
Abstract
Transport of proteins, transcription factors, and other signaling molecules between the nucleus and cytoplasm is necessary for signal transduction. The study of these transport phenomena is particularly challenging in neurons because of their highly polarized structure. The bidirectional exchange of molecular cargoes across the nuclear envelope (NE) occurs through nuclear pore complexes (NPCs), which are aqueous channels embedded in the nuclear envelope. The NE and NPCs regulate nuclear transport but are also emerging as relevant regulators of chromatin organization and gene expression. The alterations in nuclear transport are regularly identified in affected neurons associated with human neurodegenerative diseases. This review presents insights into the roles played by nuclear transport defects in neurodegenerative disease, focusing primarily on NE proteins and NPCs. The subcellular mislocalization of proteins might be a very desirable means of therapeutic intervention in neurodegenerative disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The Role of the Nuclear Envelope, Nuclear Pore Complexes, and Nucleoporins in Nucleocytoplasmic Transport
Nuclear Envelope
The nuclear envelope (NE) consists of two membranes: the outer and inner nuclear membranes (ONM and INM), which are bound by nuclear pore complexes (NPCs) and perforated by nuclear pores. Although the NE is a continuous membrane system, the protein composition differs significantly between the ONM and the INM [1]. The ONM is contiguous to the endoplasmic reticulum and sprinkled on the cytoplasmic side with ribosomes. At the same time, INM has indirect [2] interaction with a number of nuclear components, such as chromatin and nuclear lamina, which is an intermediate filament meshwork essential for the maintenance of the nuclear architecture [1, 3, 4]. The main function of NE is to protect the genome and ensure the safe transport of proteins between the cytoplasm and the nucleus [5]. Adequate functioning of the NE, in particular the INM, allows for the maintenance of the nuclear structure and position [6].
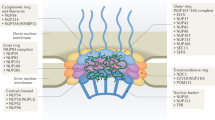
Several NE transmembrane proteins, called nesprins, are characterized by the Klarsicht/ANC-1 /Syne-1 homology (KASH) domain (Fig. 1). These proteins are localized in the ONM and directly interact with the structural components of the cytoplasm and the Sad1-UNC-84 homology (SUN) proteins [7]. SUN proteins are integral membrane proteins and contain the linker of nucleoskeleton and cytoskeleton (LINC) complex, which tends to play a crucial role in nuclear positioning and movement and cell migration [8, 9] (Fig. 1). Well-coordinated nuclear movements appear to be essential during the formation of the central nervous system, where they function in both neurogenesis and neuronal migration. For example, as neural precursor cells migrate to the developing neocortex, the centrosomes travel continuously and ahead of the nucleus, accompanied by leaps rather than smooth, gradual transitions [10].

Schematic diagram of nuclear envelope (NE) and nuclear pore complex (NPC) proteins
Nuclear lamina is an essential structural determinant of the entire nuclear envelope, connecting chromatin domains to the periphery of the nucleus and locating nuclear envelope proteins. The major components of the lamina are A-type and B-type lamins, which are members of the intermediate filament protein family. Expression of A-type laminate is controlled by development, while B-type laminate is present in all cells [11]. A-type laminate is formed by alternative splicing of the laminate A/C gene, and at least four protein types, A, AΔ10, C, and C2, are expressed. Lamin A is synthesized as prelamin, and mature lamin A/C is formed by the processing of 18 amino acids at the C-terminus. Lamin A/C interacts with chromatin and is involved in nuclear breakdown and remodeling during cell division and telomere dynamics. During somatic cell division, lamin A/C is phosphorylated and the structure of the network is degraded. As cell division comes to an end, it is dephosphorylated and reorganized into a network structure and the nuclear membrane is regenerated. Lamin A/C is known to interact not only with the nucleus but also with various proteins. It is also involved in the complex roles of the cytoskeleton and apoptosis, as well as in the replication, transcription, and transduction of DNA [12]. Alterations in A-type lamin often correspond to a selective disorder of peripheral nerves [13]. A recessively inherited missense mutation (R298C) in the LMNA gene encoding the A-type lamin rod domain causes axonal neuropathy that affects peripheral nerves, known as Charcot–Marie–Tooth type 2B1 disease. Sciatic nerves of LMNA-null mice display reduced axon density, increased axon enlargement, and the presence of unmyelinated axons; these characteristics are close to those seen in human patients and are consistent with the possibility that the R298C mutation may affect any aspect of A-type lamin function.
Nuclear Pore Complexes
The majority of enzymes and their substrates that are engaged in cell proliferation, differentiation, and other vital functions are shuttled between intranuclear and cytosolic compartments through NPCs [14, 15]. The NPCs are aqueous channels formed by multicomponent protein complexes of the nuclear envelope, regulating the movement of cell components from the cytosol to the nucleus and vice versa. Consequently, defective NPC function could lead to inappropriate localization of a large number of nuclear and cellular components.
The NPC composition and structure are age-dependent, as cells lose essential nucleopore proteins with age. Cell cultures exposed to oxidative stress also show marked changes in phosphorylation and O-glycosylation of nucleoporins and alterations in the localization of these proteins and their interaction with other transport components. These types of changes in the structure and function of the NPC could lead to aberrant intracellular trafficking of crucial proteins involved in signaling and cell cycle regulation [4].
The NPCs show a broad degree of compositional and structural conservation in all eukaryotes. It has a doughnut-shaped structure consisting of eight spokes, which are arranged radially around a central channel that serves as the conduit for macromolecular transport [16,17,18]. Additionally, NPCs can interact with chromatin and contribute to the formation of specific genomic loops [19].
A growing body of evidence, both from human brain tissue and model animal studies, indicates that the disturbances of NPC structure and function resulting from neuronal oxidative stress are typical features of degenerating neurons. Thus, it can be hypothesized that abnormal structure and disturbed function of NPC underlie the pathogenesis of neurodegeneration.
Nucleoporins
Nucleoporins (NUPs) are proteins that make up the components of NPCs [20,21,22,23]. Around 30 different NUPs are assembled together [24] (Fig. 1). The NUPs can usually be subdivided into two groups. The first category is the structural scaffold of the NPC, which is permanently embedded in the NE and contains the NUP107/160 complex and the NUP205/188/93 complex. NUP133, a component of the stable complex NUP107/160, is necessary for neuronal differentiation. Genetic deletion of NUP133 in mice causes lethality on approximately embryonic day 9.5 and a neural tube that does not normally close [25, 26]. The second class consists of “mobile” NUPs, including approximately 15 NUPs, which form peripheral components of the NPC and which shuttle off and on the NPC. Most of these NUPs have disordered phenylalanine–glycine (FG) repeat region [27], which binds directly to soluble transport receptors and facilitates transport via nuclear pore [28]. The disordered domains that distinguish FG repeats are characterized by a net positive charge, indicating that they represent a critical element of the selective barrier [2, 20].
In differentiated cells, such as muscle fibers and neurons, the oxidation of a long-lived NUP subset reduces the NPC turnover, which in turn increases nuclear leakage and may be responsible for age-related events [29, 30]. The NUP93 complex is selectively depleted from NPC in older cells. Its absence implies a potential mechanism for the degradation of the nuclear membrane barrier [31]. Nuclei purified from age-differentiated cells display defects in nuclear permeability and the accumulation of cytoplasmic tubulin, a finding consistent with the loss of nucleocytoplasmic compartmentalization.
The recessively inherited missense mutation of the NUP gene, NUP62, causes infantile bilateral striatal necrosis [32]. By comparison, the dominantly inherited missense mutation in NUP358/RanBP2 leads to an infection-induced, acute, necrotizing encephalopathy [33]. Although these are distinct diseases, they both include the acute development of bilateral necrotic lesions of deep brain structures. Interestingly, severe necrotizing encephalopathy is often caused by viral or other infections, and NUP96+/− mice have innate and adaptive immunity defects [34]. These intriguing reports indicate that the composition of the NPC can vary throughout development and in different tissues, and that particular proteins have tissue-specific functions, such as mediating nucleocytoplasmic transport of particular cargoes.
