
Abstract
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a monogenic disease caused by NOTCH3 mutations and characterized by typical clinical, neuroradiological, and pathological features. NOTCH3 belongs to a family of highly conserved transmembrane receptors rich of epidermal growth factor repeats, mostly expressed in vascular smooth muscle cells and pericytes, which perform essential developmental functions and are involved in tissues maintenance and renewal. To date, no therapeutic option for CADASIL is available except for few symptomatic treatments. Novel in vitro and in vivo models are continuously explored with the aim to investigate underlying pathogenic mechanisms and to test novel therapeutic approaches. In this scenario, knock-out, knock-in, and transgenic mice studies have generated a large amount of information on molecular and biological aspects of CADASIL, despite that they incompletely reproduce the human phenotype. Moreover, the field of in vitro models has been revolutionized in the last two decades by the introduction of induced pluripotent stem cells (iPSCs) technology. As a consequence, novel therapeutic approaches, including immunotherapy, growth factors administration, and antisense oligonucleotides, are currently under investigation. While waiting that further studies confirm the promising results obtained, the data reviewed suggest that our therapeutic approach to the disease could be transformed, generating new hope for the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a monogenic disease caused by NOTCH3 mutations [1]. Age of onset is variable from infancy to elderly, even though CADASIL patients usually develop first symptoms during early adulthood [2]. Main clinical aspects include migraine typically with aura [3], recurrent subcortical ischemic strokes and transient ischemic attacks presenting as lacunar syndromes [4], apathy and a number of psychiatric disturbances [5, 6], cognitive impairment [7], and acute encephalopathy [8]. Neuroimaging features, as detected by magnetic resonance imaging (MRI), are supportive of the diagnosis even 10–15 years before the onset of clinical manifestations and consist of white matter hyperintensities (WMH), lacunes, microbleeds, and dilated perivascular spaces, as in many other small vessel diseases (SVDs) [9]. WMH on T2-weighted or fluid-attenuated inversion recovery might be detected in the first phases of disease only in periventricular areas and in the centrum semiovale; subsequently, they spread symmetrically involving also the external capsule, the anterior portion of the temporal lobes, and the superior frontal gyrus [10].
CADASIL represents a monogenic disease with autosomal dominant inheritance caused by mutations of NOTCH3 which maps on chromosome 19p13.1 [11]. NOTCH3, a large gene containing 33 exons, encodes for a single-pass transmembrane receptor rich of epidermal growth factor repeats (EGFRs) in the extracellular domain, expressed in vascular smooth muscle cells (VSMCs) and pericytes [12, 13]. More than 200 NOTCH3 pathogenic variants have been described in CADASIL families so far. Over 95% of them are represented by missense mutations involving a cysteine residue in one of the EGFRs, mostly located in exons 3 and 4 [11], even though they can be found in all EGFR coding exons (2–24) [14]. The consequence is an increased number of cysteine residues in one of the 34 EGFR domains and the presence of one unpaired cysteine residue, resulting in an incomplete disulfide bridge with an increased NOTCH3 ectodomain (NOTCH3ECD) multimerization potential [15]. NOTCH3 mutations lead to VSMCs degeneration that is accompanied by the accumulation of granular osmiophilic material (GOM) in the extracellular space surrounding VSMCs, detected by electron microscopy [16]. Even though the whole composition of GOM is still debated, studies showed that NOTCH3ECD represents a significant constituent of these deposits [17]. Although VSMCs degeneration represents a pathological hallmark of CADASIL, an increasing number of studies has showed a significant role of endothelial cells in its pathogenesis. Endothelial cell injury seems to take part in vessel thrombosis, resulting in blood flow decrease, endothelial cell functional impairment, blood–brain barrier disruption, and, ultimately, brain parenchyma damage [18, 19]. Even though the monogenic cause differentiates it from sporadic SVDs, CADASIL represents a unique opportunity to investigate features of SVDs. An effective therapy for CADASIL is still not available, but recent findings deriving from experimental models might help considerably in this direction.
The purpose of this review is to provide an update of in vitro and in vivo models of CADASIL and to illustrate the most recent experimental therapeutic approaches currently under investigation.
Disease Models
Different disease models have been developed to investigate CADASIL pathogenic mechanisms and to assess the safety and effectiveness of novel therapeutic strategies. Here, we provide a review of the most relevant in vivo and in vitro disease models realized over the last decades.
In Vivo Models
Drosophila Lethal-Abruptex
NOTCH3 belongs to an evolutionarily conserved family of transmembrane receptors which includes Drosophila melanogaster Notch homologs 1 to 4 [20]. Drosophila Abruptex alleles represent a specific category of mutations within the Drosophila Notch gene associated with a typical phenotype, which includes interrupted wing veins and a defective number of dorsal bristle; this phenotype is due to amino acid substitutions within the extracellular domain of the Notch receptor, affecting the binding affinity for ligands, including Delta [21,22,23,24,25]. Among this group of mutations, Drosophila lethal-Abruptex resembles main features of CADASIL NOTCH3 variants by altering the number of cysteines in EGFR domains. It seems reasonable that the Drosophila lethal-Abruptex gene product inhibits Notch signaling [26] by interacting with the normal gene product at the cell surface and impairing the secondary structure of its extracellular domain so that it cannot cross-link adequately to binding proteins. Even though Drosophila lethal-Abruptex represents an in vivo model of CADASIL due to molecular similarities, Drosophila owns only one Notch gene, while mammals have four NOTCH genes [27], each one showing tissue-specific expression, thus producing significant differences between Abruptex and CADASIL.
Zebrafish Notch3 Mutants
Zebrafish transgenic models have been successfully generated. Specifically, Zaucker et al. [28] analyzed the effects of two mutations in the zebrafish Notch3 gene: the st51, which is known to reduce the expression of myelin basic protein [29], thus decreasing the number of oligodendrocyte precursor cells in larvae; the second mutation consisted of an insertional allele produced by a retroviral insertion. Histological studies performed on the Notch3 homozygous mutants survived to adulthood and, on adult zebrafish carrying both mutant alleles, showed impairment of vascular structure and function, including dilated and disorganized vessels and gaps in the arterial wall which produced hemorrhages, in association with reduced expression of Notch3 target genes such as hey1. On the other side, the presence of high Notch3 and hey1 expression in heterozygotes demonstrated the occurrence of modification in gene expression after incomplete loss of Notch3 [28]. Unfortunately, mutant Notch3 zebrafish does not recapitulate completely CADASIL pathology, thus highlighting the need for further models.
NOTCH3 Knock-Out Mice
Although NOTCH3 knock-out (NOTCH3KO) mice do not represent a genetic model of CADASIL, they provide essential evidence of NOTCH3 role in cerebrovascular maturation and homeostasis. Homozygous NOTCH3KO mice, despite being viable, fertile, and presenting no patent anatomic aberrations [30], display abnormalities of differentiation in small systemic and cerebral resistance arteries. Structural arterial deficits recorded in homozygous NOTCH3KO mice include a venous pattern of maturation and an altered location and orientation of VSMCs in the tunica media and around the lumen of small arteries due to compromised remodeling of their final shape [31]. Arboleza-Velasquez et al. [32] failed to detect the same aberrations in brain vessels and aorta of homozygous NOTCH3KO mice but found a downregulation of the conventional NOTCH3 downstream targets appointed to muscle contraction and maturation. Another work [33] showed that NOTCH3 deficiency produces functional deficits exclusively in cerebral small and tail caudal arteries, with a severe impairment of myogenic tone, thus compromising autoregulation of cerebral blood flow. Nevertheless, the lack of myogenic response restricted to isolated cerebral and tail arteries does not affect systemic blood pressure, which appears regulated mostly by larger peripheral vascular beds and hormonal vasoactive systems, including the sympathetic and renin-angiotensin systems [33]. Increased ischemia susceptibility was demonstrated in NOTCH3KO mice when occluding the proximal middle cerebral artery, which produced more than twofold larger infarcts compared to both NOTCH3 wild type (WT) and heterozygous NOTCH3KO mice [32]. The expression of WT NOTCH3 in VSMCs rescued the ischemic phenotype of NOTCH3KO mice, thus confirming the role of NOTCH3 in VSMCs ischemic susceptibility [32].
CADASIL Mutant Mice
Several CADASIL mutant mouse models have been realized and analyzed over the last decades [34,35,36,37]. Main differences among these models involve transgenic strategy, levels of expression of mutant NOTCH3, expression of endogenous NOTCH3, and consequences on NOTCH3 function.
The progressive development of arterial defects was analyzed by Ruchoux et al. [37] in a transgenic mouse model expressing the R90C mutation in the NOTCH3 human gene (TgNotch3R90C), an archetypal CADASIL mutation located in the EGFR2 and responsible for the addition of a cysteine residue. TgNotch3R90C mice were developed through a smooth muscle-specific promoter (SM22α), which drives the expression of a full-length human NOTCH3 cDNA carrying the R90C mutation in VSMCs and pericytes. The presence of signs of VSMCs degeneration, such as cytoskeleton changes and defective anchorage to extracellular matrix and cells before the deposition of NOTCH3ECD and GOM accumulation, suggests that these deposits might not be the trigger of VSMCs deterioration [37]. The functional consequences of the disruption of adhesion of VSMCs to the surrounding micro-environment consist of an impaired myogenic response to shear and tensile stress, while agonist or receptor-induced tone remains unchanged [38]. The increased actin polymerization in VSMCs might be the responsible of higher myogenic tone of arteries in this model; on the other side, the altered flow-mediated dilation might result from an indirect effect on endothelial cells [37]. Furthermore, Lacombe et al. [39] described an impaired cerebral vasoreactivity consisting of decreased relaxation and increased resistance of cerebral arteries, probably due to VSMCs dysfunction rather than VSMCs degeneration. Direct consequences are an altered cerebral blood flow autoregulation and an increased susceptibility to hypotension, thus promoting ischemic events [39]. In another work, focused on pericytes in TgNotch3R90C mice, their mitochondrial injury and autophagic degeneration were observed [40]. In TgNotch3R90C mice, NOTCH3 activity was not altered and NOTCH3ECD aggregates did not inhibit WT NOTCH3 function [41]. Actually, NOTCH3 activity levels remain constant in brain arteries of TgNotch3R90C mice, especially RBP-Jκ-mediated signaling activity which regulates the transcription of target genes in response to the binding of ligands belonging to the Delta/Jagged family and expressed on surrounding cells [41]. WT NOTCH3 removal did not influence the burden of white matter lesions in TgNotch3R90C mice [42]. Taken all together, these findings suggest that CADASIL pathogenesis might be associated with NOTCH3 gain of function rather that signaling loss.
