
Abstract
Glutamate is one of the major excitatory neurotransmitters of the CNS and is essential for numerous key neuronal functions. However, excess glutamate causes massive neuronal death and brain damage owing to excitotoxicity via the glutamate receptors. Metabotropic glutamate receptor 5 (mGluR5) is one of the glutamate receptors and represents a promising target for studying neuroprotective agents of potential application in neurodegenerative diseases. Pu-erh tea, a fermented tea, mainly produced in Yunnan province, China, has beneficial effects, including the accommodation of the CNS. In this study, pu-erh tea markedly decreased the transcription and translation of mGluR5 compared to those by black and green teas. Pu-erh tea also inhibited the expression of Homer, one of the synaptic scaffolding proteins binding to mGluR5. Pu-erh tea protected neural cells from necrosis via blocked Ca2+ influx and inhibited protein kinase C (PKC) activation induced by excess glutamate. Pu-erh tea relieved rat epilepsy induced by LiCl-pilocarpine in behavioural and physiological assays. Pu-erh tea also decreased the expression of mGluR5 in the hippocampus. These results show that the inhibition of mGluR5 plays a role in protecting neural cells from glutamate. The results also indicate that pu-erh tea contains biological compounds binding transcription factors and inhibiting the expression of mGluR5 and identify pu-erh tea as a novel natural neuroprotective agent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glutamate, the primary excitatory neurotransmitter in the brain, plays various physiological roles in cognition, memory and perception via the activation of multiple receptors [1]. An increase in the extracellular levels of glutamate leads to disorganised neuronal spike activity, impairing the processing of relevant information in the brain [2]. Glutamate binding metabotropic glutamate receptors (mGluRs) are one of G-protein-coupled receptors and exert its physiological roles through intracellular chemical-messenger signalling cascades [3]. Eight mGluRs (mGluR1–8) have been discovered and are grouped into three categories: group I (mGluR1 and 5), group II (mGluR2 and 3) and group III (mGluR4, 6, 7 and 8) according to sequence identity, pharmacological properties and downstream activation [4]. In general, mGluR5 combined with glutamate activates the G-protein Gq/11, and subsequently the activated G-protein activates phospholipase C (PLC) which hydrolyses membrane phosphoinositides to form inositol 1, 4, 5-triphosphate (IP3) and diacylglycerol (DAG). IP3 causes the release of intracellular calcium, which activates PKC [5]. The Homer family binds to the intracellular C-terminal tail of mGluR5 and also excites downstream effectors (Fig. 1) [6]. It is noteworthy that mGluR5 regulates various mechanisms implicated in neurogenesis and synaptic maintenance, and the abnormal regulation of mGluR5 has been implicated in the pathophysiology of many neurological and psychiatric disorders, including pain, epilepsy, schizophrenia, drug addiction and Alzheimer’s disease [7]. Thus, mGluR5 represents a pathophysiological hallmark for neuroprotective agents of potential application in neurodegenerative disorders [8].

Schematic of mGluR5 signalling pathway
Epilepsy is the second most common neurological disorder after stroke and a major burden for public health systems, afflicting worldwide approximately about 65 million people [9]. Although current antiepileptic drugs have achieved complete seizure control, they do not prevent or eliminate the substantial behavioural, cognitive and somatic comorbidities in many epilepsy patients [10]. New drugs with fewer side reactions and greater efficacy are urgently needed. Epilepsy researchers have recently embarked on a revitalise effort to move from targeting the control of symptoms to strategies of prevention and cure. One important direction for new therapeutics is identifying new molecular targets [11].
Tea is one of the most popular beverages in the world owing to its refreshing taste and attractive aroma. Its possible beneficial health functions have recently received much research attention. Pu-erh tea, a fermented tea, produced mainly in Yunnan province of China, is obtained by first drying crude green tea leaves (Camellia sinensis var. assamica (L.) O. Kuntze; Theaceae) and then subjecting them to secondary fermentation by microorganisms, such as Aspergillus sp., resulting in a unique type of tea [12]. Several studies have reported that pu-erh tea has shown a wide range of biological effects, such as inhibiting nitric oxide production [13], promoting weight loss [14], reducing blood lipids [15], exerting antimicrobial and antitumor activities [16, 17] and scavenging free radicals [18]. We recently also found that pu-erh tea may reduce the incidence of epilepsy and amyotrophic lateral sclerosis [19, 20]. These results suggest that the potential efficacy of pu-erh tea in the regulation of the nervous system.
