
Abstract
Both sporadic variably protease-sensitive prionopathy (VPSPr) and familial Creutzfeldt-Jakob disease linked to the prion protein (PrP) V180I mutation (fCJDV180I) have been found to share a unique pathological prion protein (PrPSc) that lacks the protease-resistant PrPSc glycosylated at residue 181 because two of four PrP glycoforms are apparently not converted into the PrPSc from their cellular PrP (PrPC). To investigate the seeding activity of these unique PrPSc molecules, we conducted in vitro prion conversion experiments using serial protein misfolding cyclic amplification (sPMCA) and real-time quaking-induced conversion (RT-QuIC) assays with different PrPC substrates. We observed that the seeding of PrPSc from VPSPr or fCJDV180I in the sPMCA reaction containing normal human or humanized transgenic (Tg) mouse brain homogenates generated PrPSc molecules that unexpectedly exhibited a dominant diglycosylated PrP isoform along with PrP monoglycosylated at residue 181. The efficiency of PrPSc amplification was significantly higher in non-CJDMM than in non-CJDVV human brain homogenate, whereas it was higher in normal TgVV than in TgMM mouse brain homogenate. PrPC from the mixture of normal TgMM and Tg mouse brain expressing PrPV180I mutation (Tg180) but not TgV180I alone was converted into PrPSc by seeding with the VPSPr or fCJDV180I. The RT-QuIC seeding activity of PrPSc from VPSPr and fCJDV180I was significantly lower than that of sCJD. Our results suggest that the formation of glycoform-selective prions may be associated with an unidentified factor in the affected brain and the glycoform-deficiency of PrPSc does not affect the glycoforms of in vitro newly amplified PrPSc.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prions are infectious pathogens that are associated with a group of fatal transmissible spongiform encephalopathies or prion diseases affecting both animals and humans. They are composed mainly of the pathologic scrapie conformers (PrPSc) that originate from their normal cellular prion protein (PrPC) through a conformational transition of the largely α-helical form to predominantly β-sheets by a seed- or template-assisted mechanism [1, 2]. Human prion diseases are highly heterogeneous: they can be familial, sporadic, or acquired by infection, and include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker (GSS) disease, fatal insomnia, kuru, variant CJD (vCJD), and variably protease-sensitive prionopathy (VPSPr) [3].
PrPSc detected in the brain of all sporadic and acquired CJD and in most familial CJD has been well recognized to be mainly composed of three typical PrP glycoforms including diglycosylated, monoglycosylated, and un-glycosylated forms. However, Zou and co-workers recently discovered that the sporadic VPSPr along with familial Creutzfeldt-Jakob disease (fCJD) associated with a PrP mutation Val to Ile at residue 180 (fCJDV180I) generates a PrPSc molecule with a unique electrophoretic gel profile and is characterized by the accumulation in the brain of PrPSc that lacks diglycosylated PrPSc and PrPSc monoglycosylated at residue 181 [4,5,6]. Unlike fCJD linked to the PrP mutation Thr to Ala at residue 183 (fCJDT183A) that eliminates the N-linked glycosylation at residue 181 [7,8,9], the overall cellular PrP from VPSPr and fCJDV180I shows a normal PrP profile with the highest amount being diglycosylated PrP, followed by monoglycosylated PrP, and the lowest amount being un-glycosylated PrP prior to digestion with proteinase K (PK). Therefore, at least based on Western blotting, PrP from VPSPr and fCJDV180I seems to have an intact glycoform profile similar to that from normal controls but somehow the diglycosylated PrP and PrP monoglycosylated at residue 181 are not converted into the PK-resistant PrPSc; only un-glycosylated PrP and PrP monoglycosylated at residue 197 are converted into PK-resistant PrPSc [4,5,6]. Interestingly, two subsequent studies revealed that PrPSc from VPSPr patients exhibited very low transmissibility in the first passage of a transmission study in humanized transgenic mice and no transmission was observed at all in the second passage [10, 11].
To further understand the molecular mechanism underlying the conversion of PrPC into this unique PrPSc in VPSPr and fCJDV180I, we employed two advanced technologies termed serial protein misfolding cyclic amplification (sPMCA) and real-time quaking-induced conversion (RT-QuIC) assays to investigate the in vitro seeding activity of PrPSc from patients with these two conditions, respectively. We found that PrPSc from VPSPr and fCJDV180I can be amplified very efficiently using the brain homogenate from non-CJD individuals carrying PrP-129MM but poorly using the brain homogenate from non-CJD individuals carrying PrP-129VV. With PrP substrates from humanized transgenic mice expressing human wild-type PrP-129MM (TgMM) or PrP-129VV (TgVV) or expressing human mutant PrP with V180I mutation coupled with PrP-129MM polymorphism (TgV180I), amplification of PrPSc from VPSPr and fCJDV180I was highly efficient using TgVV and poor using TgMM or TgV180I alone. Interestingly, PrPSc was very efficiently amplified using a mixture of brain homogenate derived from both TgMM and TgV180I. As a control, we found that PrPSc from fCJDT183A that lacks the N-linked glycosylation at residue 181 could be amplified in non-CJD human brain homogenate with either PrP-129MM or PrP-129VV. PrPSc from fCJDT183A was also very efficiently amplified using brain homogenates from TgMM and TgMM + TgV180I, but not amplified using TgVV or TgV180I alone. Surprisingly, diglycosylated PrPSc was the predominant form in all amplified PrPSc species. Moreover, RT-QuIC assay demonstrated that PrPSc from VPSPr, fCJDV180I, and fCJDT183A was all amplifiable using recombinant bank vole PrP as a substrate; the seeding activity of PrPSc from VPSPr and fCJDV180I was approximately 102- to 105-fold lower than that of PrPSc from sCJDMM1 and sCJDVV2.
Materials and Methods
Reagents and Antibodies
PK was purchased from Sigma Chemical Co. (St. Louis, MO). Reagents for enhanced chemiluminescence (ECL Plus) were from Amersham Pharmacia Biotech, Inc. (Piscataway, NJ). Anti-PrP antibodies 3F4 against human PrP residues 107-112 [12, 13], 1E4 against human PrP97-106 [14], Tohoku 2 (T2) [15], Bar209, and V14 [4, 16] were used.
Preparation of Humanized TgVV, TgMM, and TgV180I Mice
The humanized TgVV mice expressing human wild-type PrP with VV at residue 129 (also termed TgWV) were prepared as described previously [17]; the TgMM mice (also called Tg40h) expressing human wild-type PrP with MM at residue 129 were generated by self-breeding of the previously reported Tg40 mice [18, 19]. For preparation of TgV180I mice, the transgene construct was generated by introducing the V180I mutation via PCR-based mutagenesis into the pHGHuPrP-129M plasmid, which was microinjected into fertilized FVB/NJ eggs, and planted into the oviducts of pseudopregnant CD-1 mice at the transgenic mouse facility of Case Western Reserve University (Cleveland, OH). Founder pups were screened by PCR of tail DNA. All founder mice carrying the transgenes were bred with FVB/Prnp0/0 mice to obtain Tg mice in PrP-null background. Transgenic PrP expression in the brain and other tissues of the Tg mice was examined by Western blot analysis using monoclonal antibody 3F4. All animal experiments in this study were approved by the Institutional Animal Use and Care Committee and the Institutional Biosafety Committee.