Unlike TgNotch3R90C mice, those expressing the C428S mutation in the NOTCH3 human gene under the control of murine SM22α promoter (TgNotch3C428S) showed a loss of WT NOTCH3 activity and a mild dominant negative effect [36]. The authors [36] speculated that the consequences produced by the C428S mutation might be related to its location in the ligand-binding domain EGFR10, perhaps by the addition of an unpaired cysteine residue able to titrate WT NOTCH3 receptor into non-active complexes, thus reducing the RBP-Jκ-mediated signaling activity. In both the last two mouse models (TgNotch3R90C and TgNotch3C428S mice), Monet-Lepretre et al. [43] demonstrated that NOTCHECD accumulation induces the abnormal recruitment of extracellular matrix proteins, including tissue inhibitor of metalloproteinases 3 (TIMP3) and vitronectin, whose dysregulation contributes to these aggregates’ toxicity on small vessels.
Contrary to the transgenic mice above mentioned, those expressing the R142C mutation of the NOTCH3 mouse gene (TgNotch3R142C), corresponding to the very common human R141C, did not show the typical CADASIL pathological and neuroradiological features up to 20 months of age [35]. This mouse model was generated by employing a targeting construct containing the R142C for homologous recombination in embryonic stem cells (knock-in). The authors [35] excluded that the absence of CADASIL phenotype was due to an altered expression or processing of R142C NOTCH3 at RNA or protein levels. They hypothesized that a species difference between human and murine NOTCH3 could explain why mice carrying the murine gene mutated at the Arginine 142 site did not develop a CADASIL phenotype, unlike CADASIL patients with NOTCH3 R141C mutation and TgNotch3R90C mice, which carried the human NOTCH3 mutated gene [35].
CADASIL pathological hallmarks, such as NOTCH3ECD aggregations and GOM, were detected at younger age in the transgenic mice expressing the R169C mutation of NOTCH3 rat gene (TgNotch3R169C) compared to previous models, respectively at 1–2 months and 5 months of age in TgNotch3R169C mice [34] and at 14–16 months in TgNotch3R90C ones [37]. The TgNotch3R169C mice were developed by introducing the NOTCH3 R169C mutation into a large P1-derived artificial chromosome (PAC) and showed a significantly higher expression of NOTCH3 mutant gene [34] compared to previous models. Furthermore, the distribution of transgene expression resembled that of the endogenous gene. The authors reported the presence of early isolated white matter lesions and hypoperfusion not only in the areas of brain damage but also in those free from lesions [34], consistent with human findings [44]. Previous works showed a strong relation between hypoperfusion and white matter damage both in human and in mouse models [45,46,47]. In this model, the presence of significantly decreased resting cerebral blood flow, especially in white matter, even months before the appearance of brain damage, corroborates the hypothesis that hypoperfusion could be involved in white matter lesions pathogenesis [34]. The origin of severe and early reduction in cerebral blood flow is uncertain, but it might be secondary to decreased myogenic response, altered mechanical arterial functions, reduced vessel diameter resulting in increased vascular resistance, and capillary rarefaction [34]. The altered myogenic response was proved to be caused by abnormal hyperpolarization in parenchymal and pial arterioles secondary to an increased number of voltage-gated potassium (KV1) channels, which are involved in vascular smooth muscle membrane potential regulation, induced by disruption of TIMP3-sensitive pathway [48]. Indeed, TIMP3 inhibits the metalloprotease ADAM17 and HB-EGF, an EGFR agonist, in order to maintain cerebral vascular tone [49]. Reduced vasoconstriction secondary to increased luminal pressure was observed not only in pial arteries and cortical parenchymal arterioles [48, 49] but also in hippocampal microcirculation of TgNotch3R169C mice [50]. HB-EGF, which restores normal depolarization of VSMCs membrane, rescued cerebrovascular reactivity [50]. Since TgNotch3R169C mice developed NOTCH3ECD accumulation, GOM deposition, and white matter lesions in the absence of infarcts or motor deficits [34], this model is considered suitable to reproduce the early pre-symptomatic phases of disease. Segmental intramyelinic edema, represented by microvacuoles with focal myelin degradation without oligodendrocyte loss and axonal injury, was suggested to be an early alteration of white matter in CADASIL [51]. TgNotch3R169C mice showed also an impairment of NOTCH3 function in hippocampal precursor cells regulation, which depends on a balance between NOTCH1 and NOTCH3 binding to RBPJκ [52], producing respectively an activation or inhibition of hairy and enhancer of split (HES) and HES-related with YRPW motif (HEY) gene transcription [53]. Consistent with this evidence, an impairment in hippocampal neurogenesis at 12 months of age was described, probably due to an age-dependent suppression of cell proliferation by NOTCH3 over-expression [52]. Impaired hippocampal neurogenesis was confirmed both in TgNotch3R169C and TgNotch3WT by failure of environmentally enriched or running wheel cages, which simulate physiological neurogenesis stimuli [54]. Although reduced if compared with that of WT mice, physical activity of TgNotch3R169C mice was higher [54] than the rate of running wheel activity required to induce neurogenesis [55]. In a following work, the R169C NOTCH3 mutation was showed to be associated with decreased maximal dilation of cerebral arteries secondary to a higher NOTCH3 activity [56]. These findings seem to differentiate the R169C NOTCH3 mutation from those located in the ligand-binding domain of NOTCH3, which are predicted to reduce NOTCH3 activity and provide an explanation to the phenotype of R169C, which shows a more severe cognitive decline than the others according to genotype-phenotype correlation studies [36]. Furthermore, this transgenic mouse model was employed to confirm the role of endoplasmic reticulum stress and RhoA/Rho kinase in CADASIL pathogenesis, as proved by the normalization of vascular dysfunction through the employment of 4-PBA and fasudil, two pharmacological agents that inhibit endoplasmic reticulum stress and Rho kinase [57]. In this TgNotch3R169C mice model, it was found a reduced number of pericytes and a decreased pericytes coverage of cortical vessels which causes blood–brain barrier disruption and plasma proteins extravasation [58].
NOTCH3 transgenic mice expressing the mutated human NOTCH3 transgene C455R and R1031C by Cre-mediated recombination (TgNotch3R1031C and TgNotch3C455R mice), inserted into the ROSA locus [59], were realized by using the SM22α promoter [60]. TgNotch3R1031C and TgNotch3C455R mice showed the expected pathological CADASIL features without developing ischemic events [60]. Even though the age-dependent development of deposits in transgenic mice resembles what happens in CADASIL patients, GOM localization is quite different. Indeed, it is found not only in the extracellular space near plasma membrane of VSMCs [16] but also inside cells, in association with vesicles and inclusions [60]. The R1031C and C455R mutations, firstly detected in Colombian kindreds, are located respectively in the EGFR26 and in the EGFR11, which contains a ligand-binding domain [61]. While the first has been associated with a typical onset in the fourth decade of life, the second one is carried by patients who usually develop ischemic events earlier [61]. Experiments on mouse embryonic fibroblasts expressing a NOTCH3 ligand and WT NOTCH3 or mutant NOTCH3 receptors were performed, supporting the hypothesis of an age-dependent hypomorphic phenotype, with a stronger loss of function mechanism associated with the C455R rather than with the R1031C mutation [60]. Intriguingly, proteomic analysis revealed that clusterin and collagen 18α1 (COL18A1), which is cleaved post translationally into the antiangiogenic endostatin, were highly expressed in CADASIL samples rather than controls and were detected in GOMs of CADASIL-affected arteries [60]. TgNotch3R1031C and TgNotch3C455R mice were subsequently analyzed to identify candidate CADASIL biomarkers such as increased plasma levels of COL18A1, endostatin and HtrA serine protease 1 (HTRA1), reduced levels of NOTCH3ECD, and loss of VSMC in retinal vessels [62]. The increased plasma levels of HTRA1 acquire an intriguing meaning if we consider that the HTRA1 gene represents the genetic cause of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) [63]. Even though CADASIL and CARASIL share similar clinical, neuroradiological, and pathological features [64], the role and the origin of HTRA1 accumulation in CADASIL remain uncovered.
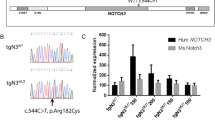
TgNotch3R182C mice, developed by using a BAC clone containing the human NOTCH3 gene and subsequently introducing the R182C variant by two-step Red-mediated recombination, showed progressively increasing age- and NOTCH3 RNA expression level-dependent vascular accumulation of NOTCH3 and GOM deposits, which progress over time and are continuously being formed, without brain parenchyma lesions occurrence [65]. This finding led authors to realize the “NOTCH3 score” as quantitative biomarker for CADASIL, attempting to transform transgenic CADASIL mice in a proper model for pre-clinical testing of therapeutic approaches [66].
Clinical and pathological features of CADASIL were partially but not exhaustively reproduced by TgNotch3R170C mice [67]. Since the age of 8 months only a minority of mice developed GOM deposition, VSMCs abnormalities with enlargement of intercellular spaces, microbleeds, thrombosis, fibrillar gliosis, microinfarctions, and neurological features, including ataxia and paresis [67]. TgNotch3R170C mice did not show an altered Notch3 signaling [67], and expression levels of Notch3 target genes were not reduced when compared with WT mice [42], thus supporting the hypothesis that CADASIL pathogenesis might be secondary to a NOTCH3 gain of function rather that a signaling inhibition. Unlike previous models [36, 37], evidence of NOTCH3ECD accumulation was detected only in one study [42], while others failed to confirm it [67]. These discoveries excluded that the lack of typical pathological findings usually found in mice could be explained by their short lifespan, which should have not allowed the development of cell degeneration. Perhaps, the significant differences of murine brain vessels compared to human ones might reduce their vulnerability to hypoperfusion, thus explaining the incomplete phenotype developed by mice. Nevertheless, their role in providing increasing knowledge about CADASIL pathogenesis and relevant features is irreplaceable.
Main features of knock-out, knock-in, and transgenic mouse models are summarized in Table 1.
In Vitro Models
The first in vitro studies were based on primary cell models, which included VSMCs [68, 69], skin fibroblasts [70], and myoblasts [71] derived from CADASIL patients; HS683 oligodendrocytes transfected with mutant NOTCH3 [72]; human embryonic kidney 293 cells (HEK293) and neuroblastoma cells (SH-SY5Y) in which the expression of mutant NOTCH3 was inducible using the tetracycline (Tet)-on regulatory system [15, 73,74,75,76,77]; primary mouse embryonic fibroblasts (MEFs) from mice carrying mutant NOTCH3 transgenes [78]; and Lec1-Chinese hamster ovary (CHO Lec1) cell lines transfected with plasmids encoding murine Notch3 fragments bearing CADASIL-like mutants [78].
