doi.org/10.1038/nrmicro.2015.25
Dhar D, Mohanty A (2020) Gut microbiota and Covid-19—possible link and implications. Virus Res 285:198018. https://doi.org/10.1016/j.virusres.2020.198018
Cersosimo MG, Benarroch EE (2012) Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol Dis 46(3):559–564. https://doi.org/10.1016/j.nbd.2011.10.014
Dutta SK, Verma S, Jain V, Surapaneni BK, Vinayek R, Phillips L, Nair PP (2019) Parkinson’s disease: the emerging role of gut dysbiosis, antibiotics, probiotics, and fecal microbiota transplantation. J Neurogastroenterol Motil 25(3):363–376. https://doi.org/10.5056/jnm19044
Tedesco D, Thapa M, Chin CY, Ge Y, Gong M, Li J, Gumber S, Speck P, Elrod EJ, Burd EM, Kitchens WH, Magliocca JF, Adams AB, Weiss DS, Mohamadzadeh M, Grakoui A (2018) Alterations in intestinal microbiota lead to production of interleukin 17 by intrahepatic gammadelta T-cell receptor-positive cells and pathogenesis of cholestatic liver disease. Gastroenterology 154(8):2178–2193. https://doi.org/10.1053/j.gastro.2018.02.019
Groves HT, Cuthbertson L, James P, Moffatt MF, Cox MJ, Tregoning JS (2018) Respiratory disease following viral lung infection alters the murine gut microbiota. Front Immunol 9:182. https://doi.org/10.3389/fimmu.2018.00182
Edouard S, Million M, Bachar D, Dubourg G, Michelle C, Ninove L, Charrel R, Raoult D (2018) The nasopharyngeal microbiota in patients with viral respiratory tract infections is enriched in bacterial pathogens. Eur J Clin Microbiol Infect Dis 37(9):1725–1733. https://doi.org/10.1007/s10096-018-3305-8
Hensley-McBain T, Zevin AS, Manuzak J, Smith E, Gile J, Miller C, Agricola B, Katze M, Reeves RK, Kraft CS, Langevin S, Klatt NR (2016) Effects of fecal microbial transplantation on microbiome and immunity in simian immunodeficiency virus-infected macaques. J Virol 90(10):4981–4989. https://doi.org/10.1128/JVI.00099-16
Park MK, Ngo V, Kwon YM, Lee YT, Yoo S, Cho YH, Hong SM, Hwang HS, Ko EJ, Jung YJ, Moon DW, Jeong EJ, Kim MC, Lee YN, Jang JH, Oh JS, Kim CH, Kang SM (2013) Lactobacillus plantarum DK119 as a probiotic confers protection against influenza virus by modulating innate immunity. PLoS ONE 8(10):e75368. https://doi.org/10.1371/journal.pone.0075368
Wang J, Wang Y, Zhang X, Liu J, Zhang Q, Zhao Y, Peng J, Feng Q, Dai J, Sun S, Zhao Y, Zhao L, Zhang Y, Hu Y, Zhang M (2017) Gut microbial dysbiosis is associated with altered hepatic functions and serum metabolites in chronic hepatitis B patients. Front Microbiol 8:2222. https://doi.org/10.3389/fmicb.2017.02222
Sulzer D, Antonini A, Leta V, Nordvig A, Smeyne RJ, Goldman JE, Al-Dalahmah O, Zecca L, Sette A, Bubacco L, Meucci O, Moro E, Harms AS, Xu Y, Fahn S, Ray Chaudhuri K (2020) COVID-19 and possible links with Parkinson’s disease and parkinsonism: from bench to bedside. NPJ Parkinsons Dis 6:18. https://doi.org/10.1038/s41531-020-00123-0
Menendez CM, Jinkins JK, Carr DJ (2016) Resident T cells are unable to control herpes simplex virus-1 activity in the brain ependymal region during latency. J Immunol 197(4):1262–1275. https://doi.org/10.4049/jimmunol.1600207
Wangensteen KJ, Krawitt EL, Hamill RW, Boyd JT (2016) Hepatitis C virus infection: a risk factor for Parkinson’s disease. J Viral Hepat 23(7):535. https://doi.org/10.1111/jvh.12517
Costa Sa AC, Madsen H, Brown JR (2019) Shared molecular signatures across neurodegenerative diseases and herpes virus infections highlights potential mechanisms for maladaptive innate immune responses. Sci Rep 9(1):8795. https://doi.org/10.1038/s41598-019-45129-8
Caggiu E, Paulus K, Galleri G, Arru G, Manetti R, Sechi GP, Sechi LA (2017) Homologous HSV1 and alpha-synuclein peptides stimulate a T cell response in Parkinson’s disease. J Neuroimmunol 310:26–31. https://doi.org/10.1016/j.jneuroim.2017.06.004
Lake MA (2020) What we know so far: COVID-19 current clinical knowledge and research. Clin Med (Lond) 20(2):124–127. https://doi.org/10.7861/clinmed.2019-coron
Wu Y, Guo C, Tang L, Hong Z, Zhou J, Dong X, Yin H, Xiao Q, Tang Y, Qu X, Kuang L, Fang X, Mishra N, Lu J, Shan H, Jiang G, Huang X (2020) Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol Hepatol 5(5):434–435. https://doi.org/10.1016/s2468-1253(20)30083-2
Yeoh YK, Zuo T, Lui GC-Y, Zhang F, Liu Q, Li AYL, Chung ACK, Cheung CP, Tso EYK, Fung KSC, Chan V, Ling L, Joynt G, Hui DS-C, Chow KM, Ng SSS, Li TC-M, Ng RWY, Yip TCF, Wong GL-H, Chan FKL, Wong CK, Chan PKS, Ng SC (2021) Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 70(4):698. https://doi.org/10.1136/gutjnl-2020-323020