Mechanism of mRNA Transport Through the Nuclear Pore
Nuclear export of mRNA is an essential process in eukaryotic gene expression. Immediately after initiation of transcription, mRNA binds to various proteins to form an mRNA–protein complex (messenger ribonucleoprotein: mRNP) [35, 36]. The mRNA precursor transcribed in the nucleus matures through complex processing such as capping, splicing, and 3′-terminal cleavage and poly(A) chain addition (cleavage and polyadenylation: CPA) [37]. Over 800 diverse protein groups function in the mRNA processing process such as transcription elongation, splicing, and CPA, and the binding of these many protein groups to mRNA is regulated in an orderly manner [35].
mRNA is recognized as cargo by nuclear export receptors by interacting with a group of RNA-binding proteins called adapter proteins of the mRNP components [37]. Premature mRNA precursors such as mRNA fragments during transcription and precursors that have not undergone splicing or CPA are eliminated from the nuclear export receptor cargo by coupling the recruitment cycle of the adapter protein with transcription and processing. Thus, the nuclear export receptor recognizes only mature mRNA as cargo, and the mRNA nuclear export mechanism plays an important role as one of the quality controls for mRNA [38].
Various RNAs expressed in cells are transported from the nucleus to the cytoplasm by their specific nuclear export receptors. Relatively small untranslated RNAs such as transfer RNA (tRNA), microRNA, and uridine-rich small nuclear RNA (UsnRNA) are transported from the nucleus to the cytoplasm by transport receptors belonging to the importin β family depending on Ran GTPase [39]. On the other hand, mRNA nuclear export depends on nuclear transport factor 2 (NTF2)–related export protein-1, Nxf1: Nxt1 heterodimer, a nuclear export receptor [40]. Nxf1 has RNA recognition motif (RRM), leucine-rich repeat (LRR), and NTF2-like domain (NTF2L) arranged in this order from the N-terminal. There is a ubiquitin-associated–like domain (UBAL) at the C-terminus. While NTF2L binds to Nxt1 to form a heterodimer, C-terminal UBAL shows the binding ability of NUP constituting NPC to the FG repeat sequence. Nxf1: Nxt1 heterodimer promotes mRNA passage through NPC by interacting with cargo and NPC via their respective domains [41].
Protein Transport Between Cytosol and Nucleus
Two translocation signals are known to control the import of cargo proteins into the nucleus or vice versa: the nuclear localization signal (NLS) and the nuclear export signal (NES). Classical nuclear import requires the identification of NLS-bearing cargo by an import adapter. The importin α-cargo complex then travels through the NPC together with the importin β through NUP FG repeat interactions.
The small G protein, Ran, plays an important role in both import and export cycles. RanGTP binds to importin β on the nuclear side of the NPC in order to remove the cargo complex and release the cargo into the nucleus. Transport cycles include the replacement of the RanGTP nuclear pool, which is carried out by the import of RanGDP by NTF2 [42, 43]. The NTF2 binds to the NUP62 and is then actively transported to the nucleus via the NPCs [44]. Several NUPs in higher organisms are distinguished by the covalent addition of O-linked N-acetylglucosamine (O-GlcNAc) to serine and threonine residues [45]. O-GlcNAc glycosylation of NUPs is produced by oxidative stress, and this oxidant-dependent increase in O-GlcNAc modification can be accomplished by complex regulation of O-GlcNAc transferase and β-N-acetylglucosaminidase. This sugar content can mediate some of the interactions with the importins. For example, wheat germ agglutinine (WGA) is a lectin that binds to N-acetylglucosamine. When added to isolated rat liver nucleus, WGA can inhibit nuclear import by associating with sugar-modified NUPs, thereby blocking the channel [46, 47]. As a consequence, the modification of O-GlcNAc results in a nucleocytoplasmic import deficiency, especially in differentiated cells.
A 70-kDa heat shock protein (HSP70) is a cytoplasmic import factor for importin-independent nuclear imports [48]. The chaperone mechanism of Hsp70 acts to recover from protein denaturation in both the cytoplasmic and nuclear compartments of mammalian cells. Hikeshi, which interacts specifically with HSP70, can bind directly to NUPs, including FG repeats, and migrate into the nucleus via NPCs. Hikeshi-mediated nuclear import of Hsp70 is required to protect cells from heat shock damages so that the deficiency can lead to neurological disorders due to inadequate cell stress response [49].
Liquid–Liquid Phase Separation in Nuclear Transport
The functional unit in the cell is an organelle surrounded by a lipid membrane. Membrane-less structures such as nucleoli that produce ribosomal RNA were thought to be rare; however recently, structures without membrane such as stress granules have been found. Furthermore, accumulated information revealed that proteins and nucleic acids are concentrated and compartmentalized in each membrane-less structure, and most of them are involved in neurodegenerative diseases [50].
The mechanism of protein and nucleic acid compartmentalization without the membrane partition is explained by liquid–liquid phase separation [51]. A sequence that easily causes phase separation is known as a low-complexity sequence (LC sequence) and is formed from a limited number of repeated amino acids. The LC sequence forms a naturally denatured region that does not have a fixed conformation and can be a structure without a membrane [52, 53]. Well-known proteins with LC sequences are the proteins that form the FET family (FUS, fused in sarcoma; EWS, Ewing sarcoma; TAF15, TATA-binding protein-associated factor 2N) [54]. Each of the FET family proteins has a similar domain structure, serine-, tyrosine-, glycine-, and glutamine-rich (SYGQ-rich) domain, called prion-like domain, and RGG repeat, which is a repeating sequence of arginine, glycine, and glycine. Also, it has an RNA-binding motif, a zinc finger, and a nuclear localization signal. The SYGQ-rich domain alone causes liquid–liquid phase separation [55]. Under physiological conditions, the LC sequence of FUS, which is the causative protein of amyotrophic lateral sclerosis (ALS), causes liquid–liquid phase separation due to the formation of a polymer that is unstable and easily dissociates in a concentration-dependent manner. On the other hand, if there is a disease-related mutation in this part, it changes from an unstable polymer to a stable polymer, causing aggregation formation [56].
FET family proteins accumulate in the cytosol when stress occurs in cells such as heat shock and DNA damage and becomes constituents of stress granules (SGs). SGs contain many proteins involved in neurodegenerative diseases. In addition, increased formation of SGs and breakdown of SG degradation mechanism are associated with neurodegenerative diseases such as ALS and frontotemporal dementia (FTD). A repeat sequence due to an abnormal extension of 6 bases (GGGGCC) in the untranslated region of the C9orf72 gene identified as the causative gene of ALS/FTD produces a cytotoxic dipeptide repeat (DPR). Recently, Zhang et al. revealed that the impaired nucleocytoplasmic transport in C9-ALS/FTD is due to the accumulation of nucleocytoplasmic transport factors such as Ran, importin, exportin, and NUPs into SGs [57]. Gasset-Rosa et al. found that endogenous levels of TDP-43 cause liquid–liquid phase separation in the nucleus. They also found that long-lived TDP-43 droplets formed in the cytoplasm were formed independently of conventional stress granules [58]. Furthermore, Kang and colleagues recently showed that tau, the cause of frontotemporal lobar degeneration (FTLD), condenses on the NE and inhibits nuclear–cytoplasmic transport. Interestingly, they also found that in living cells, tau on the nuclear envelope behaves like a droplet [59].
Nucleoporins and Nuclear Envelopes in Chromatin Organization and Gene Expression
Although NPCs are known as the critical regulators of nucleocytoplasmic transport, more recent data suggest that these structures are essential for nuclear organization and that NUPs are extensively involved in genome organization and modulation of genome activity. Within the nucleus, chromosomes occupy distinct regions from which actively transcribing genes loop into structurally distinct interchromatin compartments. Genes at the nuclear periphery tend to be inactive, and alteration of their partitioning to the interior results in gene inactivation. Topological constraints required for looping are provided through the associations of discrete regions of the genome with the nuclear scaffold/matrix attachment regions (S/MARs), which provide anchorage for higher-order genome structure. The crucial factors for DNA processing, e.g., topoisomerases, are often found associated with the nuclear scaffold/matrix, and S/MARs function in augmenting transcription, facilitating replication and DNA repair, and insulating genic domains [60, 61].