The possibility to induce pluripotent stem cells (iPSCs) from blood cells and skin fibroblasts has recently transformed the field of disease-specific in vitro models. iPSCs reproduce main features of embryonic stem cells and can be differentiated in different cell types [79]. The first human iPSC line from a CADASIL patient (IDISi001-A) was generated by Fernández-Susavila et al. in 2018 [80], starting from peripheral blood mononuclear cells of a patient carrying the R1242C NOTCH3 mutation. iPSCs from a R1076C CADASIL patient were subsequently differentiated in VSMCs, embryonic, and mesenchymal stem cells by Ling et al. in 2019 [81], thus representing all three layers of vessel wall. The authors reported several aberrations of VSMCs structure and function, including increased proliferation rate and altered cytoskeleton features [81]. Notably, they described constitutive activation of NOTCH signaling, responsible for NF-κB pathway stimulation [81] consistently with previous works [82,83,84]. NF-κB regulates expression of key components of the inflammatory response, such as cytokines, chemokines, and adhesion molecules, but also of genes implicated in extracellular matrix remodeling [85,86,87,88,89], whose upregulation in CADASIL VSMCs was demonstrated by transcriptomic analysis and resulted in vascular dysfunction [81]. These findings were even more relevant in case of TNFα-induced inflammatory stimuli, suggesting an excessive cerebral blood vessels susceptibility to damage within an inflammatory scenario [81]. Furthermore, NF-κB induces expression of vimentin [90], a component of intermediate filaments, whose structure is altered in CADASIL VSMCs. Abnormalities were also detected in microfilament structure of CADASIL VSMCs, already reported in CADASIL mice and primary VSMCs from patients [31, 69], and probably associated with NOTCH3-mediated expression of smooth muscle α-actin and Rho kinase [91, 92]. Apparently, endothelial cells showed only altered innate immunity and cellular adhesion and upregulated target genes of NF-κB in presence of TNFα-induced inflammatory factors [81]. On the other side, the adventitia presented only an age-dependent loss of mesenchymal stem cells [81]. In another work, Kelleher and colleagues [93] generated iPSCs from two CADASIL patients, respectively, with R153C and C224T NOTCH3 mutations, subsequently differentiated in vascular mural cells. iPSC-derived vascular mural cells represent an in vitro pericyte-like model, as demonstrated by the expression of pericyte markers like platelet-derived growth factor receptor beta (PDGFRβ) and NG2 [94] and by their capillary supporting function. PDGFRβ and VEGF are known stabilizing factors for microvessels [95,96,97]. Consequently, decreased PDGFRβ and reduced secretion of VEGF found in iPSC-derived vascular mural cells appear involved in impaired stabilization of capillary structures, partially rescued by the administration of VEGF and by siRNA knockdown of NOTCH3, supporting the hypothesis of a gain-of-function mechanism [93]. On the contrary, Yamamoto et al. [98] described an increased level of PDGFRβ in iPSC-derived vascular mural cells from three CADASIL patients with R182C, R41C, and C106R NOTCH3 variants, which was reduced by DAPT, a NOTCH3 pathway inhibitor. Discrepancies across studies might be the consequence of differences in differentiation protocols, particularly because platelet-derived growth factor-BB (PDGF-BB) suppresses the expression of PDGFRβ. Anyway, the different expression of PDGFR-β, either incremented or reduced, destabilizes the structure of new blood vessels, resulting in an increased susceptibility to mechanical stress. Furthermore, iPSC-derived vascular mural cells show NOTCH3ECD accumulation, which co-localize with latent transforming growth factor β binding protein-1 (LTBP-1) and HTRA1, known components of GOM [99,100,101].
Even though in vitro models represent a promising approach to investigate diseases’ pathogenesis, limitations associated with cell lines and iPSCs artificial reprogramming and transcriptional, morphological, and functional modifications in cell culture should be outlined. As a consequence, definite conclusions are hardly reached by in vitro models, at least without discoveries validation by in vivo approach.
Development of Therapeutic Strategies
To date, only few therapeutic approaches are applied in clinical practice for CADASIL. Hereafter, after a brief overview of these common strategies in CADASIL, we focus the review on novel therapeutic approaches, such as immunotherapy, growth factors administration, and antisense oligonucleotides, which are currently under investigation and are illustrated in Fig. 1.

Novel therapeutic approaches under evaluation by in vitro and in vivo models. Growth factors: the administration of hematopoietic growth factors (SCF and G-CSF) in TgNotch3R90C mice, after irradiation of bone marrow, is effective in (1) preventing VSMCs degeneration in small arteries and loss of cerebral capillaries by inhibition of apoptotic cascade; (2) reducing cerebral thrombosis; and (3) implementing neurogenesis, especially in the subventricular zone. Immunotherapy: agonist antibodies (A13 and 5E1) targeting NOTCH3 domains have been tested in NOTCH3KO and transgenic mice (respectively TgNotch3C455R and TgNotch3R169C), showing respectively (1a) increased SMA coverage in retinal arterioles; (1b) increased NOTCH3ECD/COL18A1/endostatin levels; and (2) restoration of adequate vasodilatory responses and myogenic tone. AONs: exon skipping through AONs excludes mutant EGFRs with no impairment of NOTCH3 structure or function and produces a novel “EGFR fusion domain,” with correctly spaced cysteines, derived from parts of EGFR domains encoded by exons located before and after those skipped through AONs (except for exon 2–3 skipping)
Pharmacological Approaches and Control of Risk Factors
Stroke
A single report of thrombolysis for stroke in a CADASIL patient has been reported [102], without sign of intracranial hemorrhages, a feared side effect in this population known to have microbleeds [9, 103]. Similarly, the benefit of antiplatelet drugs in CADASIL ischemic stroke prevention is unclear [104], and cases of aspirin-associated intraparenchymal bleeding are described [105]; despite of this uncertainty, most neurologists administer aspirin or clopidogrel, according to the guidelines applied for sporadic stroke. The usual prevention measures employed for stroke are applied also in CADASIL patients, including avoiding cigarette smoking, because of its relation with an earlier age of onset and incremented risk of stroke and migraine [106, 107]; managing hypertension, trying not to reach excessively low blood pressures, which are associated with an increased risk of dementia [108, 109]; and controlling diabetes [110], alcohol intake, obesity, and hypercholesterolemia with the help of statins. As above reviewed, chronic cerebral hypoperfusion might be involved in ischemic events in CADASIL patients; however, a few trials targeting vascular dysfunction failed to reveal any benefit of atorvastatin [111] and sapropterin, which acts as cofactor in nitric oxide synthesis [112]. After successful results in small studies [44, 113, 114], further investigations are required to demonstrate clinical benefits of acetazolamide and lomerizine.
Migraine
Common analgesic medications can be employed during CADASIL migraine attacks, especially a fixed combination of acetaminophen, acetylsalicylic acid, and caffeine [115]. Anecdotal reports describe also effectiveness of sodium valproate in acute setting [116]. Safety of triptans and ergot derivatives should be demonstrated before their use is implemented in clinical practice, due to the risk of vasoconstriction and injury of capillary endothelium [117, 118]. Many anecdotal reports have recorded a quite striking effect of acetazolamide for migraine prophylaxis with ensuing hypothesis on this drug way of action in CADASIL [119]. Amitriptyline, beta-blockers, flunarizine, and topiramate should be used with caution because of their impact on mood and cognitive disturbances [120]. Detection of higher homocysteine levels in CADASIL patients with migraine, especially in case of early age of onset [121], suggests the possibility to employ homocysteine lowering therapies with B vitamin supplementation to reduce migraine severity and frequency [122], but further investigations are warranted.
Cognitive Impairment
The identification of cholinergic deficit in CADASIL [123, 124] led to start clinical trials aimed to test acetylcholinesterase inhibitors. The use of donepezil in a multicenter randomized controlled trial with 168 patients did not show to improve the primary endpoint (the Vascular Dementia Assessment Scale cognitive subscale) significantly but produced some differences in secondary endpoints concerning executive function [125]. However, the presence of a small number of patients with dementia and the brief follow-up suggests that, again, larger experiences are needed [126].
Psychiatric Disturbances
Psychiatric symptoms seem to respond to common treatments used in sporadic disturbances. Anecdotal cases described the efficacy of quetiapine in a bipolar disorder [127] and of risperidone, sodium valproate, and flupentixol in schizophreniform organic psychosis [128].
Immunotherapy
Recently, NOTCH3KO and TgNotch3C455R mice were used to assess the impact of agonist antibodies (A13) targeting the NOTCH3 receptor’s negative regulatory region (NRR) located in the extracellular domain on preventing mural cell loss, a pathological hallmark of SVDs involved in CADASIL pathogenesis [129]. NRR stabilizes noncovalent bonds, thus maintaining NOTCH3 in an autoinhibited configuration in the absence of the ligand (Delta or Jagged). A S2 cleavage site, which undergoes ADAM-mediated proteolysis after the interaction of NOTCH3 with its ligands, is located within the NRR [130]. As above mentioned, unlike other NOTCH3 mutations, C455R impairs ligand-mediated NOTCH3 signaling [60]. Authors chose to evaluate mural cell coverage of retinal vessels, due to their blood barrier similarities with the cerebral one, by α-smooth muscle actin (SMA) staining, whose expression is not influenced by NOTCH3 activity modifications [129]. Firstly, after ascertaining a severe reduction of mural cell coverage in retinal vessels of NOTCH3KO and TgNotch3C455R mice of 6 months of age, authors observed that expressing WT human NOTCH3 transgene, but not C455R NOTCH3 transgene, was able to avoid mural cell loss both in NOTCH3KO and in TgNotch3C455R mice [129]. In vitro the A13 antibody succeeded in activating not only the WT but also the C455R mutant receptor, by exposing the S2 cleavage site and subsequently destabilizing the NRR independently from the Jagged 1 ligand [129]. Simultaneously, NOTCH3ECD, which is known to be reduced in TgNotch3C455R mice [62], was increased in the cell culture supernatant of WT and C455R cells incubated with A13, and this result was not affected by the addition of compound E, an A13 inhibitor, even though NOTCH3 signaling was reduced [129]. The effect of agonist antibody was investigated in vivo by injecting A13 in TgNotch3C455R mice for 5 weeks after the 8th day of life. At 6 weeks of age, SMA coverage in retinal arterioles was duplicated, whereas no differences were observed in large retinal arteries, thus suggesting a NOTCH3-mediated mural cell loss prevention [129]. Concurrently, NOTCH3ECD and COL18A1/endostatin levels were found increased as consequence of NOTCH3 signaling activation, unlike HTRA1 and insulin-like growth factor binding protein-1 (IGFBP-1) [129]. This finding suggests a potential role of NOTCH3ECD and COL18A1/endostatin plasma levels as potential biomarkers of NOTCH3 activity.