In this study, we discovered that pu-erh tea downregulated the transcription and translation of mGluR5 by Affymetrix Expression microarrays experiments and Western blot. Pu-erh tea also protected SH-SY5Y cells against apoptosis induced by L-glutamate (L-Glu) and alleviated epilepsy behaviour by inhibiting the expression of mGluR5. The results identified mGluR5 as a target for the treatment of neurological and psychiatric disorders and suggested that pu-erh tea might be a potential natural source of protection against neurodegenerative diseases associated with mGluR5. Accordingly, the aim of this study was to identify the protective mechanisms of pu-erh tea against injury to neuronal cells in vitro and in vivo.
Materials and Methods
Materials
Adult male Wistar rats (4–5 weeks old) were obtained from the Animal Resource Center of Jilin University and housed at 22 ± 2 °C in a controlled environment with a 12-h light and 12-h dark cycle. All rats were treated humanely and in compliance with the recommendations of the animal care committee of Jilin University. We used the minimum number of mice required to draw a conclusion and tried to minimise their suffering as much as possible.
3-(4, 5- Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), and dimethyl sulfoxide (DMSO), L-Glu were purchased from Sigma. Cell culture media, Dulbecco’s modified Eagle’s medium (DMEM), heat-inactivated foetal bovine serum (FBS) and trypsin were supplied by Gibco. Antibodies were purchased from Santa Cruz (CA93446, USA). A Luciferase Reporter Gene Assay Kit was purchased from Beyotime. (RS)-2-Chloro-5-hydroxyphenylglycine (CHPG), a selective mGluR5 agonist and 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a selective mGluR5 antagonist, were purchased from Selleckchem.
Cell Culture
The SH-SY5Y, HEK293T and 3 T3 cell lines were cultured in DMEM, and the Jurkat cell line was cultured in RPMI1640 medium supplemented with 10 % FBS, 100 units/mL penicillin and 100 units/mL streptomycin (Invitrogen).
Cytotoxicity Assay
In the cytotoxicity assay, cells were seeded at a density of 1 × 104 cells/well in 96-well plates and incubated for 24 h. Cells were treated with pu-erh tea at various concentrations for 24 h. MTT (5 mg/mL, 20 μL) was added to the medium 24 h later. After incubation at 37 °C for 4 h, the supernatant was aspirated and 100 μL of DMSO was added to each well, followed by vibration for 10 min. The absorbance in the experimental wells was measured at 570 nm with a microplate reader.
Affymetrix Microarrays
RNA was extracted with TRI reagent (Sigma) and DNAse I (Invitrogen) from Jurkat in triplicate with or without pu-erh tea treatment for 12 h. RNA quality and concentration were assessed with an Agilent 2100 Bioanalyser and Nanodrop ND-1000. Data from Affymetrix 430 2.0 microarray chips (SCIBLU, Affymetrix) was analysed with Arraystar 3 software (DNA STAR Inc.), which was quantile-normalised and processed by the RMA (Affymetrix) algorithm.
Luciferase Assay
Promoter regions were amplified by PCR amplification and subcloned into the KpnI/HindIII sites of the luciferase reporter vector pGL3-Basic (Promega) upstream of the firefly luciferase gene (Fig. 2). SH-SY5Y and HEK293T cells were transfected with mGluR5 promoter-pGL3 recombinant plasmid by electroporation according to the manufacturer’s instructions. A monolayer of 80 % confluent cells was trypsinised, washed with appropriate media and washed again in ice-cold PBS buffer. Cell suspension for the electroporation was first prepared in 400 μL ice-cold PBS buffer (2.5 × 106 cells/mL) in a 0.4-cm electric rotary cup, followed by adding 10 μg plasmid. The electroporation conditions were adopted from previous work, as follows: U = 225 V, R = ∞, Ω = 1050 μF. After electroporation, cells were seeded in 12-well plates at a density of 4 × 104 cells per well. After 12 h, transfected cells were incubated with pu-erh, black and green teas. Cells were then harvested and firefly luciferase activity was determined using a Sirius luminometer (Berthold Detection System). Experiments were performed in triplicates and repeated at least three times independently.