Preparation of Recombinant Bank Vole PrP109M or PrP109I
The cloning of the bank vole PrP109M or PrP109I (BVPrP109M or BVPrP109I) genes were carried out based on the previously described [20]. The DNA coding for full-length BVPrP-109M and BVPrP-109I was amplified by PCR using a template plasmid of BVPrP-109M/pOPINE or BVPrP-109I/pOPINE. The amplification was carried out using oligonucleotides 5′ CGCGGATCCATGAAGAAGCGGCCAAAGCCTGG 3′ and 5′ CCCAAGCTTTTAGGAACTTCTCCCTTCGT 3′. The PCR product was digested with BamHI and HindIII and inserted into pET-28a (Novagen). The bank vole PrP109M or PrP109I recombinant protein expression and purification was done according to our previous study [21]. The Escherichia coli Rossetta (DE3) pLysS were transformed with full-length BVPrP-109M or BVPrP-109I for a large-scale production. The bacteria were induced at A600 = 0.6 by adding 1 mM isopropyl-b-d-thiogalactopyranoside (IPTG) and then subsequently grown at 37 °C for 5–6 h. Cells were collected by centrifugation (15 min at 15,000g). The bacterial pellets were re-suspended as 0.1 g of cell paste/milliliter in lysing buffer (10 mM Tris-HCl, 100 mM Na-PO4 buffer, pH 8.0). Mechanical disruption was used to lyse the cells and followed by centrifugation at 4 °C for 30 min at 15,000×g. The inclusion bodies were re-suspended in 6 M guanidine hydrochloride (GdnHCl), 10 mM Tris-HCl, 100 mM Na-PO4 buffer, and 10 mM β-mercaptoethanol (βME), pH 8.0, and sonicated on ice until completely solubilized. The recombinant bank vole PrP109M or PrP109I was refolded on the Ni-NTA column by running a GdnHCl gradient (from 6 M GdnHCl, 10 mM Tris-HCl, and 10 mM βME, pH 8.0, to 10 mM Tris-HCl and 100 mM Na-PO4 buffer, pH 8.0) at 1 mL/min. After wash, recombinant PrP was eluted with 10 mM Tris-HCl, 100 mM Na-PO4 buffer, and 500 mM imidazole, pH 5.8. The eluted soluble BVPrP fractions were loaded on a SDS/PAGE to evaluate protein purity and then pooled. The fractions only containing BVPrP were collected and dialyzed against 10 mM sodium phosphate buffer pH 5.8 and concentrated to 0.7 mg/mL. Protein aliquots were stored at − 80 °C until use.
Preparation of Human or Mouse Brain Samples for Western Blotting, sPMCA, and RT-QuIC Assays
Frozen brain tissues from patients with VPSPr, fCJDV180I, fCJDT183A, sCJDMM1, and sCJDVV2, or normal subjects with PrP129-MM or PrP129-VV and from humanized transgenic mice of TgVV, TgMM, or TgV180I perfused, were collected and kept at − 80 °C. The samples were carefully cleaned with PBS for three times before the processing of each specimen in order to avoid blood contamination. For Western blotting, or sPMCA assay, Beads Beater was used to prepare tissue homogenates and the samples were then centrifuged at 500g for 5 min. The supernatant (S1) was transferred to a clean tube for future use while the pellet (P1) was discarded. For RT-QuIC analysis, the S1 fraction was diluted at 1:1 with 2 × conversion buffer containing 300 mM NaCl, 2% Triton X-100, and a complete protease inhibitor in PBS without Ca2+ and Mg2+ to prepare a 5% brain homogenate and then make serial dilution with 1 × N2 in 0.05%SDS/PBS as described [22].
Serial PMCA Procedures
The preparation of PrP seeds and substrates as well as sPMCA was conducted as previously described [17, 23]. In brief, human or Tg mouse brain tissues were carefully dissected to avoid cerebellum and blood contamination as much as possible. Brain homogenate substrates from normal frozen brains were homogenized (10% w/v) in PMCA conversion buffer containing 150 mM NaCl, 1% Triton X-100, and 8 mM EDTA pH 7.4 and the complete protease inhibitor mixture cocktail (Roche) in PBS. The seeds of the brain tissue homogenates were prepared as the brain substrates described above. Tissue homogenates were centrifuged at 500g for 10 min at 4 °C and the supernatant (S1) fraction was collected as the substrate or centrifuged at 500g for 3 min for the seeds of brain samples. The substrates and seeds were kept at − 80 °C until use. Each seed was diluted in the substrate at the ratios from 1:12.5 to 1:100 (1 μL or 8 μL seed + 99 μL or 92 μL substrate) into 200-μL PCR tubes with 1 PTFE beads (diameter 3/32″) (Teflon, APT, RI). Twenty microliters of each mixture was taken and kept at − 20 °C as a non-PMCA control. The remaining mixture was subjected to serial PMCA (sPMCA). Each cycle comprised a 20-s elapse time of sonication at amplitude 85 (250 W; Misonix S3000 sonicator) followed by an incubation period of 29 min 40 s at 37 °C and each round of sPMCA consisted of 96 cycles. For the serial PMCA, 15 μL sample was taken from the last cycle and placed into 85-μL fresh normal brain substrates for a new round of amplification.
RT-QuIC Analysis
RT-QuIC assay was conducted as previously described [20, 22, 24]. Briefly, the reaction mix was composed of 10 mM phosphate buffer (pH 7.4), 300 mM NaCl, 0.1 mg/mL recombinant bank vole PrP23-231, 10 μM thioflavin T (ThT), 1 mM ethylenediaminetetraacetic acid tetrasodium salt hydrate (EDTA), and 0.001% SDS. Aliquots of the reaction mix (98 μL) were loaded into each well of a 96-well plate (Nunc) and seeded with 2 μL of brain homogenate spinning at 2000g for 2 min at 4 °C as previously described [22]. The plate was then sealed with a plate-sealer film (Nalgene Nunc International) and incubated at 42 °C in a BMG FLUOstar Omega plate reader with cycles of 1-min shaking (700 rpm double orbital) and 1-min rest throughout the indicated incubation time. ThT fluorescence measurements (450 ± 10-nm excitation and 480 ± 10-nm emission; bottom read) were taken every 45 min. Four replicate reactions were seeded with the same dilution of an individual sample. The average fluorescence values per sample were calculated using fluorescence values from all four replicate wells regardless of whether these values crossed the threshold described below. At least 2 of 4 replicate wells must cross this threshold for a sample to be considered positive.