In a following work, another antibody targeting the extracellular domain of NOTCH3 (5E1) was chronically administered to TgNotch3R169C mice [131], which show the main pathological features of CADASIL, as previously mentioned [34]. The results consisted of restoration of regular vasodilatory responses and myogenic tone, without effect on NOTCH3ECD and GOM deposition even if therapy was begun before the achievement of a plateau of deposits [131]. Compared to Alzheimer’s disease mouse models, in which extracellular ß amyloid plaques disappear after treatment with antibodies against amyloid ß peptide, perhaps as consequence of microglial and macrophage activation [132,133,134], in CADASIL mice microglial might be dysfunctional and so unable to remove NOTCH3ECD and GOM deposits. An alternative hypothesis suggests that brain dysfunction might be related to soluble species rather than aggregates. In the previously reported experiments, 5E1 might have acted by eliminating these soluble toxin assemblies or by delaying their release from extensive aggregates [131].
Even though the potential of immunotherapy appears intriguing, further investigations and considerable improvements are required in vivo before moving to humans. First, the choice of antibodies should consider the affinity for NOTCH3 receptor and the ability to cross the blood–brain barrier, thus allowing to reach the brain in a more elevated percentage when peripherally administered; secondly, mice with pre-existing cerebrovascular features should be involved, thus providing a complete therapeutic context [131].
Growth Factors
Different works recently showed a potential benefit of administering hematopoietic growth factors such as stem cell factor (SCF), granulocyte colony-stimulating factor (G-CSF), and their combination in the acute, subacute, and even chronic phase of ischemic stroke in animal models [135,136,137,138]. SCF and G-CSF are deeply involved in blood cell generation as well as bone marrow cell renewal and mobilization [139]. Starting from these evidences, Liu et al. [140] tried to replicate these positive results in a CADASIL mouse model (TgNotch3R90C mice), which was irradiated to destroy bone marrow at 8 months, transplanted with the bone marrow of UBC-GFP transgenic mice for tracking bone marrow-derived cells in the brain, and treated with recombinant mouse SCF and recombinant human G-CSF after a 1-month recovery. The results were promising. Compared to control group, the treated arm displayed a long-term improvement of cognitive function in terms of spatial learning and memory, which are severely affected in TgNotch3R90C mice, as demonstrated by the water maze test performed at 11 and 19 months of age [140]. This effect might be related to the evident prevention of VSMCs degeneration in small arteries and loss of cerebral capillaries, whose pathogenic role in CADASIL evolution is well established [34, 37,38,39, 44, 141, 142], in addition to the inhibition of GOM formation. The explanation for this finding is not fully clear, even though it is thought to be related to VSMCs survival via AKT pathway and inhibition of caspase-3 cascade during an exposure to antiapoptotic factors. The authors highlighted the presence of altered capillaries in the brains of CADASIL mice made of bone marrow-derived endothelial cells [140]. This finding would explain the continuous consumption of endothelial progenitor cells observed in CADASIL patients, as consequence of their role in the replacement of damaged endothelium in course of vascular degeneration [143]. These abnormal capillaries are decreased in brains of SCF + G-CSF-treated CADASIL mice [140]. Furthermore, treatment with SCF + G-CSF was found to prevent the apoptosis cascade in mice; to implement neurogenesis, specifically in the subventricular zone, perhaps as consequence of the reduced loss of neural stem cells and neural progenitor cells compared to CADASIL mice, which are probably involved in vascular cells renewal in conditions of VSMCs degeneration, brain capillaries damage, and chronic brain ischemia; and to expand the number of bone marrow-derived cells especially in the hippocampus [140], which are thought to differentiate in neuronal-like cells in the brain, as consistent with previous works [135, 136, 144].
Subsequently, the same group repeated experiments on TgNotch3R90C mice to assess the impact of SCF + G-CSF administration at 9, 10, 12, 15, and 20 months of age on cerebral thrombosis occurrence [145]. The higher rate of thrombosis with associated IgG extravasation was detected in small arteries and in cerebral capillaries, especially near bifurcations, which represent areas of complex blood flow, predisposed to injury [145]. As strictly linked to this evidence, the authors described endothelial cell loss and replacement by bone marrow-derived cells all over the thrombotic regions [145], as previously reported in a mouse model of ischemic stroke [146]. However, these novel cerebral vessels show blood–brain barrier leakage, thus suggesting a potentially dysfunctional nature. Small vessel thrombosis appeared deeply reduced in the treatment group at 22 months of age compared to the control one [145].
VEGF and VEGF-A have been found to be notably reduced in TgNotch3R90C mice brains of 10–11 months of age compared to aged-matched WT controls [147]. Their role in angiogenesis is well known [148, 149]. Even though the relation between NOTCH3 mutations and decreased VEGF and VEGF-A levels is undetermined, the result is a reduction in cerebral vessel density, significantly associated with the reduction of dendrites, axons, and synapses in the somatosensory and motor cortex layer 2/3 and in the hippocampal CA1, and decreased neurogenesis in the subventricular zone and subgranular zone, resulting in impaired spatial learning and memory [147], as previously reported [140]. The administration of SCF + G-CSF at 9 and 10 months of age, before the occurrence of pathological alterations in VSMCs of TgNotch3R90C mice [37], was able to prevent all these processes, thus improving cognitive function in TgNotch3R90C mice. Since the use of Avastatin, a VEGF-A inhibitor, was able to impede the pro-angiogenetic and neurogenetic effect of SCF + G-CSF, the authors hypothesized that VEGF-A was required for SCF + G-CSF-mediated brain repair. Indeed, the administration of SCF + G-CSF in TgNotch3R90C mice enhanced VEGF and VEGF-A levels. In agreement with findings of previous studies in experimental models of chronic stroke [150, 151], the potential role of NF-κB, a transcription factor which induces VEGF expression and angiogenesis [152, 153], on SCF + G-CSF-enhanced cerebral VEGF and VEGF-A production needs to be clarified.
Even though subcutaneous injection of cell growth factors appears a suitable approach and has been already employed for bone marrow recovery after chemotherapy in patients with cancer, the beneficial effects in CADASIL patients remain largely unknown, thus requiring further investigations prior to human administration.
Antisense Oligonucleotides
The dramatic impact of small synthetic RNA or DNA oligomers to bind specific sequences of pre-mRNA of target genes, thus excluding specific exons from splicing and, consequently, from the mature mRNA, has been already highlighted for monogenic neurologic diseases, such as Duchenne muscular dystrophy [154] and spinal muscular atrophy [155].
Tikka et al. [69] obtained NOTCH3 silencing by employing short hairpin RNA (shRNA) in CADASIL and control VSMCs to assess the aforementioned vasoregulatory role of NOTCH3. The result was a statistically significant reduction of NOTCH3 target HES5 expression and signaling, as well as actin aberrations, both in VSMCs from controls and CADASIL patients [69].
In 2016, Rutten et al. [156] provided a first experimental exon skipping approach for CADASIL to prevent NOTCH3ECD toxic accumulation in vitro. Even though it could be used for 18 of 23 exons encoding for EGFR domains, embracing 94% of all CADASIL mutations, the authors selected antisense oligonucleotides (AONs) to skip specifically exons 2–3, 4–5, and 6, based on in silico predictions of the effect of EGFRs exclusion on primary, secondary, and tertiary structure. As expected, mutant EGFR was excluded with no impairment of NOTCH3 structure or function. Except for exon 2–3 skipping, the idea is not only to eliminate the mutant EGFR domain but also to realize a novel “EGFR fusion domain,” with six correctly spaced cysteines, derived from parts of EGFR domains encoded by exons located before and after those skipped through AONs. The result is a shorter NOTCH3 protein with no pathogenic unpaired cysteines. Skip proteins were proved to have a normal subcellular localization in transfected fibroblasts, predominantly perinuclear, reflecting an accumulation in RE and Golgi network [73, 74], and secondary at the cell surface where they co-localized with the ligand jagged 1, ensuring a correct protein processing and ligand-binding ability. Furthermore, a luciferase report assay confirmed a conserved binding activation rate. Subsequently, exon skipping revealed to be successful also in patient-derived VSMCs, and, at the same time, a reduction of NOTCH3 and downstream target genes (HES1, HEYL, PDGFR-β and JAG1) expression was excluded. However, the consequences of exon skipping on NOTCH3 aggregation in vessels, such as NOTCHECD deposits, GOM and VSMCs loss, could not be assessed because it requires in vivo studies, thus highlighting the importance of mouse models. A potential alternative to exon skipping could be NOTCH3 downregulation through AONs inducing RNase H-mediated degradation of RNA, something that remains to be done [156].
The importance of exon skipping as therapeutic approach in CADASIL has been recently suggested by the detection of a NOTCH3 mutation (G498C), which induces exon 9 skipping due to its location in a donor splice site, in a 63-year-old proband and in three siblings showing only WMH on MRI and no other CADASIL neuroradiological features. Overall, they presented a mild phenotype, with non-migraine headaches or initial cognitive impairment, and familiarity for ischemic stroke. Skin biopsies failed to reveal GOM and showed only mild NOTCH3ECD accumulation. In this case, transfected fibroblasts showed normal localization of NOTCH3 protein at the cell surface, but impaired ligand-dependent signaling. To confirm the translatability of their finding, the authors realized both an exon skipping by AONs in transfected VSMCs and a gene editing through CRISPR/Cas9 in HEK293 cells [157].
Obviously, in vitro studies represent only the first step in the process of validation of cysteine corrective exon skipping as efficacious therapeutic approach for CADASIL. In vivo studies on animal models are required before moving to the human setting. However, these encouraging preliminary data seem to extend the growing list of potential therapeutic applications of exon skipping [158].
Conclusions
CADASIL is a unique model of SVDs, with typical clinical and neuroradiological features as well as pathological hallmarks represented by NOTCH3 accumulation and GOM deposits in abnormal VSMCs. Despite the efforts, the well-known genetic landscape and the growing number of reliable disease models, its pathogenesis and the specific NOTCH3 pathway involved remain elusive. In vivo models, especially knock-out, knock-in, and transgenic mice, have provided a large amount of data, expanding our molecular and biological knowledge of underlying pathogenic mechanisms which involve altered myogenic responses secondary to a potassium channelopathy-like defect, which result in impaired cerebral blood flow autoregulation, hypoperfusion, and ischemia susceptibility. The most evident limit of mouse models is surely represented by the lack of pathological and clinical CADASIL features, thus making necessary the development of different disease models. Over the last years, iPSC technology has revolutionized the field of in vitro models. The possibility to investigate disease underlying processes and to test novel therapeutic approaches in a safe and truthful manner is opening new unexplored avenues for so many neurological disorders. In this scenario, available therapeutic strategies for CADASIL remain inadequate. Novel approaches have been recently tested in vitro and in vivo, including immunotherapy [129, 131], growth factors administration [140, 145, 147], and exon skipping through AONs [156]. All these techniques have shown promising results in preliminary studies and have been already employed as revolutionary therapy for other neurological and non-neurological diseases [154, 155]. To prevent side effects of novel therapies, alterations of NOTCH3 signaling involved in CADASIL pathogenesis, which are still uncertain, should be cautiously evaluated. Even though the feeling of lack of knowledge remains, we are moving to an increased availability of potential therapeutic targets, so that we can hope for phase 1 clinical trials in the next future.