Mounting evidence suggests that proteins of the INM, lamins and NE transmembrane proteins (NETs), play critical roles in chromatin tethering and regulation of gene expression [62, 63]. NE and NPCs can recruit chromatin with specific epigenetic marks of silenced gene expression; for instance, the hypoacetylation state of nucleosomal core histones is a well-known mark of silent chromatin, whereas acetylated histones mark active chromatin [64, 65]. NPC proteins also recruit gene-silencing factors or transcription-activating factors to chromatin sequestered at the periphery of the nucleus [66, 67]. NE and NPCs interact with chromatin with a preponderance of gene-silencing effects [34, 68], although several studies have reported NPC-proximal transcriptional activation with concomitant recruitment of induced genes to the nuclear periphery [34, 68, 69]. The role of NUP93 protein interaction with histone acetyltransferase (HAT) in gene silencing is well established [65, 68, 69], and activation of the interferon α and γ gene expression and regulation of interferon α- and γ-induced genes (MHC class I and class II) were documented for NUP96 [34, 68]. Thus, division of labor within different members of the NUP family, between silencing and activating gene expression, determines the complex role of the NPCs in gene regulation [65], and the NPC function should be perceived as a bridge between nuclear transport and gene regulation [70].
Neurons display a complex, highly polarized morphology; consequently, they could be particularly sensitive to age-related disruptions of cell nucleocytoplasmic trafficking and gene expression [71, 72]. Deterioration of the NPC composition in aging neurons could seriously change chromatin organization and function. For instance, NUP93 associated with the global histone acetylation profile is frequently damaged and lost during aging [16]. One could expect that significant changes in genome organization and gene expression, specifically those present in aged or degenerating neurons, will be revealed and quantified with modern molecular methodology.
In recent years, liquid–liquid phase separation of proteins has also been found on chromatin, which has a great impact on the research fields of chromosomes and chromatin. It is important that HP1 (heterochromatin protein 1) undergoes liquid–liquid phase separation for the formation of heterochromatin, a condensed and inactive chromosomal region [73]. In addition, it was revealed that the liquid–liquid phase separation involves phosphorylation of the naturally denatured region on the N-terminal side of HP1 [59]. In the transcriptionally active region, SAF-A (scaffold attachment factor A/hnRNP-U), an RNA-binding protein, forms oligomers through its ATPase activity and binding to RNA, and functions to preserve the decondensed chromatin structure. SAF-A has an RGG motif that is an LC sequence, causing liquid–liquid phase separation, but RNA binding prevents it [61].
Nucleocytoplasmic Transport Impairment
Polyglutamine Diseases
Polyglutamine (polyQ) diseases encompass a group of heritable neurodegenerative disorders, including Huntington disease (HD) and several spinocerebellar ataxias characterized by the pathogenic expansion of existing CAG trinucleotide repeats in the coding region of disease genes, which are translated into expanded polyQ domains in disease proteins [74, 75]. HD is a form of a polyQ repeat disease that leads to the formation of aggregates of polyQ-expanded huntingtin (Htt) protein in cell nuclei [76]. In the pathogenesis of HD, the mislocalization and aggregation of NUPs, as well as impaired nucleocytoplasmic transport, play an essential role [77]. Gasset-Rosa et al. found that age-related cellular characteristics, such as decreased nuclear envelope integrity, impaired nuclear–cytoplasmic transport, and accumulation of DNA double-strand breaks, were found in the cerebral cortex and striatum of HD model mice, depending on the amount of mutant protein and aging [78]. They also found that the accumulation of huntingtin-bound polyglutamine causes the structure of the nuclear envelope to collapse. At this time, Gle1, which is a major component of mRNA export necessary for nuclear–cytoplasmic transport [79], and RanGAP1, which is a Ran GTPase–activating protein [80], were partially segregated and mRNA accumulation occurred in the nucleus. In addition, major changes in nuclear morphology, irregular position of RanGAP1, and nuclear accumulation of mRNA have been observed in the cerebral cortex of HD patients as well as in the mice model. This indicates that polyglutamine-dependent inhibition of nuclear transport and altered nuclear integrity are central components of HD. Nuclear accumulation of Htt is associated with increased phosphorylation of Ser13 and Ser16 [81, 82] of the N-terminal of Htt and decreased interaction with the nuclear pore protein Tpr, which is involved in the nuclear export of proteins [7, 81, 83]. Huntingtin-lowering strategies may become promising therapeutic options in the future [84].
Multiple Sclerosis
Axonal swellings, impaired axonal transport, and inflammatory demyelination are the hallmarks of multiple sclerosis (MS). The morphological changes are characterized by a succession of enlargements and constrictions along the axon and the detection of “ovoids” or “endbulbs” that resemble the terminal stumps of axons [85, 86]. Axonal damage has been associated with mitochondrial malfunction as well as related to calcium entry due to aberrant activation of sodium channels or activation by excitatory amino acids and cytokine production [87, 88]. Calcium-mediated nuclear export of histone deacetylase 1 (HDAC1) is a critical modulator of impaired mitochondrial transport [85] and the induction of axonal damage in inflammatory demyelination. Also, HDAC1 nuclear export is induced by pathological stimuli before impaired mitochondrial transport and the onset of morphological changes [89].
Triple A Syndrome
AAA (triple A) is an autosomal recessive neuroendocrine disease in which esophagus achalasia, anhidrosis, and adrenocortical insufficiency are associated with muscular atrophy and weakness [90]. Adult triple A syndrome can have progressive neurodegeneration, Parkinson’s syndrome, and cognitive impairment [91]. ALADIN has been identified as the causative gene for triple A syndrome. It has been shown to be involved in many of the known aspects of the NPC function, including the assembly of NPC subdomains and the mediation of transport complex nucleation. No morphological abnormalities in the nucleus, nuclear membrane, and NPCs were observed in patients with triple A syndrome, suggesting that NPC dysfunction causes the disease. ALADIN-deficient NPCs are impaired in importin-dependent nuclear imports that prevent DNA ligase I and aprataxin, the protein required to repair DNA single-strand breakage. As a consequence, cells become susceptible to oxidative stress, resulting in cell death [92].
Ataxia Telangiectasia
Ataxia telangiectasia (AT) is a hereditary disease with a major symptom of neurodegeneration and immunodeficiency. The causative gene is ataxia telangiectasia mutated (ATM), which is a member of the PI3-kinase family of serine/threonine protein kinase. The ATM protein is predominantly located in the nucleus at approximately 350 kDa. Histone deacetylase (HDAC) is an enzyme that eliminates acetyl groups from lysine residues present at the N-terminal of histones. Since the histone terminal from which the acetyl group has been removed binds to DNA, the chromatin structure becomes compact and the expression of the gene is suppressed. HDACs therefore play a central role in epigenetic transcriptional repression. To date, 18 forms of HDACs have been described in mammals and are categorized into groups I to IV on the basis of their structural differences. HDAC4, which belongs to class II, is highly expressed in the brain, especially in Purkinje cells. Typically, HDAC4 is located in the cytosol of neuronal cells since it binds to 14-3-3 proteins. On the other hand, brain tissues of patients with AT and Atm−/− mice have been shown to be localized in the nucleus. HDAC4 in the nucleus binds to myocyte enhancer factor-2 and cyclic AMP response element-binding protein (CREB) to cause histone deacetylase of chromatin, thus altering gene expression in neurons. In experiments using ATM-deficient mice, HDAC4 nuclear accumulation inhibition suppressed neurodegeneration and improved behavioral abnormalities [93].