Western Blotting
sPMCA-treated brain samples were subjected to treatment with PK at 100 μg/mL for 70 min at 45 °C with agitation prior to Western blotting. Samples were resolved either on 15% Tris-HCl Criterion pre-cast gels (Bio-Rad) for SDS-PAGE as described previously [25]. The proteins on the gels were transferred to Immobilon-P membrane polyvinylidene fluoride (PVDF, Millipore) for 2 h at 350 mA. For probing of PrP, the membranes were incubated for 2 h at room temperature with anti-PrP antibodies 3F4 at 1:40,000, 1E4 at 1:500, T2 at 1:8000, Bar209 at 1:6000, and V14 at 1:6000 dilution, as the primary antibody. Following incubation with horseradish peroxidase-conjugated sheep anti-mouse IgG at 1:4000 or donkey anti-rabbit IgG (for T2 only) at 1:6000 dilution, the PrP bands were visualized on Kodak film by ECL Plus as described by the manufacturer. PrP protein bands were measured by densitometric analysis and quantified using a UN-SCAN-IT Graph Digitizer software (Silk Scientific, Inc., Orem, Utah).
Statistical Analysis
The statistical differences in intensity of PrPSc amplified by sPMCA among different groups detected by Western blotting were statistically analyzed using Student’s T test or ANOVA test to obtain p values for comparisons between two groups or multiple groups.
Results
PrPSc from VPSPr and fCJDV180I to be examined for prion seeding activity lacks diglycosylated PrPSc
We first wanted to confirm our previous finding in the samples to be examined in this study that PrPSc from the brain of patients with VPSPr and fCJDV180I lacks the PK-resistant diglycosylated glycoform, detected by the 3F4 and 1E4 anti-PrP antibodies [4,5,6, 9]. On the 3F4 blot, in contrast to sCJD, the brain homogenates from VPSPr and fCJDV180I patients exhibited only mono- and un-glycosylated PK-resistant PrP bands migrating at approximately 26 kDa and 20 kDa upon the treatment of brain homogenates with different amounts of PK ranging from 5 through 100 μg/mL (Fig. 1a). However, in the samples without PK treatment (0 μg/mL), the diglycosylated PrP was readily detectable not only in sCJD but also in fCJDV180I and VPSPr (Fig. 1a). Our previous studies demonstrated that the lack of the PK-resistant diglycosylated PrPSc results from the missing PrP species diglycosylated and monoglycosylated at residue 181 that are not converted into the PK-resistant PrPSc molecule [4, 6]. The PK-resistant PrPSc from fCJDT183A exhibited a predominant monoglycosylated PrP band as well as a barely detectable diglycosylated and an un-glycosylated PrP bands (Fig. 1a).

Comparison of PrPSc from VPSPr, fCJDV180I, fCJDT183A, sCJDMM1, and sCJDVV2. Representative Western blotting of untreated and treated PrPSc with different amounts of PK from sCJDMM1, sCJDVV2, fCJDT183A, VPSPrMV, and fCJDV180I probed with 3F4 (a) and 1E4 (b). PrPSc from fCJDV180I and VPSPr lacks the PK-resistant diglycosylated PrPSc on the Western blotting probed with 3F4 while it has diglycosylated PrP species prior to PK treatment. It contains monoglycosylated PrP migrating at ~ 26 kDa and un-glycosylated PrP migrating at ~ 20 kDa. On the 1E4 blot (b), additional small fragments migrating at ~ 23 kDa, ~ 17 kDa, and ~ 7 kDa were detected in the PK-treated samples from fCJDV180I and VPSPr. Molecular weight markers are shown in kDa on the right side of the blots
The epitopes of the 1E4 and 3F4 antibodies are next to each other on the protein, the former being composed of human PrP97-105 and the latter covering human PrP106-112 [13, 14], as shown in the schematic diagram of PrP structure and modifications in Fig. S1 [26,27,28]. We previously observed that the two antibodies have different immunoreactivity with distinct PrPSc molecules. For instance, compared to 3F4, 1E4 has a higher affinity for the sCJD PrPSc type 2 and a lower affinity for sCJD PrPSc type 1, in addition to its higher affinity for the unique ladder-like PrPSc in VPSPr and fCJDV180I [3,4,5,6]. According to the previous sequencing study, the PK-resistant PrPSc type 2 fragment starts at residue 97 [29], the first amino acid of the 1E4 epitope. Thus, it is most likely that the higher immunoreactivity of the 1E4 antibody than that of the 3F4 antibody is directly attributable to the well-exposed 1E4 epitope on the PrPSc type 2 fragment.
To confirm the particular gel profiles of PrPSc from VPSPr and fCJDV180I, we next probed the blots with the 1E4 antibody. Indeed, although the equal amounts of brain homogenates were loaded, compared to 3F4, 1E4 showed a greater intensity for sCJD PrPSc type 2 while a weaker intensity for sCJD PrPSc type 1 (Fig. 1b, left two panels). Moreover, in addition to the two PK-resistant PrPSc bands migrating at ~ 26 and ~ 20 kDa, 1E4 detected three additional PK-resistant PrP fragments migrating at ~ 23, ~ 17, and ~7 kDa in the samples from fCJDV180I and VPSPr, exhibiting the unique ladder-like PK-resistant PrPSc bands (Fig. 1b). They were not detectable in the samples from sCJDMM1 and sCJDVV2 and also not in the samples from fCJDT183A (Fig. 1b). Since the PrPT183A mutation has been shown to completely abolish the N-linked glycosylation at residue 181 [7,8,9], the faint diglycosylated PrPSc detected by both 3F4 and 1E4 is expected to be from the wild-type allele (Fig. 1a, b) [7, 9].
PrPSc from VPSPr and fCJDV180I Preferentially Seeds Human Brain–Derived PrPC-129MM Substrate by sPMCA, Forming Diglycoform-Containing PrPSc
Given the unique PrPSc profile and low transmissibility of VPSPr and fCJDV180I, it would be important to determine whether the PrPSc molecules from these diseases can convert normal PrPC from healthy human brain homogenates and what types of PrPSc can be generated in vitro compared to PrPSc from the most common sCJD. We conducted serial protein misfolding cyclic amplification (sPMCA) of PrPSc from brain homogenates of VPSPr-129MM (VPSPrMM), VPSPr-129VV (VPSPrVV), VPSPr-129MV (VPSPrMV), and fCJDV180I-129MM using two types of PrPC from healthy human brain homogenate with either 129MM or 129VV polymorphism as the substrates. sPMCA is a highly efficient in vitro amplification assay that has been shown to be able to faithfully mimic prion conversion by continuously seeding PrPSc in normal PrPC substrate [23]. PrPSc from sCJDMM1, sCJDVV2, and fCJDT83A was used as controls. It is known that the PrP polymorphism at residue 129 of the protein can significantly affect the efficiency of PrPC-PrPSc conversion. Thus, the PrPSc seeds from VPSPr carrying different 129-polymorphisms and the PrPC substrates from healthy human brain homogenates with either 129MM (hMM) or 129VV (hVV) were examined by sPMCA.