Data Availability
Not applicable.
References
Wang MM (2018) CADASIL. pp 733–743
Cramer J, White ML (2020) Cerebral autosomal dominant arteriopathy (CADASIL)
Guey S, Mawet J, Hervé D, Duering M, Godin O, Jouvent E, Opherk C, Alili N et al (2016) Prevalence and characteristics of migraine in CADASIL. Cephalalgia 36:1038–1047. https://doi.org/10.1177/0333102415620909
Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG (2009) CADASIL. Lancet Neurol 8:643–653. https://doi.org/10.1016/S1474-4422(09)70127-9
Reyes S, Viswanathan A, Godin O, Dufouil C, Benisty S, Hernandez K, Kurtz A, Jouvent E et al (2009) Apathy: a major symptom in CADASIL. Neurology 72:905–910. https://doi.org/10.1212/01.wnl.0000344166.03470.f8
Valenti R, Poggesi A, Pescini F, Inzitari D, Pantoni L (2008) Psychiatric disturbances in CADASIL: a brief review. Acta Neurol Scand 118:291–295. https://doi.org/10.1111/j.1600-0404.2008.01015.x
Charlton RA, Morris RG, Nitkunan A, Markus HS (2006) The cognitive profiles of CADASIL and sporadic small vessel disease. Neurology 66:1523–1526. https://doi.org/10.1212/01.wnl.0000216270.02610.7e
Schon F, Martin RJ, Prevett M, Clough C, Enevoldson TP, Markus HS (2003) “CADASIL coma”: an underdiagnosed acute encephalopathy. J Neurol Neurosurg Psychiatry 74:249–252. https://doi.org/10.1136/jnnp.74.2.249
Jouvent E, Duering M, Chabriat H (2020) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 51:21–28. https://doi.org/10.1161/STROKEAHA.119.024152
Chabriat H, Levy C, Taillia H, Iba-Zizen MT, Vahedi K, Joutel A, Tournier-Lasserve E, Bousser MG (1998) Patterns of MRI lesions in CADASIL. Neurology 51:452–457. https://doi.org/10.1212/WNL.51.2.452
Joutel A, Corpechot C, Ducros A et al (1997) Notch3 mutations in cerebral autosomal dominant Arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a Mendelian condition causing stroke and vascular dementia. Ann N Y Acad Sci 826:213–217. https://doi.org/10.1111/j.1749-6632.1997.tb48472.x
Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, Battail N, Piga N, Chapon F et al (2000) The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest 105:597–605. https://doi.org/10.1172/JCI8047
Morroni M, Marzioni D, Ragno M, di Bella P, Cartechini E, Pianese L, Lorenzi T, Castellucci M et al (2013) Role of electron microscopy in the diagnosis of Cadasil syndrome: a study of 32 patients. PLoS One 8:e65482. https://doi.org/10.1371/journal.pone.0065482
Federico A, Bianchi S, Dotti MT (2005) The spectrum of mutations for CADASIL diagnosis. Neurol Sci 26:117–124. https://doi.org/10.1007/s10072-005-0444-3
Duering M, Karpinska A, Rosner S, Hopfner F, Zechmeister M, Peters N, Kremmer E, Haffner C et al (2011) Co-aggregate formation of CADASIL-mutant NOTCH3: a single-particle analysis. Hum Mol Genet 20:3256–3265. https://doi.org/10.1093/hmg/ddr237
Baudrimont M, Dubas F, Joutel A, Tournier-Lasserve E, Bousser MG (1993) Autosomal dominant leukoencephalopathy and subcortical ischemic stroke. A clinicopathological study. Stroke 24:122–125. https://doi.org/10.1161/01.STR.24.1.122
Ishiko A, Shimizu A, Nagata E, Takahashi K, Tabira T, Suzuki N (2006) Notch3 ectodomain is a major component of granular osmiophilic material (GOM) in CADASIL. Acta Neuropathol 112:333–339. https://doi.org/10.1007/s00401-006-0116-2
Ruchoux M (1998) Endothelial changes in muscle and skin biopsies in patients with CADASIL. Neuropathol Appl Neurobiol 24:60–65. https://doi.org/10.1046/j.1365-2990.1998.00087.x
Stenborg A, Kalimo H, Viitanen M, Terent A, Lind L (2007) Impaired endothelial function of forearm resistance arteries in CADASIL patients. Stroke 38:2692–2697. https://doi.org/10.1161/STROKEAHA.107.490029
Artavanis-Tsakonas S (1999) Notch signaling: cell fate control and signal integration in development. Science (80- ) 284:770–776. https://doi.org/10.1126/science.284.5415.770
de Celis JF, Garcia-Bellido A (1994) Modifications of the notch function by Abruptex mutations in Drosophila melanogaster. Genetics 136:183–194
Kelley MR, Kidd S, Deutsch WA, Young MW (1987) Mutations altering the structure of epidermal growth factor-like coding sequences at the Drosophila Notch locus. Cell 51:539–548. https://doi.org/10.1016/0092-8674(87)90123-1
Lieber T, Wesley CS, Alcamo E, Hassel B, Krane JF, Campos-Ortega JA, Young MW (1992) Single amino acid substitutions in EGF-like elements of notch and delta modify drosophila development and affect cell adhesion in vitro. Neuron 9:847–859. https://doi.org/10.1016/0896-6273(92)90238-9
Portin P (1975) Allelic negative complementation at the Abruptex locus of Drosophila melanogaster. Genetics 81:121–133
Sirén M, Portin P (2008) Gene dosage studies of a temperature sensitive Abruptex mutation of the Notch locus of Drosophila melanogaster. Hereditas 110:175–178. https://doi.org/10.1111/j.1601-5223.1989.tb00439.x
Spinner NB (2000) CADASIL: Notch signaling defect or protein accumulation problem? J Clin Invest 105:561–562. https://doi.org/10.1172/JCI9511
Williams R, Lendahl U, Lardelli M (1995) Complementary and combinatorial patterns of Notch gene family expression during early mouse development. Mech Dev 53:357–368. https://doi.org/10.1016/0925-4773(95)00451-3
Zaucker A, Mercurio S, Sternheim N, Talbot WS, Marlow FL (2013) notch3 is essential for oligodendrocyte development and vascular integrity in zebrafish. Dis Model Mech 6:1246–1259. https://doi.org/10.1242/dmm.012005
Pogoda H-M, Sternheim N, Lyons DA, Diamond B, Hawkins TA, Woods IG, Bhatt DH, Franzini-Armstrong C et al (2006) A genetic screen identifies genes essential for development of myelinated axons in zebrafish. Dev Biol 298:118–131. https://doi.org/10.1016/j.ydbio.2006.06.021
Krebs LT, Xue Y, Norton CR et al (2003) Characterization ofNotch3-deficient mice: Normal embryonic development and absence of genetic interactions with aNotch1 mutation. Genesis 37:139–143. https://doi.org/10.1002/gene.10241
Domenga V (2004) Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 18:2730–2735. https://doi.org/10.1101/gad.308904
Arboleda-Velasquez JF, Zhou Z, Shin HK, Louvi A, Kim HH, Savitz SI, Liao JK, Salomone S et al (2008) Linking Notch signaling to ischemic stroke. Proc Natl Acad Sci 105:4856–4861. https://doi.org/10.1073/pnas.0709867105
Belin de Chantemèle EJ, Retailleau K, Pinaud F et al (2008) Notch3 is a major regulator of vascular tone in cerebral and tail resistance arteries. Arterioscler Thromb Vasc Biol 28:2216–2224. https://doi.org/10.1161/ATVBAHA.108.171751
Joutel A, Monet-Leprêtre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, Lemaire-Carrette B, Domenga V et al (2010) Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest 120:433–445. https://doi.org/10.1172/JCI39733
Lundkvist J, Zhu S, Hansson EM, et al (2005) Mice carrying a R142C Notch 3 knock-in mutation do not develop a CADASIL-like phenotype. genesis 41:13–22. https://doi.org/10.1002/gene.20091
Monet-Leprêtre M, Bardot B, Lemaire B, Domenga V, Godin O, Dichgans M, Tournier-Lasserve E, Cohen-Tannoudji M et al (2009) Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain. Brain 132:1601–1612. https://doi.org/10.1093/brain/awp049
Ruchoux MM, Domenga V, Brulin P, Maciazek J, Limol S, Tournier-Lasserve E, Joutel A (2003) Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am J Pathol 162:329–342. https://doi.org/10.1016/S0002-9440(10)63824-2
Dubroca C, Lacombe P, Domenga V et al (2005) Impaired vascular mechanotransduction in a transgenic mouse model of CADASIL arteriopathy. Stroke 36:113–117. https://doi.org/10.1161/01.STR.0000149949.92854.45
Lacombe P, Oligo C, Domenga V et al (2005) Impaired cerebral vasoreactivity in a transgenic mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy arteriopathy. Stroke 36:1053–1058. https://doi.org/10.1161/01.STR.0000163080.82766.eb
Gu X, Liu X-Y, Fagan A, Gonzalez-Toledo ME, Zhao LR (2012) Ultrastructural changes in cerebral capillary pericytes in aged Notch3 mutant transgenic mice. Ultrastruct Pathol 36:48–55. https://doi.org/10.3109/01913123.2011.620220
Monet M, Domenga V, Lemaire B, Souilhol C, Langa F, Babinet C, Gridley T, Tournier-Lasserve E et al (2007) The archetypal R90C CADASIL–NOTCH3 mutation retains NOTCH3 function in vivo. Hum Mol Genet 16:982–992. https://doi.org/10.1093/hmg/ddm042
Cognat E, Baron-Menguy C, Domenga-Denier V, Cleophax S, Fouillade C, Monet-Leprêtre M, Dewerchin M, Joutel A (2014) Archetypal Arg169Cys mutation in NOTCH3 does not drive the pathogenesis in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy via a loss-of-function mechanism. Stroke 45:842–849. https://doi.org/10.1161/STROKEAHA.113.003339
Monet-Leprêtre M, Haddad I, Baron-Menguy C, Fouillot-Panchal M, Riani M, Domenga-Denier V, Dussaule C, Cognat E et al (2013) Abnormal recruitment of extracellular matrix proteins by excess Notch3ECD: a new pathomechanism in CADASIL. Brain 136:1830–1845. https://doi.org/10.1093/brain/awt092
Chabriat H, Pappata S, Ostergaard L, Clark CA, Pachot-Clouard M, Vahedi K, Jobert A, le Bihan D et al (2000) Cerebral hemodynamics in CADASIL before and after acetazolamide challenge assessed with MRI bolus tracking. Stroke 31:1904–1912. https://doi.org/10.1161/01.STR.31.8.1904