Construction strategy and structure of the recombinant plasmid mGluR5 promoter-pGL3
Western Blot
Cells were prepared in ice-cold lysis buffer (100 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 10 mM MgCl2, pH 7.2) containing 1 % Triton X-100, phosphatase and protease inhibitor cocktail (Dinggguo, China) for 10 min. Cell lysates were centrifuged at 12,000g for 10 min at 4 °C, and protein concentrations of supernatants were determined with BCA assay. Lysates were added 5 × SDS containing 100 mM DTT. Equal amounts of protein were loaded onto and separated on 12 % SDS-PAGE gels and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were incubated with primary antibodies overnight, washed three times in washing buffer (0.1 % Tween-20 in PBS), incubated with HRP-coupled secondary antibodies for 1 h, washed three times in washing buffer and developed using chemiluminescent HRP detection substrate. When proteins were extracted from hippocampus tissue, hippocampus tissues were placed in lysis buffer (as above) and homogenised for 1 min. The homogenate was then centrifuged at 12,000g for 10 min at 4 °C. The final supernatant was collected and processed as above.
Nuclear Staining with Hoechst 33342
SH-SY5Y cells were seeded in 6-well plates (1.5 × 105 cells/well) for 24 h. Cells in a 6-well plate were incubated with pu-erh, black or green teas for 12 h before treatment with L-Glu for 24 h. Cells were washed with PBS and stained with 500 μL of Hoechst 33342 solution for 15 min at room temperature in the dark. The nuclear morphology of the cells was examined under a fluorescent microscope.
Flow Cytometry Analysis of Apoptosis in SH-SY5Y
SH-SY5Y cells were seeded in 6-well plates (1.5 × 105 cells/well) for 24 h. Cells in each 6-well plate were incubated with pu-erh, black or green teas for 12 h before treatment with L-Glu for 24 h. Cells treated with medium alone were used negative control. Apoptosis of cells was evaluated by measurement of the exposure of phosphatidylserine on the cell membranes using Apoptosis Detection Kits. Cell pellets were resuspended in a staining solution containing propidium iodide (PI) for 5 min and Annexin V-FITC for 15 min at room temperature in the dark. The cells were assessed by fluorescence-activated cell sorting (FACS) using the Cell Quest software (BD, Pharmingen).
Calcium Imaging
SH-SY5Y cells were seeded in 6-well plates (1.5 × 105 cells/well) for 24 h. Cells in a 6-well plate were incubated with pu-erh, black, or green teas for 12 h before treatment with L-Glu for 24 h. Cells treated with medium alone were used as negative control. Cells were loaded with 2 μM Fluo-3 AM at 37 °C for 1 h. Cells were suspended in PBS after being washed three times and analysed by a flow cytometry device equipped with the Cell Quest software at 488 nm.
Epileptic Rat Model
Status epilepticus (SE) was induced in adult Wistar rats by using Licl-pilocarpin [21]. Rats were randomly divided into 6 groups with 6 rats in each group. Three groups were administered intragastricly with pu-erh tea at a dose of 0.5 g/Kg/day, the other three groups were treated with saline. After 20 days, lithium chloride was intraperitoneally injected into the four groups rats that two of them were dministered intragastricly with pu-erh tea, the other two were treated with saline at a 127 mg/kg dosage. After 18 h, the rats were treated with a 40 mg/kg dose of pilocarpine, administered intraperitoneally. Two of the groups that one was treated with pu-erh tea and Licl-pilocarpin, the other was treated with saline and Licl-pilocarpin were treated with 4 mg/kg diazepam, before pilocaroine administered intragastricly. Saline was administered to the control group at the same dosage.
Behavioural Study
Rats were video monitored from day 0 to day 15 post status epilepsy (SE) and were scored by an individual blind to the experimental design. Behavioural seizure was recorded according to a modification of the classification of Racine [22]: 0, exploring; 1, immobility; 2, rigid posture; 3, head nodding; 4, bilateral forelimb clonus and falling; 5, continued clonus and falling; and 6, generalised tonus. Three behavioural patterns of SE could be recognised: I, initial (class 1–2), M, middle (class 3) and C, critical (class 4–6). Seizure number and type were recorded within 1 h of the seizure in day 0. Then, seizure number and type were recorded 30 min every 24 h from day 1 to day 15.
Measurements of IP3 and DAG
The measurement of the contents of IP3 and DAG was conducted using IP3 and DAG ELISA kits (RD, USA). Hippocampus tissues were placed in ice-cold PBS and homogenised for 1 min. The homogenate was then centrifuged at 12,000g for 10 min at 4 °C. The final supernatant was collected.