Amplification of PrPSc from sCJDMM1 or sCJDVV2 was observed in all three rounds of sPMCA-treated samples but not in non-sPMCA-treated control samples conducted with the seed-substrate polymorphism-matched and unmatched sPMCA (Fig. 2a, b). The amplification efficiency of sCJDMM1 PrPSc was significantly greater in seed-substrate-matched than unmatched sPMCA (MM-MM vs MM-VV, p < 0.001) while there was no significant difference in the amplification efficiency of sCJDVV2 between the seed-substrate-matched and unmatched sPMCA (VV-VV vs VV-MM, p > 0.05) (Fig. 2a, b, Table S1).

Serial PMCA of PrPSc from sCJD, VPSPr, fCJDV180I, and fCJDT183A in human brain homogenate substrates. Representative Western blotting of PrPSc from sCJDMM1 and sCJDVV2 (a), VPSPr, fCJDV180I, and fCJDT183A (c, d) amplified with 1–3, 1–6, or 1–8 rounds of sPMCA in human brain homogenate substrates from non-CJD MM (hMM) or VV (hVV) probed with the 3F4 antibody. sPMCA-R, sPMCA rounds. Molecular weight markers are shown in kDa on the left side of the blots. PK, proteinase K. b, e Bar graph showing the quantitative analyses of the intensity of PrPSc amplified by sPMCA after densitometric scanning. *p < 0.05; **p < 0.01; ***p < 0.001; NS, not statistically significant
After 6 rounds of sPMCA, PrPSc from three genotypes of VPSPr including 129-MM, 129-VV, and 129-MV was all amplified in normal human brain homogenates hMM (Fig. 2c, e). In contrast, in hVV, PrPSc from VPSPrMM or VPSPrVV exhibited significantly low efficiency while the PrPSc amplification from VPSPrMV or fCJDV180I varied (Fig. 2c, e). The amplification of PrPSc from VPSPrMM and VPSPrVV was significantly higher in hMM than in hVV substrate (p < 0.001), whereas no significant difference was observed for VPSPrMV (p > 0.05) (Fig. 2c, e). PrPSc from fCJDV180I was amplified in both hMM and hVV substrates while it showed significantly stronger amplification in hMM than in hVV (Fig. 2c, e). Notably, PrPSc from fCJDT183A was amplified efficiently with 5–8 rounds of sPMCA in both hMM and hVV; the amplification efficiency was significantly higher in hVV than in hMM (p < 0.01) (Fig. 2d, e), which was different from VPSPr, fCJDV180I, or sCJD. ANOVA analysis showed no differences in PrPSc amplification between different seeds but significant differences between substrates (Table S1).
In sum, like sCJDMM1, PrPSc from three genotypes of VPSPr and fCJDV180I was all amplified in the hMM substrate while no or less amplification in the hVV substrate. PrPSc from sCJDVV2 or fCJDT183A showed increased amplification in hVV than in hMM. In contrast to sCJD whose PrPSc amplification could be observed at the first round of sPMCA, PrPSc was not amplified until 4 or 5 rounds of sPMCA in VPSPr and fCJD, suggesting that the prion seeding activity in glycoform-deficient PrPSc from VPSPr and the two fCJD cases was lower than that from sCJD. Most surprisingly, all amplified PK-resistant PrPSc showed a predominant diglycosylated PrP isoform, although the PrPSc seeds display no, or significantly decreased, such isoform in VPSPr, fCJDV180I, and fCJDT183A.
PrPSc of VPSPr and fCJDV180I Favorably Seeds Tg Mice–Derived Human PrPC-129VV Substrate by sPMCA, Also Forming Diglycoform-Containing PrPSc
Human PrPC from the brain homogenate of humanized transgenic (Tg) mice expressing human PrP has been widely used as a substrate for amplification of human PrPSc by sPMCA in vitro [17, 30]. To determine whether the glycoform-deficient PrPSc from VPSPr and fCJDV180I is amplifiable in different humanized Tg mouse brain homogenates, their PrPSc molecules were subjected to sPMCA in four different Tg mouse brain homogenates, respectively, expressing human PrP-129VV (TgVV), PrP-129MM (TgMM), PrPV180I (Tg180), or in vitro mixed brain homogenate from TgMM and Tg180 mice (Tg180/TgMM). Since the PrPV180I mutation is characterized with the deposition in the brain of fCJDV180I of the unique PK-resistant PrPSc that has the gel profile similar to that of PrPSc from VPSPr, we generated humanized Tg mice expressing human PrPV180I to determine how the mutation affects the PrPSc formation in vitro.
We first determined amplification of classic PrPSc from sCJDMM1 or sCJDVV2 using sPMCA with the Tg mouse brain homogenate substrates. PrPSc from both sCJDMM1 and sCJDVV2 was amplified in the TgVV and TgMM substrates, respectively. The efficiency of amplification was significantly higher in TgVV than in TgMM substrate from both sCJDMM1 and sCJDVV2 PrPSc seeds (p < 0.01 or p < 0.0001) (Fig. 3a, b, c, Table S1). Interestingly, although PrPSc of sCJDMM1 or sCJDVV2 was virtually not amplified in the Tg180 substrate and less amplifiable in the TgMM substrate along, it was highly efficiently amplified in the in vitro mixed substrate of TgMM and Tg180 brain homogenate (p < 0.0005 or p < 0.0001) (Fig. 3a, b, c, Table S1).

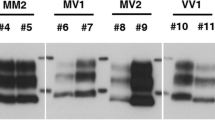
Serial PMCA of PrPSc from sCJD, VPSPr, fCJDV180I, and fCJDT183A in transgenic mouse brain homogenates. Representative Western blotting of PrPSc from sCJDMM1 (a); sCJDVV2 (b); and VPSPr, fCJDV180I, and fCJDT183A (d, e, f) amplified with 1–3, 1–6, or 1–8 rounds of sPMCA in humanized transgenic mouse brain homogenates from TgVV, TgMM, Tg180, or TgMM + Tg180 mouse line prior to PK digestion and Western blotting probed with the 3F4 antibody. sPMCA-R, sPMCA rounds. PK, proteinase K. c, g Bar graph showing the quantitative analyses of the intensity of PrPSc amplified by sPMCA after densitometric scanning. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; NS, not statistically significant
Similar to PrPSc from sCJD, the amplification of PrPSc of all three genotypes of VPSPr and fCJDV180I was highly efficient in the TgVV than in the TgMM substrate (p < 0.0001) (Fig. 3d, e, g, Table S1). Also no amplification was found using the brain homogenate as the substrate from Tg180 mice, although the level of PrPC in the brain of Tg180 was similar to that of PrPC in the brain of TgVV and TgMM (Fig. S2). Interestingly, amplification was rescued when TgMM substrate was mixed with the Tg180 mouse brain homogenate substrate (Fig. 3d, e). We also noticed that although the gel profiles of PrPSc seeds are different among different genotypes of VPSPr, sCJDMM1, and sCJDVV2, a similar pattern of PrPSc was generated by sPMCA using PrPC from the TgVV brain homogenate as the same substrate, (Fig. 3a, b, d, e, Table S1), suggesting that the PrPC substrate may determine the gel profile of newly generated PrPSc by sPMCA.