Amyotrophic Lateral Sclerosis
ALS is an intractable neurological disorder. The upper motor neurons of the cerebral cortex and the lower motor neurons of the brain stem and spinal cord are gradually degenerated and destroyed. Approximately 90% of patients are sporadic, with an unknown cause. Copper- and zinc-dependent superoxide dismutase (SOD1) gene mutations are responsible for the disease in patients with familial onset [94]. In experiments using mutant SOD1 (G93A) transgenic mice, the localization of nuclear–cytoplasmic transport proteins, such as importin α and β, changed their localization from the nucleus to the cytoplasm in the lumbar spinal cord. In addition, spinal cord anterior horn cells (AHC) have demonstrated a further increase in cytoplasmic localization of these proteins as the disease progresses [95, 96].
TDP-43 (TAR DNA-binding protein of 43 kDa), an RNA-binding protein, undergoes and accumulates modifications such as ubiquitination, abnormal phosphorylation, and fragmentation in the cytoplasmic inclusion bodies of denatured neurons. Approximately 90% of TDP-43 is initially found in the nucleus, but almost all of it is accumulated in cytoplasmic inclusion bodies in degenerated neurons. On this basis, research on the pathophysiology of the ALS cell death mechanism has been established on both sides due to the loss of function of the TDP-43 in the nucleus or the gained toxicity of the TDP-43 aggregate itself. FUS protein is an RNA-binding protein such as TDP-43 [97, 98].
Mutations in the FUS gene have also been identified in FTLD, indicating that RNA-binding proteins could be generally involved in the pathophysiology of ALS and FTLD [99, 100]. Many FUSs are located in the nucleus and participate in various RNA functions, such as splicing and stabilizing mRNA [101, 102]. It is known that the gene mutation of FUS identified in ALS is abundant near the prion-like domain on the N-terminal side and the nuclear localization signal on the C-terminal side [103]. Mutations that occur on the side of the nuclear localization signal can also change the localization of FUS from the nucleus to the cytoplasm, and patients with these mutations tend to develop earlier. FUS has a prion-like domain like TDP-43, so aggregates are easily formed, hence FUS-positive inclusion bodies are found in ALS with mutations in the FUS gene [99, 100].
As mentioned above (liquid–liquid phase separation in nuclear transport), the familial FTD-ALS linkage analysis reported a number of associations with the 21st region of the short arm of chromosome 9, and C9ORF72 was identified as a causative gene [104].
An abnormal extension of the hexanucleotide repeat sequence (GGGGCC) of the intron between exons 1a and 1b of C9ORF72 was discovered, and the number of GGGGCC repeats in healthy subjects is 2–23, while the number of patients has increased to more than 700 [69]. In this case, the symptoms are FTD, ALS, or a combination of both. As a clinical disease type of FTD, disinhibition type bvFTD is the most common. The primary pathology is a combination of FTLD-TDP and ALS, where positive TDP-43 inclusions are widely distributed in the cerebral cortex, hippocampus, basal ganglia, substantia nigra, brainstem, and spinal cord motor neurons. In addition to TDP-43-positive neuronal cytoplasmic inclusions (NCIs), TDP-43-negative and p62-positive NCIs also appear in the pyramidal cells of the hippocampus and cerebellar granule cells.
Parkinson’s Disease
Parkinson’s disease (PD) is a progressive, age-related neurodegenerative motor system disorder that arises due to the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc). Several examples point to an improper localization of various transcription factors and signaling molecules in dopaminergic neurons of diseased patients. Postmortem PD brains exhibit an increased nuclear translocation of nuclear factor-кB in the substantia nigra and ventral tegmental area neurons [105]. Activating transcription factor 2 (ATF2) is significantly downregulated in SNc neurons of PD brains [106], possibly indicating a decreased nuclear/cytoplasmic ratio of ATF2.
CREB is a family of transcription factors that formed the dimerized leucine zipper and is a transcription factor involved in memory, plasticity, and survival [103]. CREB is one of the most widely studied transcription factors in neurons, as more than 100 genes important for neuronal function contain the cAMP response element (CRE) in the promoter region [107]. In PD brain SNc neurons, pCREB aggregates are detected in the cytoplasm, while they are located in the control brain nucleus [108]. Since the nuclear activity of CREB plays a central role in neuronal adaptation and survival, segregation of this transcription factor in the cytoplasm may be a mechanism that contributes to neuronal degeneration and death.
TDP-43, which is usually a nuclear protein, was reported to accumulate in cytoplasmic inclusions in cases of FTLD and ALS but was also found in SNc neurons of PD patients, where it co-existed with Lewy bodies [109]. Overexpression of TDP-43 in the substantia nigra induced dopaminergic neuron death and potentiated α-synuclein toxicity to dopaminergic neurons in the PD mouse model [109]. The data suggest that cleavage of TDP-43 by caspases leads to its toxic gain of function; the truncated protein redistributes from the nucleus to the cytoplasm, where it joins with Lewy bodies [110].
Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common type of dementia which causes memory, thinking, and behavioral problems. Improper subcellular locations of various transcription factors and signaling proteins and karyopherins have been identified in the neurons of the AD brain. In AD brain neurons, the interface between the cytoplasm and the nucleus is significantly altered. Evidence of abnormalities in the nuclear membrane and aggregation of nuclear pores, intranuclear position of tubulin-positive filaments, accumulation of Nrf2 in the cytoplasm, reduced nuclear content of Nrf2 in hippocampal neurons, and decreased pCREB levels in AD brains [45, 71, 111, 112] strongly suggest impaired nucleocytoplasmic trafficking in affected AD neurons. Importin alpha was found in the Hirano body of AD neurons in human hippocampal CA1 neurons, but not in amyloid β plaques or neurofibril entanglement [98]. In addition, it was not observed in Lewy’s bodies in PD or in Pick’s bodies in Pick’s disease, indicating that this occurrence is unique to AD [112, 113].
Incorrect localization of TDP-43 in AD has also been found in the cytoplasm of inferior olive neurons where intracellular inclusion bodies are formed [114]. TDP-43 mislocation was detected in 25–50% of AD cases, particularly those with more serious clinical pathology [115]. It is worth noting that the existence of TDP-43 was also reported in healthy elderly people at increased risk of AD [115]. Postmortem brain study of nine patients with early mild AD showed elevated cytoplasmic ATF2 levels. Detection of the incorrect position of this primarily nuclear protein may be useful in the distinction between healthy and slightly impaired neurons [116].
GAPDH has emerged as an enzyme involved in various cellular processes [117, 118]. It not only functions in cytoplasm glycolysis but also plays an important role in other cell compartments, including the nucleus [117, 119, 120]. Oxidative stress causes GAPDH to undergo S-nitrosylation. In AD, GAPDH expression and nitrosylation are increased, presumably leading to elevated levels of GAPDH in the nucleus, which in turn promotes apoptosis [118]. As a result, oxidant-induced changes in GAPDH enzyme activity and intracellular distribution reduce energy supply and advance apoptosis in the brain of AD patients. Since GAPDH is an existing target for oxidative damage in many neurodegenerative diseases, oxidant-dependent changes in nuclear transport and the resulting increase in cell death may be common to multiple types of neurodegeneration.
Pathological protein tau, which is aggregated in AD patients, can directly affect NUPs, leading to disturbance of their structure, mislocalization, and impaired function [121]. Pathological tau-induced neurotoxicity is associated with impaired nuclear–cytoplasmic transport due to tau interaction with NPCs [122].
Perspectives of Enhancing Nucleocytoplasmic Transport in Neurodegeneration
Impaired nucleocytoplasmic transport is widespread in neurodegenerative disorders, and the possibility of therapeutic approaches for improving transport abnormalities is widely discussed. Since nucleocytoplasmic transport control is complex, however, more basic research is still required to thoroughly elucidate the mechanism of action of drug candidates targeting nucleocytoplasmic transport disorders currently being studied [23].