Haematoxylin and Eosin (HE) Staining
Brains were prepared from control and post-SE rats. Animals were anaesthetised using 20 % urethane at a dose of 0.5 mL/kg. Once deeply anesthetised, animals underwent heart perfusion with ice-cold modified PBS followed by 4 % poly-formaldehyde. The brain was removed and dehydrated in 20 and 30 % sucrose. The brains were then embedded in optimal cutting temperature compound (OCT). Six-micrometre-thick slices were made with a tissue slicer and stained in HE solutions to examine morphological changes.
Immunofluorescence
To detect the expression of mGluR5 in rat brains, 6-μm paraffin sections were subjected to antigen retrieval and blocking of endogenous peroxidase activity, followed by incubation with mGluR5 monoclonal antibody (diluted at 1:200) overnight at 4 °C. The sections were incubated with FITC-conjugated anti-mouse IgG (1:250) for 2 h at room temperature. All images were acquired with a fluorescence microscope.
Statistical Analysis
All values were expressed as mean ± S.D. A One-way analysis of variance (ANOVA) was used to detect statistical significance followed by post hoc multiple comparisons (Dunn’s test) by GraphPad Prism 6.0. A value of P < 0.05 was considered to be significant [23, 24].
Results
Pu-erh Tea Down-Regulated the Transcription of mGluR5
To investigate the cytotoxicity of pu-erh tea, cell viability assays were performed. Our finding demonstrated that pu-erh did not inhibit the proliferation of Jurkat or SH-SY5Y at concentrations lower than 250 μg/mL, whereas it exerted a marked inhibitory effect on the proliferation of Jurkat and SH-SY5Y at concentrations greater than 0.5 mg/mL. No inhibition was observed in 3 T3 (Fig. 3).

a Jurkat cell viability after incubation with various concentration of pu-erh tea for 24 h. b 3 T3 cell viability after incubation with various concentration of pu-erh tea for 24 h. c SH-SY5Y cell viability after incubation with various concentration of pu-erh tea for 24 h. Data were analysed using the GraphPad Prism software (n = 6)
The messenger RNA (mRNA) expression differences between the Jurkat cells treated and untreated with pu-erh tea were detected with Affymetrix expression microarrays. The results showed that pu-erh tea could downregulate mGluR5, one of the G protein-coupled receptors implicated in the nervous system (Tables 1, 2).
Pu-erh Tea Inhibited the Activity of mGluR5 Promoter
To assess the role of pu-erh tea in transcriptional regulation and cell-specific expression of mGluR5, we constructed mGluR5 promoter-pGL3 recombinant plasmid The PCR products of the mGluR5 promoter were examined by 1 % agarose gel electrophoresis (Fig. 4a). PCR product is 1167-bp specific fragment consistent with the mGluR5 promoter. The promoter region was then subcloned into the KpnI/HindIII sites of pGL3-Basic. Double enzyme digestion reaction yielded a clear obvious 1167-bp fragment, showing that a recombinant plasmid, mGluR5 promoter-pGL3 had been constructed (Fig. 4b).

a Agarose gel electrophoresis of mGluR5 promoter. M: 2000 DNA marker; 1:mGluR5 promoter PCR product. b Agarose gel electrophoresis of mGluR5 promoter-pGL3 double restriction enzyme digestion. M: 2000 bp; 1: mGluR5 promoter-pGL3 double restriction enzyme digestion; 2: mGluR5 promoter-pGL3 KpnI restriction enzyme digestion; 3: mGluR5 promoter-pGL3 HindIII restriction enzyme digestion. c Luciferase activity after treatment with various concentrations of pu-erh tea for 12 h. d Luciferase activity after treatment with 62.5 μg/mL pu-erh tea for various times. e Luciferase activity after treatment with 62.5 μg/mL pu-erh, black, green teas for various times. f Luciferase activity after treatment with 62.5 μg/mL pu-erh, black, green teas for 6 h. Data were analysed using the GraphPad Prism software (***p < 0.001 vs. vehicle control, n = 6)
Thus, the following detection on the activity of mGluR5 promoter to drive the expression of luciferase was assessed in HEK293T and SH-SY5Y cells. Luciferase activity driven by mGluR5 promoter was decreased significantly at a concentration of 62.5 μg/mL (Fig. 4c). The transfected cells were then incubated at this concentration for various times, with the result that at 6 h luciferase activity was decreased significantly (Fig. 4d). Compared to black and green teas, treatment with pu-erh tea for 6 h had a greater suppressive effect on transcriptional regulation of mGluR5 in both HEK293T and SH-SY5Y cells (Fig. 4e, f).