O’Sullivan M, Lythgoe DJ, Pereira AC et al (2002) Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology 59:321–326. https://doi.org/10.1212/WNL.59.3.321
Otori T, Katsumata T, Muramatsu H, Kashiwagi F, Katayama Y, Terashi A (2003) Long-term measurement of cerebral blood flow and metabolism in a rat chronic hypoperfusion model. Clin Exp Pharmacol Physiol 30:266–272. https://doi.org/10.1046/j.1440-1681.2003.03825.x
Shibata M, Ohtani R, Ihara M, Tomimoto H (2004) White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 35:2598–2603. https://doi.org/10.1161/01.STR.0000143725.19053.60
Dabertrand F, Krøigaard C, Bonev AD, Cognat E, Dalsgaard T, Domenga-Denier V, Hill-Eubanks DC, Brayden JE et al (2015) Potassium channelopathy-like defect underlies early-stage cerebrovascular dysfunction in a genetic model of small vessel disease. Proc Natl Acad Sci 112:E796–E805. https://doi.org/10.1073/pnas.1420765112
Capone C, Dabertrand F, Baron-Menguy C, et al (2016) Mechanistic insights into a TIMP3-sensitive pathway constitutively engaged in the regulation of cerebral hemodynamics. Elife 5:. https://doi.org/10.7554/eLife.17536
Fontaine JT, Rosehart AC, Joutel A, Dabertrand F (2020) HB-EGF depolarizes hippocampal arterioles to restore myogenic tone in a genetic model of small vessel disease. Mech Ageing Dev 192:111389. https://doi.org/10.1016/j.mad.2020.111389
Cognat E, Cleophax S, Domenga-Denier V, Joutel A (2014) Early white matter changes in CADASIL: evidence of segmental intramyelinic oedema in a pre-clinical mouse model. Acta Neuropathol Commun 2:49. https://doi.org/10.1186/2051-5960-2-49
Ehret F, Vogler S, Pojar S, Elliott DA, Bradke F, Steiner B, Kempermann G (2015) Mouse model of CADASIL reveals novel insights into Notch3 function in adult hippocampal neurogenesis. Neurobiol Dis 75:131–141. https://doi.org/10.1016/j.nbd.2014.12.018
Beatus P, Lundkvist J, Öberg C, Pedersen K, Lendahl U (2001) The origin of the ankyrin repeat region in Notch intracellular domains is critical for regulation of HES promoter activity. Mech Dev 104:3–20. https://doi.org/10.1016/S0925-4773(01)00373-2
Klein C, Schreyer S, Kohrs FE, Elhamoury P, Pfeffer A, Munder T, Steiner B (2017) Stimulation of adult hippocampal neurogenesis by physical exercise and enriched environment is disturbed in a CADASIL mouse model. Sci Rep 7:45372. https://doi.org/10.1038/srep45372
Kronenberg G, Bick-Sander A, Bunk E, Wolf C, Ehninger D, Kempermann G (2006) Physical exercise prevents age-related decline in precursor cell activity in the mouse dentate gyrus. Neurobiol Aging 27:1505–1513. https://doi.org/10.1016/j.neurobiolaging.2005.09.016
Baron-Menguy C, Domenga-Denier V, Ghezali L, Faraci FM, Joutel A (2017) Increased Notch3 activity mediates pathological changes in structure of cerebral arteries. Hypertension 69:60–70. https://doi.org/10.1161/HYPERTENSIONAHA.116.08015
Neves KB, Harvey AP, Moreton F, Montezano AC, Rios FJ, Alves-Lopes R, Nguyen Dinh Cat A, Rocchicciolli P et al (2019) ER stress and Rho kinase activation underlie the vasculopathy of CADASIL. JCI Insight 4. https://doi.org/10.1172/jci.insight.131344
Ghosh M, Balbi M, Hellal F, Dichgans M, Lindauer U, Plesnila N (2015) Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann Neurol 78:887–900. https://doi.org/10.1002/ana.24512
Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21:70–71. https://doi.org/10.1038/5007
Arboleda-Velasquez JF, Manent J, Lee JH, Tikka S, Ospina C, Vanderburg CR, Frosch MP, Rodriguez-Falcon M et al (2011) Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc Natl Acad Sci 108:E128–E135. https://doi.org/10.1073/pnas.1101964108
Arboleda-Velasquez JF, Lopera F, Lopez E et al (2002) C455R notch3 mutation in a Colombian CADASIL kindred with early onset of stroke. Neurology 59:277–279. https://doi.org/10.1212/WNL.59.2.277
Primo V, Graham M, Bigger-Allen AA, Chick JM, Ospina C, Quiroz YT, Manent J, Gygi SP et al (2016) Blood biomarkers in a mouse model of CADASIL. Brain Res 1644:118–126. https://doi.org/10.1016/j.brainres.2016.05.008
Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A, Kawata H, Koyama A et al (2009) Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med 360:1729–1739. https://doi.org/10.1056/NEJMoa0801560
Tikka S, Baumann M, Siitonen M, Pasanen P, Pöyhönen M, Myllykangas L, Viitanen M, Fukutake T et al (2014) CADASIL and CARASIL. Brain Pathol 24:525–544. https://doi.org/10.1111/bpa.12181
Gravesteijn G, Munting LP, Overzier M, Mulder AA, Hegeman I, Derieppe M, Koster AJ, van Duinen SG et al (2019) Progression and classification of granular osmiophilic material (GOM) deposits in functionally characterized human NOTCH3 transgenic mice. Transl Stroke Res 11:517–527. https://doi.org/10.1007/s12975-019-00742-7
Rutten JW, Klever RR, Hegeman IM, Poole DS, Dauwerse HG, Broos LAM, Breukel C, Aartsma-Rus AM et al (2015) The NOTCH3 score: a pre-clinical CADASIL biomarker in a novel human genomic NOTCH3 transgenic mouse model with early progressive vascular NOTCH3 accumulation. Acta Neuropathol Commun 3:89. https://doi.org/10.1186/s40478-015-0268-1
Wallays G, Nuyens D, Silasi-Mansat R, Souffreau J, Callaerts-Vegh Z, van Nuffelen A, Moons L, D'Hooge R et al (2011) Notch3 Arg170Cys knock-in mice display pathologic and clinical features of the neurovascular disorder cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Arterioscler Thromb Vasc Biol 31:2881–2888. https://doi.org/10.1161/ATVBAHA.111.237859
Hanemaaijer ES, Panahi M, Swaddiwudhipong N, Tikka S, Winblad B, Viitanen M, Piras A, Behbahani H (2018) Autophagy-lysosomal defect in human CADASIL vascular smooth muscle cells. Eur J Cell Biol 97:557–567. https://doi.org/10.1016/j.ejcb.2018.10.001
Tikka S, Ng YP, Di Maio G et al (2012) CADASIL mutations and shRNA silencing of NOTCH3 affect actin organization in cultured vascular smooth muscle cells. J Cereb Blood Flow Metab 32:2171–2180. https://doi.org/10.1038/jcbfm.2012.123
Qualtieri A, Ungaro C, Bagalà A, Bianchi S, Pantoni L, Moccia M, Mazzei R (2018) Notch3 protein expression in skin fibroblasts from CADASIL patients. J Neurol Sci 390:121–128. https://doi.org/10.1016/j.jns.2018.04.027
Annunen-Rasila J, Kärppä M, Finnilä S, Ylä-Outinen H, Veijola J, Tuominen H, Peltonen J, Majamaa K (2007) Cytoskeletal structure in cells harboring two mutations: R133C in NOTCH3 and 5650G>A in mitochondrial DNA. Mitochondrion 7:96–100. https://doi.org/10.1016/j.mito.2006.11.004
Tang M, Shi C, Song B, Yang J, Yang T, Mao C, Li Y, Liu X et al (2017) CADASIL mutant NOTCH3(R90C) decreases the viability of HS683 oligodendrocytes via apoptosis. Mol Biol Rep 44:273–280. https://doi.org/10.1007/s11033-017-4107-2
Karlstrom H, Beatus P, Dannaeus K, Chapman G, Lendahl U, Lundkvist J (2002) A CADASIL-mutated Notch 3 receptor exhibits impaired intracellular trafficking and maturation but normal ligand-induced signaling. Proc Natl Acad Sci 99:17119–17124. https://doi.org/10.1073/pnas.252624099
Low W-C, Santa Y, Takahashi K, Tabira T, Kalaria RN (2006) CADASIL-causing mutations do not alter Notch3 receptor processing and activation. Neuroreport 17:945–949. https://doi.org/10.1097/01.wnr.0000223394.66951.48
Opherk C, Duering M, Peters N, Karpinska A, Rosner S, Schneider E, Bader B, Giese A et al (2009) CADASIL mutations enhance spontaneous multimerization of NOTCH3. Hum Mol Genet 18:2761–2767. https://doi.org/10.1093/hmg/ddp211
Takahashi K, Adachi K, Yoshizaki K, Kunimoto S, Kalaria RN, Watanabe A (2010) Mutations in NOTCH3 cause the formation and retention of aggregates in the endoplasmic reticulum, leading to impaired cell proliferation. Hum Mol Genet 19:79–89. https://doi.org/10.1093/hmg/ddp468
Watanabe-Hosomi A, Watanabe Y, Tanaka M, Nakagawa M, Mizuno T (2012) Transendocytosis is impaired in CADASIL-mutant NOTCH3. Exp Neurol 233:303–311. https://doi.org/10.1016/j.expneurol.2011.10.020
Arboleda-Velasquez JF, Rampal R, Fung E, Darland DC, Liu M, Martinez MC, Donahue CP, Navarro-Gonzalez MF et al (2005) CADASIL mutations impair Notch3 glycosylation by Fringe. Hum Mol Genet 14:1631–1639. https://doi.org/10.1093/hmg/ddi171
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. https://doi.org/10.1016/j.cell.2006.07.024
Fernández-Susavila H, Mora C, Aramburu-Núñez M, Quintas-Rey R, Arias S, Collado M, López-Arias E, Sobrino T et al (2018) Generation and characterization of the human iPSC line IDISi001-A isolated from blood cells of a CADASIL patient carrying a NOTCH3 mutation. Stem Cell Res 28:16–20. https://doi.org/10.1016/j.scr.2018.01.023
Ling C, Liu Z, Song M, Zhang W, Wang S, Liu X, Ma S, Sun S et al (2019) Modeling CADASIL vascular pathologies with patient-derived induced pluripotent stem cells. Protein Cell 10:249–271. https://doi.org/10.1007/s13238-019-0608-1
Ruan Z, Fu X, Li W, Ye J, Wang RZ, Zhu L (2016) Effect of notch1,2,3 genes silicing on NF-κB signaling pathway of macrophages in patients with atherosclerosis. Biomed Pharmacother 84:666–673. https://doi.org/10.1016/j.biopha.2016.09.078