After 5–8 rounds of sPMCA, amplification efficiency of PrPSc from fCJDT183A was significantly higher in TgMM than in TgVV substrate (p < 0.0001) (Fig. 3f, g), which was opposite to the amplification efficiency of PrPSc from VPSPr and fCJDV180I (Fig. 3d, e, g). Notably, the similar opposite effect of PrPT183A on the PrPSc amplification in the two Tg mouse–derived substrates between fCJDT183A and all other prion diseases examined above including sCJD, VPSPr, and fCJDV180I was also observed in human brain–derived PrPC substrates (Fig. 2). Again, no PrPSc of fCJDT183A was amplified in the Tg180 substrate but amplification was rescued when TgMM mouse brain homogenate was mixed with Tg180 brain homogenate (Fig. 3f). Although PrPSc from VPSPr, fCJDV180I, and fCJDT183A contains virtually no diglycosylated PrPSc, the same as in human brain–derived PrPC substrate, a dominant diglycosylated PrPSc was amplified in the Tg mouse brain–derived PrPC substrates as did PrPSc from sCJD (Fig. 3a, b, d, e, f, Table S1).
Taken together, as in the human brain–derived PrPC substrate, PrPSc from VPSPr and fCJDV180I was also amplified in the Tg mouse–derived human PrPC substrate and the amplified PrPSc contained a dominant diglycosylated PrP species. However, unlike the human brain PrPC substrate, the TgVV substrate was more susceptible to be recruited into PrPSc than the TgMM by sCJD, VPSPr, and fCJDV180I by sPMCA. Like in the human brain substrate, PrPSc from fCJDT183A showed the effect of the 129-polymorphsim on PrPSc amplification opposite to that from sCJD, VPSPr, and fCJDV180I. Remarkably, while PrPSc from none of the four human prion diseases was able to seed the Tg180 substrate alone, it was amplified only in the substrate of combination of TgMM and Tg180 substrates.
No Small PK-Resistant PrPSc Fragments Are Amplified by sPMCA from VPSPr and fCJDV180I in Both Human- and Humanized Tg Mouse–Derived PrPC Substrates
In addition to the lack of diglycosylated PrP and the PrP molecule monoglycosylated at residue 181 shown by the 3F4 antibody and other anti-PrP antibodies including the 1E4 antibody, another unique feature of the gel profiles of PrPSc from VPSPr and fCJDV180I is the formation of the ladder-like five PK-resistant PrPSc bands with extra small fragments detected by the 1E4 antibody [4,5,6, 31], as shown in Fig. 1. Moreover, using glycoform-specific anti-PrP antibodies Bar209 and V14, we identified the PrP molecules with N-linked glycosylation specifically at residue 181 that are not converted into PK-resistant PrPSc [4]. To determine whether the PrPSc amplified by sPMCA in TgVV, TgMM, Tg180, or the mixture of TgMM and Tg180 substrate contains those particular small PK-resistant fragments migrating at ~ 23 kDa, ~ 17 kDa, and ~ 7 kDa, we probed the amplified PrPSc with different anti-PrP antibodies.
When the PrPSc molecule amplified with the VPSPr and fCJDV180I seeds in the hMM or hVV substrate was probed with the 1E4 antibody, the PrP gel profiles observed were virtually the same as those detected by the 3F4 antibody without extra small PK-resistant PrPSc fragments (Fig. 4a). The observable differences between the two blots included that the intensity of the un-glycosylated PrPSc band from all VPSPr and fCJDV180I amplified in hMM or hVV was increased on the 1E4 blot compared to the 3F4 blot and the amplified PrPSc in the hVV substrate became readily detectable with 1E4 (Fig. 4a with 1E4 vs Fig. 2c with 3F4). When PrPSc was amplified in the TgVV or TgMM mouse substrate, notably, no typical three small PK-resistant PrPSc were detected (Fig. 4b). Since it is known that 1E4 has a higher affinity for PrPSc type 2 than PrPSc type 1 compared to 3F4 [14], we expected that more PrPSc type 2 was detected by 1E4 from PrPSc amplified in the TgVV substrate compared to 3F4. To confirm this, we used another antibody named Tohoku 2 (T2) that specifically detects PrPSc type 2 to probe the blot [15]. As shown in Fig. S3, similar to 1E4, the intensity of the PK-resistant un-glycosylated PrP was increased compared to that shown by the 3F4 antibody, although there were some smear bands migrating under ~ 19 kDa in the TgVV or TgMM + Tg180 seeded with PrPSc of VPSPrMM or VPSPrVV.

Serial PMCA of PrPSc from VPSPr and fCJDV180I in transgenic mouse brain homogenates. Representative Western blotting of PrPSc amplified with 4–6 rounds of sPMCA by seeding PrPSc of VPSPrMM, VPSPrVV, VPSPrMV, or fCJDV180I in humanized transgenic mouse brain homogenates from TgVV, TgMM, Tg180, or TgMM + Tg180 mouse line (b and c). The blots were probed with the 1E4 antibody. sPMCA-R, sPMCA rounds. Molecular weight markers are shown in kDa on the left side of the blots. PK, proteinase K
PrPSc of VPSPr and fCJDV180I Amplified in Human Brain– or Humanized Tg Mouse–Derived PrPC Substrates Contains Intact Mono- and Diglycosylated PrP Species
To determine whether there is any glycoform deficiency of PrPSc amplified by sPMCA, we detected the sPMCA-amplified PrPSc with glycoform-specific anti-PrP antibodies Bar 209 and V14 [4, 16]. The Bar209 antibody specifically recognizes the PrP molecule monoglycosylated at residue 197 while the V14 antibody specially recognizes the PrP molecule monoglycosylated at residue 181; both of them detect the un-glycosylated PrP molecule [4, 16]. When the PrPSc amplified in different brain homogenate substrates was probed with the two antibodies, the two glycoforms were detected by the two antibodies (Fig. S4), suggesting that unlike the PrPSc seeds themselves from VPSPr and fCJDV180I, both glycoforms were converted into PrPSc from various PrPC substrates by sPMCA.
In Vitro Prion Seeding Activity of Glycoform-Deficient PrPSc from VPSPr and fCJDV180I Is Detectable by RT-QuIC Assay
RT-QuIC is another in vitro ultrasensitive assay to amplify and quantify PrPSc by measuring its seeding activity [32]. Prion-seeding activity is expected to reflect the ability of PrPSc to replicate in the presence of the PrPC substrate, characteristic of infectious prions [22, 24, 32]. Unlike sPMCA, RT-QuIC assay uses the recombinant PrP as the substrate instead of non-infected brain homogenate and monitors the aggregation-triggered increase in thioflavin T fluorescence in real time [24, 32]. Our RT-QuIC end-point titration assay revealed that the seeding activity of PrPSc from VPSPr and fCJDV180I in the recombinant bank vole PrP23-231 substrate was approximately 102- to 105-fold lower than that of PrPSc from sCJDMM1 and sCJDVV2 (Fig. 5). Among the three genotypes of VPSPr, fCJDV180I, and fCJDT183A, the prion seeding activity was highest in fCJDT183A, followed by VPSPrVV, fCJDV180I, VPSPrMM, and VPSPrMV according to the seeding activity at the highest dilution (Fig. 5). The latter two (VPSPrMM and VPSPrMV) exhibited the similar prion seeding activity.