The most studied intracellular and preclinical studies are groups of small molecules, called selective nuclear export inhibitors (SINEs), that block nuclear export. This group of compounds has been shown to form covalent bonds on the exportin1 (XPO1) with cysteine (Cys-528) inhibiting exports of nuclear protein and ribonucleic acid [123]. SINE induces temporary degradation of the XPO1 protein, which is degraded reversibly when the SINE compound stops. Cytoplasmic protein aggregates resulting from inhibition of nuclear exports are lysed and returned to the nucleus via the karyopherin alpha, karyopherin beta 1 and beta 2, and transporin 1 nuclear import receptors [124]. Of the compounds in this category, KPT-276, KPT-350, and KPT-335 showed important neuroprotective effects of primary cultured neurons and induced pluripotent stem cells (iPS cells) in vitro [77, 125]. More recent studies, on the other hand, have shown that transport of TDP-43 and FUS proteins from the nucleus to the cytoplasm is primarily passive diffusion and XPO1 is not involved [126, 127]. Consequently, at the present stage of knowledge, it is difficult to explain the role of SINE in the ALS/FTD.
As another therapeutic strategy, an importin role reinforcement approach is envisaged.
There are, for example, methods of protein engineering to produce compounds that enhance the affinity of β2 karyopherin to FUS proteins and get functionally enhanced imports [124, 128].
By improving the function of these nuclear import receptors, the lysis of aggregates such as TDP-43 protein aggregates and FUS containing proline–tyrosine nuclear localization signals can be improved.
Optimizing and modifying nuclear export/import proteins that are impaired in many neurodegenerative diseases may offer unique therapeutic options for treating fatal diseases in these incurable diseases.
Final Remarks
The role of impaired nucleocytoplasmic transport in the pathogenesis of neurodegenerative disorders is essential [108]. The NE and NPC are responsible for the bidirectional trafficking of proteins, including many transcription factors, between the cytoplasm and the nucleus, as well as for the spatial organization of chromatin and the regulation of gene expression. Translocation of signaling proteins is crucially involved in the pathophysiology of neurodegenerative disorders. In postmitotic cells, such as neurons, several nuclear pore proteins, such as the NUP107/160 complex, are long-lived, and during their lifespan, they accumulate oxidative modifications [30, 129, 130]. As a consequence, progressive deterioration of the NPC structure and function is observed in aged neurons, with cytosolic proteins being leaked into the nucleus [129]. Impaired functions of the NE and NPCs are regular features of age-related neurodegeneration [30]. Recent attention has been drawn to the mislocalization process in neurodegenerative disorders like AD, PD, HD, and prion diseases. Considerable evidence has been accumulated to indicate that the earlier stages of the protein mislocalization process are more directly tied to pathogenesis than the filamentous protein aggregates. For example, the mislocalization of tau to dendritic spines has recently been reported to mediate a synaptic dysfunction that is associated with impaired brain function at the preclinical disease stages that immediately precede neurodegeneration [131]. P-tau interacts with Nup98, the displacement of this nucleoporin from the nuclear membrane into the cytosol, where it often locates in neurofibrillary tangles, while Nup98 in the cytosol enhances P-tau oligomerization and aggregation [121]. Thus, tau-induced mislocation of NPCs contributes to tau-induced neurotoxicity in AD [121]. It has also been hypothesized that an altered localization of transcription factors such as NF-kB, activating transcription factors 2, CREB, p53, E2F transcription factor, and NF-E2-related factor 2 might contribute to cell death commitment in several neurodegenerative diseases [71]. Mislocalized proteins frequently join cytosolic or intranuclear inclusion bodies, which are present in the majority of degenerating neurons. A better understanding of the mechanisms of nucleocytoplasmic transport and the establishment of the NE and NPCs’ contributions in chromatin organization and gene expression should lead to new approaches for therapeutic intervention in many diseases connected with NE and NPC malfunction, as well as in neuronal aging and neurodegeneration. Especially, from this point of view, modifying the disease-related subcellular mislocalization of proteins might be an attractive means of therapeutic intervention. In particular, cellular processes that link protein folding, cell signaling, and nuclear import and export to the subcellular localization of proteins have been proposed as targets for therapeutic intervention. For example, blocking translocation of signaling proteins in subcellular compartments such as NF-kB can be of exciting action for suppressing disease, e.g., AD in the very early stages of disease processes before clinical manifestation.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- ATF2:
-
Activating transcription factor 2
- CREB:
-
cAMP response element-binding protein
- FTLD:
-
Frontotemporal lobar degeneration
- HAT:
-
Histone acetyltransferase
- HD:
-
Huntington disease
- HDAC:
-
Histone deacetylase
- Hsp70s:
-
Heat shock proteins of 70-kDa
- Htt:
-
Huntingtin protein
- KASH:
-
Klarsicht/ANC-1/Syne-1 homology
- LINC:
-
Linker of nucleoskeleton and cytoskeleton
- MS:
-
Multiple sclerosis
- NE:
-
Nuclear envelope
- NES:
-
Nuclear export signal
- NETs:
-
Nuclear envelope transmembrane proteins
- NLS:
-
Nuclear localization signal
- NM:
-
Inner nuclear membrane
- NPCs:
-
Nuclear pore complexes
- NTF2:
-
Nuclear transport factor 2
- NUPs:
-
Nucleoporins
- O-GlcNAc:
-
O-linked N-acetylglucosamine
- ONM:
-
Outer nuclear membrane
- pCREB:
-
Phosphorylated form of CREB
- PD:
-
Parkinson’s disease
- S/MARs:
-
Nuclear scaffold/matrix attachment regions
- SNc:
-
Substantia nigra pars compacta
- SUN:
-
Sad1-UNC-84 homology
- TDP-43:
-
Transactivation response DNA-binding protein 43
- WGA:
-
Wheat germ agglutinin
References
Stewart CL, Roux KJ, Burke B (2007) Blurring the boundary: the nuclear envelope extends its reach. Science 318. https://doi.org/10.1126/science.1142034
Meinema AC, Laba JK, Hapsari RA, Otten R, Mulder FAA, Kralt A, Van Den Bogaart G, Lusk CP et al (2011) Long unfolded linkers facilitate membrane protein import through the nuclear pore complex. Science 333(6038):90–93. https://doi.org/10.1126/science.1205741
D'Angelo MA, Hetzer MW (2006) The role of the nuclear envelope in cellular organization. Cell Mol Life Sci vol 63:316–332. https://doi.org/10.1007/s00018-005-5361-3
Hutten S, Dormann D (2020) Nucleocytoplasmic transport defects in neurodegeneration—cause or consequence? Semin Cell Dev Biol 99. Elsevier Ltd:151–162. https://doi.org/10.1016/j.semcdb.2019.05.020
Martins F, Sousa J, Pereira CD, da Cruz e Silva OAB, Rebelo S (2020) Nuclear envelope dysfunction and its contribution to the aging process. Aging Cell 19. Blackwell Publishing Ltd. https://doi.org/10.1111/acel.13143
Smoyer CJ, Jaspersen SL (2019) Patrolling the nucleus: inner nuclear membrane-associated degradation. Curr Genet 65(5):1099–1106. https://doi.org/10.1007/s00294-019-00971-1
Turgay Y, Ungricht R, Rothballer A, Kiss A, Csucs G, Horvath P, Kutay U (2010) A classical NLS and the SUN domain contribute to the targeting of SUN2 to the inner nuclear membrane. EMBO J 29(14):2262–2275. https://doi.org/10.1038/emboj.2010.119
Lee JSH, Hale CM, Panorchan P, Khatau SB, George JP, Tseng Y, Stewart CL, Hodzic D et al (2007) Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys J 93(7):2542–2552. https://doi.org/10.1529/biophysj.106.102426