Shin HM, Tilahun ME, Cho OH, Chandiran K, Kuksin CA, Keerthivasan S, Fauq AH, Golde TE et al (2014) NOTCH1 can initiate NF-ÎoB activation via cytosolic interactions with components of the T cell Signalosome. Front Immunol 5. https://doi.org/10.3389/fimmu.2014.00249
Vacca A, Felli MP, Palermo R, di Mario G, Calce A, di Giovine M, Frati L, Gulino A et al (2006) Notch3 and pre-TCR interaction unveils distinct NF-κB pathways in T-cell development and leukemia. EMBO J 25:1000–1008. https://doi.org/10.1038/sj.emboj.7600996
Bonnefoy A, Moura R, Hoylaerts MF (2008) Thrombospondins: from structure to therapeutics. Cell Mol Life Sci 65:713–727. https://doi.org/10.1007/s00018-007-7487-y
Edwards D, Handsley M, Pennington C (2008) The ADAM metalloproteinases. Mol Asp Med 29:258–289. https://doi.org/10.1016/j.mam.2008.08.001
Kim W-J, Kang Y-J, Suk K, Park JE, Kwon BS, Lee WH (2008) Comparative analysis of the expression patterns of various TNFSF/TNFRSF in atherosclerotic plaques. Immunol Investig 37:359–373. https://doi.org/10.1080/08820130802123139
Penn DL, Witte SR, Komotar RJ, Sander Connolly E (2014) The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J Clin Neurosci 21:28–32. https://doi.org/10.1016/j.jocn.2013.07.004
Qi B, Newcomer R, Sang Q-X (2009) ADAM19/Adamalysin 19 structure, function, and role as a putative target in tumors and inflammatory diseases. Curr Pharm Des 15:2336–2348. https://doi.org/10.2174/138161209788682352
Xu C-Y, Qin M-B, Tan L et al (2016) NIBP impacts on the expression of E-cadherin, CD44 and vimentin in colon cancer via the NF-κB pathway. Mol Med Rep 13:5379–5385. https://doi.org/10.3892/mmr.2016.5165
Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, Karsan A (2006) Smooth muscle α-actin is a direct target of Notch/CSL. Circ Res 98:1468–1470. https://doi.org/10.1161/01.RES.0000229683.81357.26
Venkatesh D, Fredette N, Rostama B, Tang Y, Vary CPH, Liaw L, Urs S (2011) RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol 31:876–882. https://doi.org/10.1161/ATVBAHA.110.221945
Kelleher J, Dickinson A, Cain S, Hu Y, Bates N, Harvey A, Ren J, Zhang W et al (2019) Patient-specific iPSC model of a genetic vascular dementia syndrome reveals failure of mural cells to stabilize capillary structures. Stem Cell Reports 13:817–831. https://doi.org/10.1016/j.stemcr.2019.10.004
Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, del Gaudio F, Nahar K, Lebouvier T et al (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554:475–480. https://doi.org/10.1038/nature25739
Darland D, Massingham L, Smith S et al (2003) Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev Biol 264:275–288. https://doi.org/10.1016/j.ydbio.2003.08.015
Phng L-K, Gerhardt H (2009) Angiogenesis: A team effort coordinated by notch. Dev Cell 16:196–208. https://doi.org/10.1016/j.devcel.2009.01.015
Ribatti D, Nico B, Crivellato E (2011) The role of pericytes in angiogenesis. Int J Dev Biol 55:261–268. https://doi.org/10.1387/ijdb.103167dr
Yamamoto Y, Kojima K, Taura D, Sone M, Washida K, Egawa N, Kondo T, Minakawa EN et al (2020) Human iPS cell-derived mural cells as an in vitro model of hereditary cerebral small vessel disease. Mol Brain 13:38. https://doi.org/10.1186/s13041-020-00573-w
Kast J, Hanecker P, Beaufort N, Giese A, Joutel A, Dichgans M, Opherk C, Haffner C (2014) Sequestration of latent TGF-β binding protein 1 into CADASIL-related Notch3-ECD deposits. Acta Neuropathol Commun 2:96. https://doi.org/10.1186/s40478-014-0096-8
Yamamoto Y, Craggs LJL, Watanabe A, Booth T, Attems J, Low RWC, Oakley AE, Kalaria RN (2013) Brain microvascular accumulation and distribution of the NOTCH3 ectodomain and granular osmiophilic material in CADASIL. J Neuropathol Exp Neurol 72:416–431. https://doi.org/10.1097/NEN.0b013e31829020b5
Zellner A, Scharrer E, Arzberger T, Oka C, Domenga-Denier V, Joutel A, Lichtenthaler SF, Müller SA et al (2018) CADASIL brain vessels show a HTRA1 loss-of-function profile. Acta Neuropathol 136:111–125. https://doi.org/10.1007/s00401-018-1853-8
Khan MT, Murray A, Smith M (2016) Successful use of intravenous tissue plasminogen activator as treatment for a patient with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: a case report and review of literature. J Stroke Cerebrovasc Dis 25:e53–e57. https://doi.org/10.1016/j.jstrokecerebrovasdis.2015.12.006
Nannucci S, Rinnoci V, Pracucci G, MacKinnon AD, Pescini F, Adib-Samii P, Bianchi S, Dotti MT et al (2018) Location, number and factors associated with cerebral microbleeds in an Italian-British cohort of CADASIL patients. PLoS One 13:e0190878. https://doi.org/10.1371/journal.pone.0190878
del Río-Espínola A, Mendióroz M, Domingues-Montanari S, Pozo-Rosich P, Solé E, Fernández-Morales J, Fernández-Cadenas I, Montaner J (2009) CADASIL management or what to do when there is little one can do. Expert Rev Neurother 9:197–210. https://doi.org/10.1586/14737175.9.2.197
Oh J-H, Lee JS, Kang S-Y, Kang JH, Choi JC (2008) Aspirin-associated intracerebral hemorrhage in a patient with CADASIL. Clin Neurol Neurosurg 110:384–386. https://doi.org/10.1016/j.clineuro.2007.12.001
Adib-Samii P, Brice G, Martin RJ, Markus HS (2010) Clinical spectrum of CADASIL and the effect of cardiovascular risk factors on phenotype. Stroke 41:630–634. https://doi.org/10.1161/STROKEAHA.109.568402
Singhal S (2004) The influence of genetic and cardiovascular risk factors on the CADASIL phenotype. Brain 127:2031–2038. https://doi.org/10.1093/brain/awh223
Guo Z, Qiu C, Viitanen M, Fastbom J, Winblad B, Fratiglioni L (2001) Blood pressure and dementia in persons 75+ years old: 3-year follow-up results from the Kungsholmen Project. J Alzheimers Dis 3:585–591. https://doi.org/10.3233/JAD-2001-3609
Verghese J, Lipton RB, Hall CB, Kuslansky G, Katz MJ (2003) Low blood pressure and the risk of dementia in very old individuals. Neurology 61:1667–1672. https://doi.org/10.1212/01.WNL.0000098934.18300.BE
Viswanathan A (2006) Blood pressure and haemoglobin A1c are associated with microhaemorrhage in CADASIL: a two-centre cohort study. Brain 129:2375–2383. https://doi.org/10.1093/brain/awl177
Peters N, Freilinger T, Opherk C, Pfefferkorn T, Dichgans M (2007) Effects of short term atorvastatin treatment on cerebral hemodynamics in CADASIL. J Neurol Sci 260:100–105. https://doi.org/10.1016/j.jns.2007.04.015
De Maria R, Campolo J, Frontali M et al (2014) Effects of sapropterin on endothelium-dependent vasodilation in patients with CADASIL. Stroke 45:2959–2966. https://doi.org/10.1161/STROKEAHA.114.005937
Huang L, Yang Q, Zhang L, Chen X, Huang Q, Wang H (2010) Acetazolamide improves cerebral hemodynamics in CADASIL. J Neurol Sci 292:77–80. https://doi.org/10.1016/j.jns.2010.01.023
Watanabe-Hosomi A, Mizuta I, Koizumi T, Yokota I, Mukai M, Hamano A, Kondo M, Fujii A et al (2020) Effect of lomerizine hydrochloride on preventing strokes in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Clin Neuropharmacol 43:146–150. https://doi.org/10.1097/WNF.0000000000000402
Goldstein J, Hagen M, Gold M (2014) Results of a multicenter, double-blind, randomized, parallel-group, placebo-controlled, single-dose study comparing the fixed combination of acetaminophen, acetylsalicylic acid, and caffeine with ibuprofen for acute treatment of patients with severe migra. Cephalalgia 34:1070–1078. https://doi.org/10.1177/0333102414530527
Martikainen MH, Roine S (2012) Rapid improvement of a complex migrainous episode with sodium valproate in a patient with CADASIL. J Headache Pain 13:95–97. https://doi.org/10.1007/s10194-011-0400-y
Ferrari MD, Roon KI, Lipton RB, Goadsby PJ (2001) Oral triptans (serotonin 5-HT 1B/1D agonists) in acute migraine treatment: a meta-analysis of 53 trials. Lancet 358:1668–1675. https://doi.org/10.1016/S0140-6736(01)06711-3
Tfelt-Hansen P (2000) Ergotamine in the acute treatment of migraine: a review and European consensus. Brain 123:9–18. https://doi.org/10.1093/brain/123.1.9
Donnini I, Nannucci S, Valenti R, Pescini F, Bianchi S, Inzitari D, Pantoni L (2012) Acetazolamide for the prophylaxis of migraine in CADASIL: a preliminary experience. J Headache Pain 13:299–302. https://doi.org/10.1007/s10194-012-0426-9
Finocchi C, Villani V, Casucci G (2010) Therapeutic strategies in migraine patients with mood and anxiety disorders: clinical evidence. Neurol Sci 31:95–98. https://doi.org/10.1007/s10072-010-0297-2
Bousser M-G (2001) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: from stroke to vessel wall physiology. J Neurol Neurosurg Psychiatry 70:285–287. https://doi.org/10.1136/jnnp.70.3.285
Lippi G, Mattiuzzi C, Meschi T, Cervellin G, Borghi L (2014) Homocysteine and migraine. A narrative review. Clin Chim Acta 433:5–11. https://doi.org/10.1016/j.cca.2014.02.028
Keverne JS, Low WCR, Ziabreva I, Court JA, Oakley AE, Kalaria RN (2007) Cholinergic neuronal deficits in CADASIL. Stroke 38:188–191. https://doi.org/10.1161/01.STR.0000251787.90695.05