Pu-erh Tea Inhibited the Expression of mGluR5
To further validate the inhibition of the expression of the mGluR5, several concentrations of pu-erh tea, 50 μM CHPG (a selective mGluR5 agonist) and 50 μM MPEP (a selective mGluR5 antagonist) were added to SH-SY5Y cells and followed by incubation for 12 h. There were no effects on the growth of SH-SY5Y at the concentration of 62.5 μg/mL (Fig. 3c). The expression of mGluR5 was markedly reduced compared to the control when cells were treated with 62.5 μg/mL of pu-erh tea for 12 h compared to the control (Fig. 5a, b). When SH-SY5Y cells were treated with 62.5 μg/mL of pu-erh tea for various times, pu-erh tea markedly inhibited the expression of mGluR5 compared to the control (Fig. 5d, e). Compared to black and green teas, treatment with pu-erh tea for 12 h significantly inhibited the expression of mGluR5 (Fig. 5g, h).

The expression of mGluR5 and Homer were detected by Western blot and analysed by ImageJ. a–c Cells were treated with various concentrations of pu-erh tea for 12 h. d–f Cells were treated with 62.5 μg/mL pu-erh tea for 3, 6, 9, 12 h. g–i Cells were treated with 62.5 μg/mL pu-erh, black, or green teas for 12 h. Data were analysed using GraphPad Prism software (***p < 0.001 vs. vehicle control, **p < 0.01 vs. vehicle control, n = 3)
The Homer family of proteins is localised predominantly at the postsynaptic density (PSD) in mammalian neurons and binds a specific proline-rich sequence on group I mGluRs. We assayed the expression of Homer by Western blot, and found that Homer was also significantly inhibited by pu-erh tea at dose-dependently and time-dependently (Fig. 5).
Effects of Pu-erh Tea on Neuronal Injury
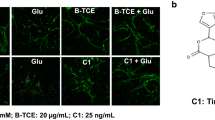
Increase in extracellular levels of glutamate leads to activation of mGluR5 and a strong excitatory neurotoxicity, resulting in neuronal swelling and necrosis. To confirm that pu-erh tea protects cells from nerve injury, SH-SY5Y cells were incubated with pu-erh tea before treatment with L-glutamate (L-Glu). The nuclear morphology of SH-SY5Y cells was examined after staining with Hoechst 33342. Compared to black and green teas, 12-h treatment with 62.5 μg/mL of pu-erh tea reduced the number of hallmarks of apoptosis induced by 160 mM L-Glu, whereas the mGluR5 agonist CHPG increased and the mGluR5 antagonist MPEP decreased the number of apoptosis (Fig. 6a).


a Image of Hoechst 33342-stained cells. b Flow cytometry analysis of apoptosis. c Flow cytometry analysis of Ca2+ influx. d Western blot analysis of PKCα activity. e Quantification of protein level from three independent experiments using ImageJ. Cells were pretreated with 62.5 μg/mL pu-erh, black, or green teas for 12 h before treatment with 160 mM L-Glu. Data were analysed using GraphPad Prism software (***p < 0.001, n = 3)
To further investigate whether pu-erh tea protects cells against apoptosis, the cells were analysed by flow cytometry after staining with Annexin V-FITC and PI. The percentage of surviving cells increased from 28.36 to 71.99 % after 12-h of treatment with pu-erh tea, whereas the mGluR5 agonist CHPG reduced the cell surviv to cells to 6.33 %. Treatment with black or green tea also increased the percentage of surviving cells (Fig. 6b).
Involvement of Ca2+ in Prevention of Nerve Injury by Pu-erh Tea
The activation of mGluR5 could increase Ca2+ influx, and excitotoxic Ca2+ elevation is an early and possibly reversible pathological event that may lead to neuronal injury. To investigate the role of pu-erh tea in Ca2+ release in nerve injury induced by L-Glu, SH-SY5Y cells were incubated with pu-erh tea before treated with L-Glu, using an intracellular Ca2+ chelator (Fluo-3 AM). Pretreatment with pu-erh tea significantly inhibited calcium influx compared to that black tea or green teas. However, mGluR5 CHPG activated the calcium influx (Fig. 6c).