Lüke Y, Zaim H, Karakesisoglou I, Jaeger VM, Sellin L, Lu W, Schneider M, Neumann S et al (2008) Nesprin-2 giant (NUANCE) maintains nuclear envelope architecture and composition in skin. J Cell Sci 121(11):1887–1898. https://doi.org/10.1242/jcs.019075
Tsai JW, Bremner KH, Vallee RB (2007) Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci 10(8):970–979. https://doi.org/10.1038/nn1934
Burke B, Stewart CL (2013) The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol 14:13–24. https://doi.org/10.1038/nrm3488
Broers JLV, Ramaekers FCS, Bonne G, Ben Yaou R, Hutchison CJ (2006) Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev vol 86:967–1008. https://doi.org/10.1152/physrev.00047.2005
De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T et al (2002) Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 70(3):726–736. https://doi.org/10.1086/339274
Beck M, Hurt E (2017) The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18. Nature Publishing Group. https://doi.org/10.1038/nrm.2016.147
Fallini C, Khalil B, Smith CL, Rossoll W (2020) Traffic jam at the nuclear pore: all roads lead to nucleocytoplasmic transport defects in ALS/FTD. Neurobiol Dis 140. Academic Press Inc. https://doi.org/10.1016/j.nbd.2020.104835
Hetzer MW, Walther TC, Mattaj IW (2005) Pushing the envelope: structure, function, and dynamics of the nuclear periphery. Annu Rev Cell Dev Biol 21:347–380. https://doi.org/10.1146/annurev.cellbio.21.090704.151152
Tran EJ, Wente SR (2006) Dynamic nuclear pore complexes: life on the edge. Cell. 125:1041–1053. https://doi.org/10.1016/j.cell.2006.05.027
Beck M, Förster F, Ecke M, Plitzko JM, Melchior F, Gerisch G, Baumeister W, Medalia O (2004) Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science 306(5700):1387–1390. https://doi.org/10.1126/science.1104808
Pascual-Garcia P, Capelson M (2019) Nuclear pores in genome architecture and enhancer function. Curr Opin Cell Biol 58. Elsevier Ltd. https://doi.org/10.1016/j.ceb.2019.04.001
Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, Chait BT (2000) The yeast nuclear pore complex: composition, architecture, and transport mechanism. J Cell Biol 148(4):635–651. https://doi.org/10.1083/jcb.148.4.635
Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MLJ (2002) Proteomic analysis of the mammalian nuclear pore complex. J Cell Biol 158(5):915–927. https://doi.org/10.1083/jcb.200206106
Sellés J, Penrad-Mobayed M, Guillaume C, Fuger A, Auvray L, Faklaris O, Montel F (2017) Nuclear pore complex plasticity during developmental process as revealed by super-resolution microscopy. Sci Rep 7(1):1–8. https://doi.org/10.1038/s41598-017-15433-2
Moore S, Rabichow BE, Sattler R (2020) The Hitchhiker’s guide to nucleocytoplasmic trafficking in neurodegeneration. Neurochem Res 45. Springer. https://doi.org/10.1007/s11064-020-02989-1
Hampoelz B, Andres-Pons A, Kastritis P, Beck M (2019) Structure and assembly of the nuclear pore complex. Annu Rev Biophys 48. Annual Reviews Inc. https://doi.org/10.1146/annurev-biophys-052118-115308
Lupu F, Alves A, Anderson K, Doye V, Lacy E (2008) Nuclear pore composition regulates neural stem/progenitor cell differentiation in the mouse embryo. Dev Cell 14(6):831–842. https://doi.org/10.1016/j.devcel.2008.03.011
Smitherman M, Lee K, Swanger J, Kapur R, Clurman BE (2000) Characterization and targeted disruption of murine Nup50, a p27Kip1-interacting component of the nuclear pore complex. Mol Cell Biol 20(15):5631–5642. https://doi.org/10.1128/mcb.20.15.5631-5642.2000
Frey S, Richter RP, Görlich D (2006) FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 314(5800):815–817. https://doi.org/10.1126/science.1132516
Macara IG (2001) Transport into and out of the nucleus. Microbiol Mol Biol Rev 65(4):570–594. https://doi.org/10.1128/MMBR.65.4.570-594.2001
Bano D, Hengartner MO, Nicotera P (2010) Nuclear pore complex during neuronal degeneration: cracking the last barrier! Nucleus 1(2):136–138. https://doi.org/10.4161/nucl.1.2.10798
D'Angelo MA, Raices M, Panowski SH, Hetzer MW (2009) Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 136(2):284–295. https://doi.org/10.1016/j.cell.2008.11.037
Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O et al (2007) The molecular architecture of the nuclear pore complex. Nature 450(7170):695–701. https://doi.org/10.1038/nature06405
Basel-Vanagaite L, Muncher L, Straussberg R, Pasmanik-Chor M, Yahav M, Rainshtein L, Walsh CA, Magal N et al (2006) Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol 60(2):214–222. https://doi.org/10.1002/ana.20902
Neilson DE, Adams MD, Orr CMD, Schelling DK, Eiben RM, Kerr DS, Anderson J, Bassuk AG et al (2009) Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am J Hum Genet 84(1):44–51. https://doi.org/10.1016/j.ajhg.2008.12.009
Faria AMC, Levay A, Wang Y, Kamphorst AO, Rosa MLP, Nussenzveig DR, Balkan W, Chook YM et al (2006) The nucleoporin Nup96 is required for proper expression of interferon-regulated proteins and functions. Immunity 24(3):295–304. https://doi.org/10.1016/j.immuni.2006.01.014
Müller-McNicoll M, Neugebauer KM (2013) How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat Rev Genet 14(4):275–287. https://doi.org/10.1038/nrg3434
Nourse J, Spada S, Danckwardt S (2020) Emerging roles of RNA 3′-end cleavage and polyadenylation in pathogenesis, diagnosis and therapy of human disorders. Biomolecules 10(6):915. https://doi.org/10.3390/biom10060915
Björk P, Wieslander L (2017) Integration of mRNP formation and export. Cell Mol Life Sci 74(16):2875–2897. https://doi.org/10.1007/s00018-017-2503-3
Rissland OS (2017) The organization and regulation of mRNA-protein complexes. Wiley Interdiscip Rev RNA 8(1):e1369. https://doi.org/10.1002/wrna.1369
Köhler A, Hurt E (2007) Exporting RNA from the nucleus to the cytoplasm. Nat Rev Mol Cell Biol vol 8:761–773. https://doi.org/10.1038/nrm2255
Xie Y, Ren Y (2019) Mechanisms of nuclear mRNA export: a structural perspective. Traffic 20(11):829–840. https://doi.org/10.1111/tra.12691
Valkov E, Dean JC, Jani D, Kuhlmann SI, Stewart M (2012) Structural basis for the assembly and disassembly of mRNA nuclear export complexes. Biochim Biophys Acta vol 1819:578–592. https://doi.org/10.1016/j.bbagrm.2012.02.017
Smith A, Brownawell A, Macara IG (1998) Nuclear import of Ran is mediated by the transport factor NTF2. Curr Biol 8(25):1403–1406. https://doi.org/10.1016/s0960-9822(98)00023-2
Bischoff FR, Ponstingl H (1991) Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature 354(6348):80–82. https://doi.org/10.1038/354080a0
Paschal BM, Gerace L (1995) Identification of NTF2, a cytosolic factor for nuclear import that interacts with nuclear pore complex protein p62. J Cell Biol 129(4):925–937. https://doi.org/10.1083/jcb.129.4.925
Sheffield LG, Miskiewicz HB, Tannenbaum LB, Mirra SS (2006) Nuclear pore complex proteins in Alzheimer disease. J Neuropathol Exp Neurol 65(1):45–54. https://doi.org/10.1097/01.jnen.0000195939.40410.08
Gasiorowski JZ, Dean DA (2003) Mechanisms of nuclear transport and interventions. Adv Drug Deliv Rev 55(6):703–716. https://doi.org/10.1016/S0169-409X(03)00048-6
Finlay DR, Newmeyer DD, Price TM, Forbes DJ (1987) Inhibition of in vitro nuclear transport by a lectin that binds to nuclear pores. J Cell Biol 104(2):189–200. https://doi.org/10.1083/jcb.104.2.189
Kose S, Furuta M, Imamoto N (2012) Hikeshi, a nuclear import carrier for Hsp70s, protects cells from heat shock-induced nuclear damage. Cell 149(3):578–589. https://doi.org/10.1016/j.cell.2012.02.058
Michels AA, Kanon B, Konings AW, Ohtsuka K, Bensaude O, Kampinga HH (1997) Hsp70 and Hsp40 chaperone activities in the cytoplasm and the nucleus of mammalian cells. J Biol Chem 272(52):33283–33289. https://doi.org/10.1074/jbc.272.52.33283