Manganelli F, Ragno M, Cacchiò G, Iodice V, Trojano L, Silvaggio F, Scarcella M, Grazioli M et al (2008) Motor cortex cholinergic dysfunction in CADASIL: a transcranial magnetic demonstration. Clin Neurophysiol 119:351–355. https://doi.org/10.1016/j.clinph.2007.10.011
Dichgans M, Markus HS, Salloway S, Verkkoniemi A, Moline M, Wang Q, Posner H, Chabriat HS (2008) Donepezil in patients with subcortical vascular cognitive impairment: a randomised double-blind trial in CADASIL. Lancet Neurol 7:310–318. https://doi.org/10.1016/S1474-4422(08)70046-2
Schneider LS (2008) Does donepezil improve executive function in patients with CADASIL? Lancet Neurol 7:287–289. https://doi.org/10.1016/S1474-4422(08)70047-4
Park S, Park B, Koh MK, Joo YH (2014) Case report: bipolar disorder as the first manifestation of CADASIL. BMC Psychiatry 14:175. https://doi.org/10.1186/1471-244X-14-175
Ho CSH, Mondry A (2015) CADASIL presenting as schizophreniform organic psychosis. Gen Hosp Psychiatry 37:273.e11-273.e13. https://doi.org/10.1016/j.genhosppsych.2015.02.006
Machuca-Parra AI, Bigger-Allen AA, Sanchez AV, Boutabla A, Cardona-Vélez J, Amarnani D, Saint-Geniez M, Siebel CW et al (2017) Therapeutic antibody targeting of Notch3 signaling prevents mural cell loss in CADASIL. J Exp Med 214:2271–2282. https://doi.org/10.1084/jem.20161715
Xu X, Choi SH, Hu T, Tiyanont K, Habets R, Groot AJ, Vooijs M, Aster JC et al (2015) Insights into autoregulation of Notch3 from structural and functional studies of its negative regulatory region. Structure 23:1227–1235. https://doi.org/10.1016/j.str.2015.05.001
Ghezali L, Capone C, Baron-Menguy C, Ratelade J, Christensen S, Østergaard Pedersen L, Domenga-Denier V, Pedersen JT et al (2018) Notch3 ECD immunotherapy improves cerebrovascular responses in CADASIL mice. Ann Neurol 84:246–259. https://doi.org/10.1002/ana.25284
DeMattos RB, Lu J, Tang Y et al (2012) A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 76:908–920. https://doi.org/10.1016/j.neuron.2012.10.029
Golde T, Das P, Levites Y (2009) Quantitative and mechanistic studies of Abeta; immunotherapy. CNS Neurol Disord - Drug Targets 8:31–49. https://doi.org/10.2174/187152709787601830
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S et al (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537:50–56. https://doi.org/10.1038/nature19323
Kawada H, Takizawa S, Takanashi T, Morita Y, Fujita J, Fukuda K, Takagi S, Okano H et al (2006) Administration of hematopoietic cytokines in the subacute phase after cerebral infarction is effective for functional recovery facilitating proliferation of intrinsic neural stem/progenitor cells and transition of bone marrow-derived neuronal cells. Circulation 113:701–710. https://doi.org/10.1161/CIRCULATIONAHA.105.563668
Piao C-S, Gonzalez-Toledo ME, Xue Y-Q, Duan WM, Terao S, Granger DN, Kelley RE, Zhao LR (2009) The role of stem cell factor and granulocyte-colony stimulating factor in brain repair during chronic stroke. J Cereb Blood Flow Metab 29:759–770. https://doi.org/10.1038/jcbfm.2008.168
Schneider A, Krüger C, Steigleder T, Weber D, Pitzer C, Laage R, Aronowski J, Maurer MH et al (2005) The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest 115:2083–2098. https://doi.org/10.1172/JCI23559
Toth ZE, Leker RR, Shahar T, Pastorino S, Szalayova I, Asemenew B, Key S, Parmelee A et al (2008) The combination of granulocyte colony-stimulating factor and stem cell factor significantly increases the number of bone marrow–derived endothelial cells in brains of mice following cerebral ischemia. Blood 111:5544–5552. https://doi.org/10.1182/blood-2007-10-119073
Zsebo KM, Wypych J, McNiece IK et al (1990) Identification, purification, and biological characterization of hematopoietic stem cell factor from buffalo rat liver-conditioned medium. Cell 63:195–201. https://doi.org/10.1016/0092-8674(90)90300-4
Liu X-Y, Gonzalez-Toledo ME, Fagan A, Duan WM, Liu Y, Zhang S, Li B, Piao CS et al (2015) Stem cell factor and granulocyte colony-stimulating factor exhibit therapeutic effects in a mouse model of CADASIL. Neurobiol Dis 73:189–203. https://doi.org/10.1016/j.nbd.2014.09.006
Ruchoux M-M, Maurage C-A (1997) CADASIL. J Neuropathol Exp Neurol 56:947–964. https://doi.org/10.1097/00005072-199709000-00001
Tuominen S, Miao Q, Kurki T, Tuisku S, Pöyhönen M, Kalimo H, Viitanen M, Sipilä HT et al (2004) Positron emission tomography examination of cerebral blood flow and glucose metabolism in young CADASIL patients. Stroke 35:1063–1067. https://doi.org/10.1161/01.STR.0000124124.69842.2d
Pescini F, Cesari F, Giusti B, Sarti C, Zicari E, Bianchi S, Dotti MT, Federico A et al (2010) Bone marrow-derived progenitor cells in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 41:218–223. https://doi.org/10.1161/STROKEAHA.109.563726
Corti S, Locatelli F, Strazzer S, Salani S, del Bo R, Soligo D, Bossolasco P, Bresolin N et al (2002) Modulated generation of neuronal cells from bone marrow by expansion and mobilization of circulating stem cells with in vivo cytokine treatment. Exp Neurol 177:443–452. https://doi.org/10.1006/exnr.2002.8004
Ping S, Qiu X, Gonzalez-Toledo ME, Liu X, Zhao LR (2018) Stem cell factor in combination with granulocyte colony-stimulating factor reduces cerebral capillary thrombosis in a mouse model of CADASIL. Cell Transplant 27:637–647. https://doi.org/10.1177/0963689718766460
Hess DC, Hill WD, Martin-Studdard A et al (2002) Bone marrow as a source of endothelial cells and NeuN-expressing cells after stroke. Stroke 33:1362–1368. https://doi.org/10.1161/01.STR.0000014925.09415.C3
Ping S, Qiu X, Kyle M, Hughes K, Longo J, Zhao LR (2019) Stem cell factor and granulocyte colony-stimulating factor promote brain repair and improve cognitive function through VEGF-A in a mouse model of CADASIL. Neurobiol Dis 132:104561. https://doi.org/10.1016/j.nbd.2019.104561
Ferrara N, Gerber H-P, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669–676. https://doi.org/10.1038/nm0603-669
Welti J, Loges S, Dimmeler S, Carmeliet P (2013) Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest 123:3190–3200. https://doi.org/10.1172/JCI70212
Cui L, Wang D, McGillis S, Kyle M, Zhao LR (2016) Repairing the brain by SCF+G-CSF treatment at 6 months postexperimental stroke. ASN Neuro 8:175909141665501. https://doi.org/10.1177/1759091416655010
Cui L, Duchamp NS, Boston DJ, Ren X, Zhang X, Hu H, Zhao LR (2015) NF-κB is involved in brain repair by stem cell factor and granulocyte-colony stimulating factor in chronic stroke. Exp Neurol 263:17–27. https://doi.org/10.1016/j.expneurol.2014.08.026
Kiriakidis S (2003) VEGF expression in human macrophages is NF-kappaB-dependent: studies using adenoviruses expressing the endogenous NF-kappaB inhibitor IkappaBalpha and a kinase-defective form of the IkappaB kinase 2. J Cell Sci 116:665–674. https://doi.org/10.1242/jcs.00286
Stoltz RA, Abraham NG, Laniado-Schwartzman M (1996) The role of NF-kappaB in the angiogenic response of coronary microvessel endothelial cells. Proc Natl Acad Sci 93:2832–2837. https://doi.org/10.1073/pnas.93.7.2832
Voit T, Topaloglu H, Straub V, Muntoni F, Deconinck N, Campion G, de Kimpe SJ, Eagle M et al (2014) Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol 13:987–996. https://doi.org/10.1016/S1474-4422(14)70195-4
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, de Vivo DC, Yamashita M, Rigo F et al (2016) Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388:3017–3026. https://doi.org/10.1016/S0140-6736(16)31408-8
Rutten JW, Dauwerse HG, Peters DJM, Goldfarb A, Venselaar H, Haffner C, van Ommen GJB, Aartsma-Rus AM et al (2016) Therapeutic NOTCH3 cysteine correction in CADASIL using exon skipping: in vitro proof of concept. Brain 139:1123–1135. https://doi.org/10.1093/brain/aww011
Gravesteijn G, Dauwerse JG, Overzier M, Brouwer G, Hegeman I, Mulder AA, Baas F, Kruit MC et al (2020) Naturally occurring NOTCH3 exon skipping attenuates NOTCH3 protein aggregation and disease severity in CADASIL patients. Hum Mol Genet 29:1853–1863. https://doi.org/10.1093/hmg/ddz285
van Roon-Mom WMC, Aartsma-Rus A (2012) Overview on applications of antisense-mediated exon skipping. pp 79–96
Acknowledgments
Images from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License (http://smart.servier.com/), were modified to generate the figure of this review. We thank Mr. Lorenzo Sdraffa for his help with graphical material.
Funding
Open Access funding provided by Università degli Studi di Milano.
Author information
Authors and Affiliations
Contributions
Conceived the idea and wrote the manuscript: AM. Critical discussion: AM and LP. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interests.
Consent to Participate
Not applicable.
Consent to Publication
Not applicable.
Code Availability
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Manini, A., Pantoni, L. CADASIL from Bench to Bedside: Disease Models and Novel Therapeutic Approaches. Mol Neurobiol 58, 2558–2573 (2021). https://doi.org/10.1007/s12035-021-02282-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02282-4