RT-QuIC analysis of PrPSc from VPSPr, fCJDV180I, fCJDT183A, sCJDMM1, and sCJDVV2 with the recombinant bank vole PrP109I. PrPSc from VPSPr, fCJDV180I, fCJDT183A, sCJDMM1, and sCJDVV2 was seeded in recombinant bank vole PrP23-231 prior to RT-QuIC assay. The prion seeding activity was measured until 60 h. Different dilution of brain homogenates from infected human brains was tested from 10−3 to 10−11 according to their seeding activity. The panel at the lower right corner of the figure is the scatter graph for the comparison of the maximal dilution to be detected by RT-QuIC
Discussion
The key molecular event in the pathogenesis of various animal and human prion diseases is the conversion of PrPC into PrPSc. The molecular mechanism underlying the structural conversion of the two isoforms remains poorly understood, which prevents not only our understanding of pathogenesis and transmission of prion diseases between different species but also the development of efficient therapeutics for prion diseases. For instance, the species barriers are widely present in transmission of prion diseases among different species and are believed to result from a complex interplay of primary amino acid sequence, glycoform patterns, and three-dimensional structure of the PrP molecule [3].
Several lines of evidence have suggested that the individual PrP glycoforms may affect the efficiency of PrPC-to-PrPSc conversion even within the same species and individuals. Our previous work has shown, for the first time, that similar to fCJDT183A, in which the PrPT183A mutation eliminates the N-linked glycosylation at residue 181, the PrP molecules with N-linked glycosylation at residue 181, including both di- and monoglycosylated species, are not converted into PK-resistant PrPSc in sporadic VPSPr and familial CJD associated with PrPV180I mutation [4, 6]. However, our study also revealed that in contrast to fCJDT183A, both VPSPr and fCJDV180I exhibit three intact PrP glycoforms prior to PK digestion [4, 9]. Indeed, the glycoform profiles of PrPV180I mutation from transfected M17 cell lines and humanized Tg mice expressing human PrP with V180I mutation also showed no difference from those of wild-type PrP expressed in similar cell lines and humanized Tg mice [4]. Notably, we observed that PK-resistant PrPV180I from the cultured M17 cells contained diglycosylated PrP species, which made us think that the deficiency in conversion of PrPC into PrPSc may not be directly attributable to the PrPV180I mutation alone and there might be other factors that are involved in mediating the formation of this unique PrPSc isoform in vivo. Our previous findings raised two possibilities: first, the glycoform-selective prion formation observed in the brain of patients with VPSPr or fCJDV180I may involve dominant-negative inhibition caused by the interaction between misfolded and normal PrP molecules; second, one or more co-factors may be operating in VPSPr and fCJDV180I and the co-factors may prevent conversion of the PrPC molecules with N-linked glycosylation at residue 181 into PrPSc including both di- and monoglycosylated species [4, 6].
In addition, it is possible that the presence of the multiple PK-resistant PrPSc fragments in VPSPr and fCJDV180I may be associated with the dysfunction of the endoproteolytic processing event in vivo. For instance, a small C-terminal PK-resistant PrPSc termed C3 migrating at ~ 7 kDa that was believed to be generated by the γ-cleavage was significantly increased in the brain of sCJD patients [33]. The ~ 7-kDa fragment, one of the three small PK-resistant PrPSc fragments detected in VPSPr and fCJDV180I, is detected by 1E4 with the epitope localized between residues 97 and 105. Thus, this fragment is expected to be different from the C3 fragment generated by the γ-cleavage but more like the 7-kDa PrP observed in GSS.
The serial PMCA is able to faithfully amplify PrPSc in vitro [23, 34]. Using this ultrasensitive method, we observed that PrPSc from VPSPr and fCJDV180I was amplified very efficiently using the brain homogenate substrates from TgVV, non-CJD patients with PrP-129MM, and mixed brain homogenates with Tg180 and TgMM as well as less efficiently using TgMM and non-CJD patients with PrP-129VV. Currently, we do not know the exact reason why the PrPC from TgVV substrate is more susceptible to sCJDMM1, VPSPr, and fCJDV180IMM than that from TgMM, except for fCJDT183A. It would be interesting to compare sPMCA assay with animal-based bioassay with these cases in the future. No PrPSc amplification was found when brain homogenate from Tg180 alone was used as the substrate. PrPSc from fCJDT183A was also amplified using human hMM or hVV, TgMM, or TgMM + Tg180. Moreover, like sCJDMM1 or sCJDVV2, PrPSc amplified from VPSPr and fCJDV180I surprisingly showed a dominant diglycosylated PrPSc isoform.
Our finding is consistent with the recent observation by Peden et al. [35] in which they observed the generation of a ~ 30-kDa band corresponding to PK-resistant diglycosylated PrPSc after PMCA of PrPSc from brain homogenates of VPSPr patients using human brain homogenate as the substrate. Notably, sPMCA of PrPSc from fCJDT183A also generated a dominant diglycosylated PrPSc isoform. The PrPT183A mutation has been shown to specifically eliminate the first N-linked glycosylation site of the protein so that the mutant PrP cannot have either the di- or the monoglycosylated form at residue 181. Bearing this in mind, it is conceivable that the PK-resistant diglycosylated PrP amplified from fCJDT183A was most likely originated from the wild-type PrPSc since most of the fCJD patients, if not all of them, carry both mutant and wild-type alleles. Indeed, a small amount of PK-resistant diglycosylated PrPSc was observed in the brain samples from a patient with fCJDT183A, which was considered to derive from the wild-type allele [8]. Although it is unclear at the present where the diglycosylated PrPSc exactly comes from and why the diglycosylated PrPSc is highly efficiently generated in sPMCA reactions seeded by VPSPr and fCJDV180I brain homogenates, it is likely that the amplified diglycosylated PrPSc could be converted from the normal wild-type PrP as does PrPSc in fCJDT183A. On the other hand, the extra small PK-resistant PrP fragments detected specifically by the 1E4 antibody were not amplified in either human or mouse brain homogenates, consistent with the observation as well by Peden et al. [35]. The finding of no seeding activity of these 1E4-detected PrPSc small fragments by sPMCA may echo the observation of the low, or lack of, transmissibility of VPSPr observed by bioassays [10, 11].
While PrPSc from VPSPr showed highly efficient amplification in the brain homogenate of TgVV, similar to that from fCJDV180I and sCJD, it had a poor amplification in the brain homogenate of TgMM compared to that from the two fCJD and two sCJD cases. Although the amplification of PrPSc from all three genotypes of VPSPr with brain homogenates from TgMM or Tg180 mice alone was poor or even not at all, it was significantly increased by mixing the brain homogenates from the two Tg mice, especially for PrPSc from VPSPr-129VV. This result seemed to contradict the observation found in the brain of patients with VPSPr, where we expected that the interaction between wild-type and mutant PrPV180I in the brain may prevent the conversion of the PrPC molecules glycosylated at residue 181 to the PK-resistant PrP. Telling et al. [36] reported that the presence of wild-type PrP could delay or prevent prion formation initiated by the mutant PrP. Also, Noble et al. [37] observed that recombinant wild-type PrP in trans inhibited the spontaneous formation of a PK-resistant recombinant mutant PrP. Therefore, it is most likely that PrPSc from VPSPr and fCJDV180I could represent a unique prion strain that behaves differently from other common human prion strains.