Aguzzi A, Altmeyer M (2016) Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol 26. Elsevier Ltd. https://doi.org/10.1016/j.tcb.2016.03.004
Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18. Nature Publishing Group. https://doi.org/10.1038/nrm.2017.7
Shin Y, Brangwynne CP (2017) Liquid phase condensation in cell physiology and disease. Science 357(6357):eaaf4382. https://doi.org/10.1126/science.aaf4382
Kato M, McKnight SL (2017) Cross-β polymerization of low complexity sequence domains. Cold Spring Harb Perspect Biol 9(3). https://doi.org/10.1101/cshperspect.a023598
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339(6125):1335–1338. https://doi.org/10.1126/science.1232927
Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, Tycko R (2017) Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 171(3):615–627.e616. https://doi.org/10.1016/j.cell.2017.08.048
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J et al (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162(5):1066–1077. https://doi.org/10.1016/j.cell.2015.07.047
Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, Bowen KE, Wadhwa H et al (2018) Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173(4):958–971.e917. https://doi.org/10.1016/j.cell.2018.03.025
Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S et al (2019) Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102(2):339–357.e337. https://doi.org/10.1016/j.neuron.2019.02.038
Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S et al (2017) Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547(7662):236–240. https://doi.org/10.1038/nature22822
Linnemann AK, Platts AE, Krawetz SA (2009) Differential nuclear scaffold/matrix attachment marks expressed genes. Hum Mol Genet 18(4):645–654. https://doi.org/10.1093/hmg/ddn394
Nozawa RS, Boteva L, Soares DC, Naughton C, Dun AR, Buckle A, Ramsahoye B, Bruton PC et al (2017) SAF-A regulates interphase chromosome structure through oligomerization with chromatin-associated RNAs. Cell 169(7):1214–1227.e1218. https://doi.org/10.1016/j.cell.2017.05.029
Hutchison CJ (2002) Lamins: building blocks or regulators of gene expression? Nat Rev Mol Cell Biol 3:848–858. https://doi.org/10.1038/nrm950
Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL (2005) The nuclear lamina comes of age. Nat Rev Mol Cell Biol vol 6:21–31. https://doi.org/10.1038/nrm1550
Kurdistani SK, Tavazoie S, Grunstein M (2004) Mapping global histone acetylation patterns to gene expression. Cell 117(6):721–733. https://doi.org/10.1016/j.cell.2004.05.023
Schirmer EC (2008) The epigenetics of nuclear envelope organization and disease. Mutat Res vol 647:112–121. https://doi.org/10.1016/j.mrfmmm.2008.07.012
Kwong LK, Neumann M, Sampathu DM, Lee VMY, Trojanowski JQ (2007) TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol vol 114:63–70. https://doi.org/10.1007/s00401-007-0226-5
Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ (2008) ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol vol 15:772–780. https://doi.org/10.1111/j.1468-1331.2008.02195.x
Zuleger N, Robson MI, Schirmer EC (2011) The nuclear envelope as a chromatin organizer. Nucleus 2. Taylor and Francis Inc. https://doi.org/10.4161/nucl.2.5.17846
Brown CR, Kennedy CJ, Delmar VA, Forbes DJ, Silver PA (2008) Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev 22(5):627–639. https://doi.org/10.1101/gad.1632708
Strambio-De-Castillia C, Niepel M, Rout MP (2010) The nuclear pore complex: bridging nuclear transport and gene regulation. Nat Rev Mol Cell Biol 11. Nature Publishing Group. https://doi.org/10.1038/nrm2928
Chu CT, Plowey ED, Wang Y, Patel V, Jordan-Sciutto KL (2007) Location, location, location: altered transcription factor trafficking in neurodegeneration. J Neuropathol Exp Neurol 66(10):873–883. https://doi.org/10.1097/nen.0b013e318156a3d7
Hetzer MW (2010) The role of the nuclear pore complex in aging of post-mitotic cells. Aging (Albany NY) 2. Impact Journals LLC. https://doi.org/10.18632/aging.100125
Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH (2017) Phase separation drives heterochromatin domain formation. Nature 547(7662):241–245. https://doi.org/10.1038/nature22989
Chan WM, Tsoi H, Wu CC, Wong CH, Cheng TC, Li HY, Lau KF, Shaw PC et al (2011) Expanded polyglutamine domain possesses nuclear export activity which modulates subcellular localization and toxicity of polyQ disease protein via exportin-1. Hum Mol Genet 20(9):1738–1750. https://doi.org/10.1093/hmg/ddr049
Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30(1):575–621. https://doi.org/10.1146/annurev.neuro.29.051605.113042
Zuccato C, Valenza M, Cattaneo E (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev 90(3):905–981. https://doi.org/10.1152/physrev.00041.2009
Grima JC, Daigle JG, Arbez N, Cunningham KC, Zhang K, Ochaba J, Geater C, Morozko E et al (2017) Mutant Huntingtin disrupts the nuclear pore complex. Neuron 94(1):93–107.e106. https://doi.org/10.1016/j.neuron.2017.03.023
Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, Atwal RS, Artates JW, Tabet R, Wheeler VC, Bang AG et al (2017) Polyglutamine-expanded Huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 94(1):48–57.e44. https://doi.org/10.1016/j.neuron.2017.03.027
Watkins JL, Murphy R, Emtage JL, Wente SR (1998) The human homologue of Saccharomyces cerevisiae Gle1p is required for poly(A)+ RNA export. Proc Natl Acad Sci U S A 95(12):6779–6784. https://doi.org/10.1073/pnas.95.12.6779
Bischoff FR, Krebber H, Kempf T, Hermes I, Ponstingl H (1995) Human RanGTPase-activating protein RanGAP1 is a homologue of yeast Rna1p involved in mRNA processing and transport. Proc Natl Acad Sci U S A 92(5):1749–1753. https://doi.org/10.1073/pnas.92.5.1749
Havel LS, Wang C-E, Wade B, Huang B, Li S, Li X-J (2011) Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation. Hum Mol Genet 20(7):1424–1437. https://doi.org/10.1093/hmg/ddr023
Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M et al (2009) IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol 187(7):1083–1099. https://doi.org/10.1083/jcb.200909067
Frosst P, Guan T, Subauste C, Hahn K, Gerace L (2002) Tpr is localized within the nuclear basket of the pore complex and has a role in nuclear protein export. J Cell Biol 156(4):617–630. https://doi.org/10.1083/jcb.200106046
Marxreiter F, Stemick J, Kohl Z (2020) Huntingtin lowering strategies. Int J Mol Sci 21(6):2146. https://doi.org/10.3390/ijms21062146
Kim JY, Shen S, Dietz K, He Y, Howell O, Reynolds R, Casaccia P (2010) HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci 13(2):180–189. https://doi.org/10.1038/nn.2471
Dutta R, Trapp BD Pathogenesis of axonal and neuronal damage in multiple sclerosis. In: 2007/05//. Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology. pp S22-S31. https://doi.org/10.1212/01.wnl.0000275229.13012.32
Trapp BD, Stys PK (2009) Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol vol 8:280–291. https://doi.org/10.1016/S1474-4422(09)70043-2
Adams JH, Graham DI, Gennarelli TA, Maxwell WL (1991) Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatry 54. BMJ Publishing Group. https://doi.org/10.1136/jnnp.54.6.481
Kim JY, Casaccia P (2010) HDAC1 in axonal degeneration: a matter of subcellular localization. Cell Cycle 9. Taylor and Francis Inc. https://doi.org/10.4161/cc.9.18.12716
Flokas ME, Tomani M, Agdere L, Brown B (2019) Triple A syndrome (Allgrove syndrome): improving outcomes with a multidisciplinary approach. Pediatric Health Med Ther 10:99–106. https://doi.org/10.2147/phmt.s173081
Yadav P, Kumar D, Bohra G, Garg M (2020) Triple A syndrome (Allgrove syndrome)–a journey from clinical symptoms to a syndrome. J Family Med Prim Care 9(5):2531–2534. https://doi.org/10.4103/jfmpc.jfmpc_237_20