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, Chesselet MF, Keshavarzian A, Shannon KM, Krajmalnik-Brown R, Wittung-Stafshede P, Knight R, Mazmanian SK (2016) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167(6):1469–1480. https://doi.org/10.1016/j.cell.2016.11.018 (e1412)
Ponsen MM, Stoffers D, Booij J, van Eck-Smit BL, Wolters E, Berendse HW (2004) Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol 56(2):173–181. https://doi.org/10.1002/ana.20160
Brai E, Hummel T, Alberi L (2020) Smell, an underrated early biomarker for brain aging. Front Neurosci 14:792. https://doi.org/10.3389/fnins.2020.00792
Brozzetti L, Sacchetto L, Cecchini MP, Avesani A, Perra D, Bongianni M, Portioli C, Scupoli M, Ghetti B, Monaco S, Buffelli M, Zanusso G (2020) Neurodegeneration-associated proteins in human olfactory neurons collected by nasal brushing. Front Neurosci 14:145. https://doi.org/10.3389/fnins.2020.00145
Alves G, Forsaa EB, Pedersen KF, Dreetz Gjerstad M, Larsen JP (2008) Epidemiology of Parkinson’s disease. J Neurol 255(5):18–32. https://doi.org/10.1007/s00415-008-5004-3
Ponsen MM, Stoffers D, Wolters E, Booij J, Berendse HW (2010) Olfactory testing combined with dopamine transporter imaging as a method to detect prodromal Parkinson’s disease. J Neurol Neurosurg Psychiatry 81(4):396–399. https://doi.org/10.1136/jnnp.2009.183715
Godoy MD, Voegels RL, Pinna Fde R, Imamura R, Farfel JM (2015) Olfaction in neurologic and neurodegenerative diseases: a literature review. Int Arch Otorhinolaryngol 19(2):176–179. https://doi.org/10.1055/s-0034-1390136
Passarelli PC, Lopez MA, Mastandrea Bonaviri GN, Garcia-Godoy F, D’Addona A (2020) Taste and smell as chemosensory dysfunctions in COVID-19 infection. Am J Dent 33(3):135–137
David P, Shoenfeld Y (2020) The smell in COVID-19 infection: diagnostic opportunities. Isr Med Assoc J: IMAJ 7(22):335–337
Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, Chance R, Macaulay IC, Chou H-J, Fletcher RB, Das D, Street K, de Bezieux HR, Choi Y-G, Risso D, Dudoit S, Purdom E, Mill J, Hachem RA, Matsunami H, Logan DW, Goldstein BJ, Grubb MS, Ngai J, Datta SR (2020) Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv 6(31):eabc5801. https://doi.org/10.1126/sciadv.abc5801
Liang F (2020) Sustentacular cell enwrapment of olfactory receptor neuronal dendrites: an update. Genes (Basel) 11(5):493. https://doi.org/10.3390/genes11050493
Marreiros R, Müller-Schiffmann A, Trossbach SV, Prikulis I, Hänsch S, Weidtkamp-Peters S, Moreira AR, Sahu S, Soloviev I, Selvarajah S, Lingappa VR, Korth C (2020) Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc Natl Acad Sci U S A 117(12):6741–6751. https://doi.org/10.1073/pnas.1906466117
Tremblay M-E, Cookson MR, Civiero L (2019) Glial phagocytic clearance in Parkinson’s disease. Mol Neurodegener 14(1):16. https://doi.org/10.1186/s13024-019-0314-8
Kurtishi A, Rosen B, Patil K, Alves G, Moller S (2018) Cellular proteostasis in neurodegeneration. Mol Neurobiol 56:3676–3689. https://doi.org/10.1007/s12035-018-1334-z
Helmich RC, Bloem BR (2020) The impact of the COVID-19 pandemic on Parkinson’s disease: hidden sorrows and emerging opportunities. J Parkinsons Dis 10(2):351–354. https://doi.org/10.3233/jpd-202038
Brown EG, Chahine LM, Goldman SM, Korell M, Mann E, Kinel DR, Arnedo V, Marek KL, Tanner CM (2020) The effect of the COVID-19 pandemic on people with Parkinson’s disease. J Parkinsons Dis 10:1365–1377. https://doi.org/10.3233/JPD-202249
Salari N, Hosseinian-Far A, Jalali R, Vaisi-Raygani A, Rasoulpoor S, Mohammadi M, Rasoulpoor S, Khaledi-Paveh B (2020) Prevalence of stress, anxiety, depression among the general population during the COVID-19 pandemic: a systematic review and meta-analysis. Glob Health 16(1):57. https://doi.org/10.1186/s12992-020-00589-w
Dolhun R (2020) Ask the MD: Coronavirus and Parkinson’s. https://www.michaeljfox.org/news/ask-md-coronavirus-and-parkinsons. Accessed 14 Sept 2020
Vistven A. https://parkinson.no/behandling-og-rehabilitering/trening. Accessed September 14, 2020
Savica R, Grossardt BR, Bower JH, Ahlskog JE, Rocca WA (2016) Time trends in the incidence of Parkinson disease. JAMA Neurol 73(8):981–989. https://doi.org/10.1001/jamaneurol.2016.0947
Author information
Authors and Affiliations
Contributions
All authors contributed equally.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflicts of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rosen, B., Kurtishi, A., Vazquez-Jimenez, G.R. et al. The Intersection of Parkinson’s Disease, Viral Infections, and COVID-19. Mol Neurobiol 58, 4477–4486 (2021). https://doi.org/10.1007/s12035-021-02408-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02408-8