RT-QuIC assay is another ultrasensitive approach for conversion of normal PrP into PrP aggregates in vitro [22, 24, 32]. It has been found that RT-QuIC assay is able to differentiate prion strains [22, 24, 35]. Unlike sPMCA, it uses recombinant PrP molecules instead of normal brain homogenates as the substrates and involves shaking instead of sonication to trigger the protein conversion process. The other feature of RT-QuIC is that the sequence barriers of the PrPSc-PrPC seeding are not so strict, which does seem to be different from sPMCA. For instance, many prion strains from different species can have fairly efficient conversion by RT-QuIC with substrates of recombinant hamster or bank vole PrP molecule. On the other hand, the conversion efficiency of PrP by sPMCA is mostly determined by the similarity in protein sequences, which is consistent with in vivo bioassays. Using the RT-QuIC assay, prion seeding activity was found using all genotypes of VPSPr, fCJDV180I, and fCJDT183A. Our RT-QuIC end-point titration assay indicated that the prion seeding activity was approximately 102- to 105-fold lower in VPSP and fCJDV180I than in sCJDMM1 and sCJDVV2, which is consistent with the observation by Peden et al. [35] in which they observed a lower prion seeding activity in VPSPr than in sCJD.
In conclusion, our current findings indicate that like sCJD, the glycoform-deficient PrPSc from VPSPr and fCJDV180I can be amplified as the typical PrPSc with intact diglycosylated species using sPMCA. It also exhibited prion seeding activity despite the lower amount than that in sCJD. The amplification efficiency of the glycoform-deficient PrPSc in vitro was enhanced by the interaction of brain homogenates from wild-type and mutant PrPC substrates. The extra three small PK-resistant PrPSc fragments were not be amplified by sPMCA in vitro, suggesting that those PrPSc species may not be highly infectious, which is consistent with the observations by in vivo animal-based bioassay reported previously. On the other hand, the inability of in vitro amplification to duplicate the unique PrPSc gel profile of VPSPr and fCJDV180I with either human or humanized Tg mouse brain substrate suggests that the formation of glycoform-selective and the ladder-like PK-resistant PrPSc may be associated with an unidentified factor in the affected human brain. It would be important to exclude the possibility in the future that the unknown factor could be a second component that is able to cause prion disease, as shown in other neurodegenerative diseases such as Alzheimer’s disease and amyotrophic lateral sclerosis in which there are more than one gene that are involved in the pathogenesis of the diseases.
Change history
01 February 2019
The original version of this article unfortunately contained a mistake. The email address Dr. Wen-Quan Zou, one of the corresponding authors should be written as “wxz6@case.edu” instead of “wxz@case.edu”.
References
Lansbury PT Jr (1997) Structural neurology: are seeds at the root of neuronal degeneration? Neuron 19:1151–1154. https://doi.org/10.1016/S0896-6273(00)80406-7
Prusiner SB (1998) Nobel Lecture: Prions. Prions Proc Natl Acad Sci U S A 95:13363–13383. https://doi.org/10.1073/pnas.95.23.13363
Das AS, Zou WQ (2016) Prions: beyond a single protein. Clin Microbiol Rev 29:633–658. https://doi.org/10.1128/CMR.00046-15
Xiao X, Yuan J, Haïk S, Cali I, Zhan Y, Moudjou M, Li B, Laplanche JL et al (2013) Glycoform-selective prion formation in sporadic and familial forms of prion disease. PLoS One 8:e58786. https://doi.org/10.1371/journal.pone.0058786
Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I, Shimoji M, Langeveld JP et al (2010) Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol 68:162–172. https://doi.org/10.1002/ana.22094
Zou WQ, Gambetti P, Xiao X, Yuan J, Langeveld J, Pirisinu L (2013) Prions in variably protease-sensitive prionopathy: an update. Pathogens 2:457–471. https://doi.org/10.3390/pathogens2030457
Grasbon-Frodl E, Lorenz H, Mann U, Nitsch RM, Windl O, Kretzschmar HA (2004) Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol 108:476–484. https://doi.org/10.1007/2Fs00401-004-0913-4
Nitrini R, Rosemberg S, Passos-Bueno MR, da Silva LS, Iughetti P, Papadopoulos M, Carrilho PM, Caramelli P et al (1997) Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann Neurol 42:138–146. https://doi.org/10.1002/ana.410420203
Zou RS, Fujioka H, Guo JP, Xiao X, Shimoji M, Kong C, Chen C, Tasnadi M et al (2011) Characterization of spontaneously generated prion-like conformers in cultured cells. Aging (Albany NY) 3:968–984. https://doi.org/10.18632/aging.100370
Diack AB, Ritchie DL, Peden AH, Brown D, Boyle A, Morabito L, Maclennan D, Burgoyne P et al (2014) Variably protease-sensitive prionopathy, a unique prion variant with inefficient transmission properties. Emerg Infect Dis 20:1969–1979. https://doi.org/10.3201/eid2012.140214
Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar-Calvo P, Kofskey D, Cali I et al (2014) Transmission characteristics of variably protease-sensitive prionopathy. Emerg Infect Dis 20:2006–2014. https://doi.org/10.3201/eid2012.140548
Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H (1987) Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 61:3688–3693
Zou WQ, Langeveld J, Xiao X, Chen S, McGeer PL, Yuan J, Payne MC, Kang HE et al (2010) PrP conformational transitions alter species preference of a PrP-specific antibody. J Biol Chem 285:13874–13884. https://doi.org/10.1074/jbc.M109.088831
Yuan J, Dong Z, Guo JP, McGeehan J, Xiao X, Wang J, Cali I, McGeer PL et al (2008) Accessibility of a critical prion protein region involved in strain recognition and its implications for the early detection of prions. Cell Mol Life Sci 65:631–643. https://doi.org/10.1007/s00018-007-7478-z
Kobayashi A, Mizukoshi K, Iwasaki Y, Miyata H, Yoshida Y, Kitamoto T (2011) Co-occurrence of types 1 and 2 PrP(res) in sporadic Creutzfeldt-Jakob disease MM1. Am J Pathol 178:1309–1315. https://doi.org/10.1016/j.ajpath.2010.11.069
Moudjou M, Treguer E, Rezaei H, Sabuncu E, Neuendorf E, Groschup MH, Grosclaude J, Laude H (2004) Glycan-controlled epitopes of prion protein include a major determinant of susceptibility to sheep scrapie. J Virol 78:9270–9276. https://doi.org/10.1128/JVI.78.17.9270-9276.2004
Yuan J, Zhan YA, Abskharon R, Xiao X, Martinez MC, Zhou X, Kneale G, Mikol J et al (2013) Recombinant human prion protein inhibits prion propagation in vitro. Sci Rep 3:2911. https://doi.org/10.1038/srep02911
Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D, Yuan J, Zheng M et al (2005) Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci 25:7944–7949. https://doi.org/10.1523/JNEUROSCI.2467-05.2005