Cronshaw JM, Matunis MJ (2003) The nuclear pore complex protein ALADIN is mislocalized in triple A syndrome. Proc Natl Acad Sci U S A 100(10):5823–5827. https://doi.org/10.1073/pnas.1031047100
Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, Herrup K (2012) Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat Med 18(5):783–790. https://doi.org/10.1038/nm.2709
Hitchler MJ, Domann FE (2014) Regulation of CuZnSOD and its redox signaling potential: implications for amyotrophic lateral sclerosis. Antioxid Redox Signal 20(10):1590–1598. https://doi.org/10.1089/ars.2013.5385
Kinoshita Y, Ito H, Hirano A, Fujita K, Wate R, Nakamura M, Kaneko S, Nakano S et al (2009) Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 68(11):1184–1192. https://doi.org/10.1097/NEN.0b013e3181bc3bec
Zhang J, Ito H, Wate R, Ohnishi S, Nakano S, Kusaka H (2006) Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol 112(6):673–680. https://doi.org/10.1007/s00401-006-0130-4
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351(3):602–611. https://doi.org/10.1016/j.bbrc.2006.10.093
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133. https://doi.org/10.1126/science.1134108
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211. https://doi.org/10.1126/science.1165942
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918):1205–1208. https://doi.org/10.1126/science.1166066
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC et al (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15(11):1488–1497. https://doi.org/10.1038/nn.3230
Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G et al (2012) Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep 2:529. https://doi.org/10.1038/srep00529
Dormann D, Haass C (2013) Fused in sarcoma (FUS): an oncogene goes awry in neurodegeneration. Mol Cell Neurosci 56:475–486. https://doi.org/10.1016/j.mcn.2013.03.006
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NCA et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2):245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y et al (1997) Nuclear translocation of NF-κb is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci U S A 94(14):7531–7536. https://doi.org/10.1073/pnas.94.14.7531
Pearson AG, Curtis MA, Waldvogel HJ, Faull RLM, Dragunow M (2005) Activating transcription factor 2 expression in the adult human brain: association with both neurodegeneration and neurogenesis. Neuroscience 133(2):437–451. https://doi.org/10.1016/j.neuroscience.2005.02.029
Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35(4):605–623. https://doi.org/10.1016/s0896-6273(02)00828-0
Chalovich EM, Zhu JH, Caltagarone J, Bowser R, Chu CT (2006) Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem 281(26):17870–17881. https://doi.org/10.1074/jbc.M602632200
Tian T, Huang C, Tong J, Yang M, Zhou H, Xia XG (2011) TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int J Biol Sci 7(2):234–243. https://doi.org/10.7150/ijbs.7.234
Kokoulina P, Rohn TT (2010) Caspase-cleaved transactivation response DNA-binding protein 43 in Parkinson’s disease and dementia with Lewy bodies. Neurodegener Dis 7(4):243–250. https://doi.org/10.1159/000287952
Woulfe JM (2007) Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathol Appl Neurobiol 33:2–42. https://doi.org/10.1111/j.1365-2990.2006.00819.x
Patel VP, Chu CT (2011) Nuclear transport, oxidative stress, and neurodegeneration. Int J Clin Exp Pathol 4:215–229 e-Century Publishing Corporation
Lee H, Ueda M, Miyamoto Y, Yoneda Y, Perry G, Smith MA, Zhu X (2006) Aberrant localization of importin α1 in hippocampal neurons in Alzheimer disease. Brain Res 1124(1):1–4. https://doi.org/10.1016/j.brainres.2006.09.084
Davidson Y, Amin H, Kelley T, Shi J, Tian J, Kumaran R, Lashley T, Lees AJ et al (2009) TDP-43 in ubiquitinated inclusions in the inferior olives in frontotemporal lobar degeneration and in other neurodegenerative diseases: a degenerative process distinct from normal ageing. Acta Neuropathol 118(3):359–369. https://doi.org/10.1007/s00401-009-0526-z
Wilson AC, Dugger BN, Dickson DW, Wang DS (2011) TDP-43 in aging and Alzheimer’s disease-a review. Int J Clin Exp Pathol 4:147–155 e-Century Publishing Corporation
Yamada T, Yoshiyama Y, Kawaguchi N (1997) Expression of activating transcription factor-2 (ATF-2), one of the cyclic AMP response element (CRE) binding proteins, in Alzheimer disease and non-neurological brain tissues. Brain Res 749(2):329–334. https://doi.org/10.1016/S0006-8993(96)01356-X
Tristan C, Shahani N, Sedlak TW, Sawa A (2011) The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal vol 23:317–323. https://doi.org/10.1016/j.cellsig.2010.08.003
Butterfield DA, Hardas SS, Lange MLB (2010) Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: many pathways to neurodegeneration. J Alzheimers Dis 20. IOS Press. https://doi.org/10.3233/JAD-2010-1375
Azam S, Jouvet N, Jilani A, Vongsamphanh R, Yang X, Yang S, Ramotar D (2008) Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J Biol Chem 283(45):30632–30641. https://doi.org/10.1074/jbc.M801401200
Hara MR, Cascio MB, Sawa A (2006) GAPDH as a sensor of NO stress. Biochim Biophys Acta vol 1762:502–509. https://doi.org/10.1016/j.bbadis.2006.01.012
Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, Cook C, Miller SJ et al (2018) Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99(5):925–940.e927. https://doi.org/10.1016/j.neuron.2018.07.039
Tripathi T, Prakash J, Shav-Tal Y (2019) Phospho-tau impairs nuclear-cytoplasmic transport. ACS Chem Neurosci 10. American Chemical Society. https://doi.org/10.1021/acschemneuro.8b00632
Ferreira PA (2019) The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell Mol Life Sci 76(12):2247–2273. https://doi.org/10.1007/s00018-019-03029-0
Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O'Donovan K, Fare CM et al (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173(3):677–692.e620. https://doi.org/10.1016/j.cell.2018.03.002
Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG et al (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21(2):228–239. https://doi.org/10.1038/s41593-017-0047-3
Ederle H, Funk C, Abou-Ajram C, Hutten S, Funk EBE, Kehlenbach RH, Bailer SM, Dormann D (2018) Nuclear egress of TDP-43 and FUS occurs independently of exportin-1/CRM1. Sci Rep 8(1):7084. https://doi.org/10.1038/s41598-018-25007-5
Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EMH, Li X, Miguez R, Dayton RD et al (2018) TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep 8(1):4606. https://doi.org/10.1038/s41598-018-22858-w
Guo L, Fare CM, Shorter J (2019) Therapeutic dissolution of aberrant phases by nuclear-import receptors. Trends Cell Biol 29(4):308–322. https://doi.org/10.1016/j.tcb.2018.12.004
Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR 3rd, Hetzer MW (2013) Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154(5):971–982. https://doi.org/10.1016/j.cell.2013.07.037
Savas JN, Toyama BH, Xu T, Yates JR 3rd, Hetzer MW (2012) Extremely long-lived nuclear pore proteins in the rat brain. Science 335(6071):942. https://doi.org/10.1126/science.1217421
Hung MC, Link W (2011) Protein localization in disease and therapy. J Cell Sci 124(Pt 20):3381–3392. https://doi.org/10.1242/jcs.089110
Funding
This work was supported by JSPS Grant Number JP19K2252 (NH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hachiya, N., Sochocka, M., Brzecka, A. et al. Nuclear Envelope and Nuclear Pore Complexes in Neurodegenerative Diseases—New Perspectives for Therapeutic Interventions. Mol Neurobiol 58, 983–995 (2021). https://doi.org/10.1007/s12035-020-02168-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02168-x