Kong Q, Zheng M, Casalone C, Qing L, Huang S, Chakraborty B, Wang P, Chen F et al (2008) Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol 82:3697–3701. https://doi.org/10.1128/JVI.02561-07
Abskharon R, Dang J, Elfarash A, Wang Z, Shen P, Zou LS, Hassan S, Wang F et al (2017) Soluble polymorphic bank vole prion proteins induced by co-expression of quiescin sulfhydryl oxidase in E. coli and their aggregation behaviors. Microb Cell Factories 16:170–181. https://doi.org/10.1186/s12934-017-0782-x
Abskharon R, Wang F, Vander Stel KJ, Sinniah K, Ma J (2016) The role of the unusual threonine string in the conversion of prion protein. Sci Rep 6:38877. https://doi.org/10.1038/srep38877
Orrú CD, Yuan J, Appleby BS, Li B, Li Y, Winner D, Wang Z, Zhan YA et al (2017) Prion seeding activity and infectivity in skin samples from patients with sporadic Creutzfeldt-Jakob disease. Sci Transl Med 9:417–427. https://doi.org/10.1126/scitranslmed.aam7785
Castilla J, Saá P, Hetz C, Soto C (2005) In vitro generation of infectious scrapie prions. Cell 121:195–206. https://doi.org/10.1016/j.cell.2005.02.011
Orrú CD, Groveman BR, Raymond LD, Hughson AG, Nonno R, Zou W, Ghetti B, Gambetti P et al (2015) Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog 11:e1004983. https://doi.org/10.1371/journal.ppat.1004983
Yuan J, Xiao X, McGeehan J, Dong Z, Cali I, Fujioka H, Kong Q, Kneale G et al (2006) Insoluble aggregates and protease-resistant conformers of prion protein in uninfected human brains. J Biol Chem 281:34848–34858. https://doi.org/10.1074/jbc.M602238200
Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L et al (2000) NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 97:145–150. https://doi.org/10.1073/pnas.97.1.145
Stahl N, Borchelt DR, Hsiao K, Prusiner SB (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51:229–240. https://doi.org/10.1016/0092-8674(87)90150-4
Baldwin MA, Stahl N, Reinders LG, Gibson BW, Prusiner SB, Burlingame AL (1990) Permethylation and tandem mass spectrometry of oligosaccharides having free hexosamine: analysis of the glycoinositol phospholipid anchor glycan from the scrapie prion protein. Anal Biochem 191:174–182. https://doi.org/10.1016/0003-2697(90)90405-X
Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ et al (2000) Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A 97:10168–10172. https://doi.org/10.1073/pnas.97.18.10168
Concha-Marambio L, Pritzkow S, Moda F, Tagliavini F, Ironside JW, Schulz PE, Soto C (2016) Detection of prions in blood from patients with variant Creutzfeldt-Jakob disease. Sci Transl Med 8:370ra183. https://doi.org/10.1126/scitranslmed.aaf6188
Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, Castellani R, Cohen M et al (2008) A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 63:697–708. https://doi.org/10.1002/ana.21420
Wilham JM, Orru CD, Vascellari S, Hughson AG, Caughey B (2013) Quaking-induced conversion assays for the detection of diagnosis of prion diseases. In: Zou WQ, Gambetti P (eds) Prions and diseases: Vol 2, animals, humans, and the environment, 1st edn. Springer Science+Business Media, New York, pp. 223–232
Lewis V, Johanssen VA, Crouch PJ, Klug GM, Hooper NM, Collins SJ (2016) Prion protein “gamma-cleavage”: characterizing a novel endoproteolytic processing event. Cell Mol Life Sci 73:667–683. https://doi.org/10.1007/s00018-015-2022-z
Morales R, Abid K, Soto C (2007) The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta 1772:681–691. https://doi.org/10.1016/j.bbadis.2006.12.006
Peden AH, Sarode DP, Mulholland CR, Barria MA, Ritchie DL, Ironside JW, Head MW (2014) The prion protein protease sensitivity, stability and seeding activity in variably protease sensitive prionopathy brain tissue suggests molecular overlaps with sporadic Creutzfeldt-Jakob disease. Acta Neuropathol Commun 2(152):152. https://doi.org/10.1186/s40478-014-0152-4
Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB (1996) Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 10:1736–1750. https://doi.org/10.1101/gad.10.14.1736
Noble GP, Walsh DJ, Miller MB, Jackson WS, Supattapone S (2015) Requirements for mutant and wild-type prion protein misfolding in vitro. Biochemistry 54:1180–1187. https://doi.org/10.1021/bi501495j
Acknowledgements
We thank all donors for brain tissues, the families affected by CJD, and the physicians for support. We also thank the CJD Foundation for its invaluable help as well as Aaron Foutz and Xiaoqing Liu for providing technique support for RT-QuIC assay. This study was supported in part by the CJD Foundation and the National Institutes of Health (NIH) NS062787 and NS087588 to W.Q.Z., NS062787 and NS109532 to W.Q.Z. and Q.K., NS088604 to Q.K., the Centers for Disease Control and Prevention Contract UR8/CCU515004 to B.S.A., and the National Natural Science Foundation of China (NNSFC) (No. 81801207) to PS, as well as NNSFC (No. 81671186) to LC.
Author information
Authors and Affiliations
Contributions
ZW, WQZ, and LC conceived and designed the study. ZW performed sPMCA and RT-QuIC analyses. JY performed animal breeding and tissue collections and Western blotting of human and animal brain samples. PS, YL, JD, AA, and LX conducted Western blotting analysis of human brain samples. RA and JM performed design and generation of recombinant bank vole PrP. QK performed design and generation of humanized transgenic mice (TgMM). RBP and WQZ performed design and generation of humanized transgenic mice (TgVV and Tg180). MM, TK, and JL provided antibodies against PrP. ZW and WQZ wrote the first draft of the manuscript. All authors critically reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
The use of human brain tissues was authorized by the Institutional Review Board, which is recognized by the Office for Human Research Protections of the US Department of Health and Human Services. The Institutional Animal Use and Care Committee and the Institutional Biosafety Committee approved all of the animal experiments used in this study.
Informed Consent
The consents were received for each case through our National Prion Disease Pathology Surveillance Center.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised due to the additional of "Hyoung-gon Lee & Yong-Sun Kim" as the 13th and 14th authors.
Electronic Supplementary Material
ESM 1
(DOC 925 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, Z., Yuan, J., Shen, P. et al. In Vitro Seeding Activity of Glycoform-Deficient Prions from Variably Protease-Sensitive Prionopathy and Familial CJD Associated with PrPV180I Mutation. Mol Neurobiol 56, 5456–5469 (2019). https://doi.org/10.1007/s12035-018-1459-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1459-0