Pu-erh Tea Inhibited PKCα Activation by L-Glu
In addition to Ca2+ release, PKCα is involved in mGluR5 signalling. After nerve injury by L-Glu, we observed that PKCα activity was increased. Moreover, pretreatment with the mGluR5 antagonist MPEP, inhibited PKCα activity. Pu-erh tea significantly reduced PKCα activity compared to black or green teas. The content of PKCα in each group did not change. (Fig. 6d, e).
Effect of Pu-erh Tea on Behaviour
The Wistar rats that injected intraperitoneally with Licl-pilocarpine only had obvious epilepsy behaviour, while the groups treated with pu-erh tea onLicl-pilocarpine induced SE significantly attenuated the maximal seizure classes and the predominant behavioural seizure patterns in SE mice compared with the control (Table 3, n = 6).
Pu-erh Tea Prevents Epilepsy by Inhibiting mGluR5
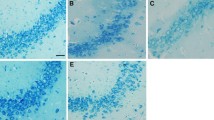
IP3 and DAG, as second messengers, play important roles on in intracellular messenger transduction. These two substances stimulate neurons and can cause neuronal damage if present in excess. We accordingly assayed the contents of IP3 and DAG in the hippocampus by ELISA to confirm the effect of pu-erh tea on prevention of epilepsy. The results showed that IP3 and DAG were increased in epilepsy and decreased under intragastric administration of pu-erh tea (Fig. 7a, b). Histological images using HE staining showed that after epilepsy rat hippocampus showed injury, whereas before intragastric administration of pu-erh tea or diazepam, rat hippocampus showed no changes compared to the control (Fig. 7c).


a Content of IP3 in the hippocampus. b Content of DAG in the hippocampus. c HE staining of hippocampus. d Immunofluorescence of hippocampus with Hochest 33,342 (blue) and anti-mGluR5 (green). e Quantification of relative fluorescence intensity of mGluR5/Hochest 33342 by ImageJ. f Western blot analysis the expression of mGluR5 in the hippocampus. g Quantification of protein levels from three independent experiments by ImageJ. Data were analysed using GraphPad Prism software (***p < 0.001, **p < 0.01, *p < 0.05, n = 3)
To determine whether mGluR5 is implicated in epilepsy, we used immunofluorescence and Western blot assays. In the immunofluorescence assays, the expression of mGluR5 was significantly upregulated in epileptic rats only administrated intraperitoneally with Licl-pilocarpin compared to others, whereas the rats before intragastric administration of pu-erh tea or diazepam showed reduced expression of mGluR5 compared to that in epilepsy (Fig. 7d, e). In Western blot, we also found the similar result (Fig. 7f, g).
Discussion
In the present study, we observed that pu-erh tea decreased the mGluR5 promoter activity and downregulated the expression of the mGluR5 at dose-dependently and time-dependently. We also discovered that pu-erh tea protected SH-SY5Y cells against apoptosis induced by L-glu via blocking Ca2+ influx and inhibiting the PKCα activation. Pu-er tea had the role of alleviating epilepsy and reducing the expression of mGluR5 in epilepsy rats.
Human epidemiological and animal research experiments suggest that the pharmacological benefits of tea drinking may help protect the brain. And tea drinking has been shown to exert neuroprotective activities in a wide array of cellular and animal models of neurological disorders [25]. The Affymetrix expression microarrays showed that 0.25 mg/mL pu-erh tea decreased the mGluR5. Our results that pu-erh tea showed no toxicity to tumour cells and normal cells at low concentrations (≤ 0.25 mg/mL) was in accordance with the report that pu-erh tea showed no signs of toxicity and no deaths in SD rats at dosages ≤10 g/kg [26]. These results indicated that the reduction of mGluR5 mRNA and protein by pu-erh tea was not the result of cell death at low concentration (≤ 0.25 mg/mL).
Excessive mGluR5 activation has already been alluded to as a potential contributing factor in synaptic disorders, and a number of studies are currently testing the therapeutic potential of drugs that modify mGluR5 [27]. The modification of particular mGluR5 has been implicated in synaptic diseases and psychiatric disorders [28]. Despite progress in drug treatment of schizophrenia, cognitive and other negative symptoms remain a major issue of the disease, often representing residual symptoms of resistant schizophrenia and being worsened by the side effects of antipsychotics effects [29].
The mGluR5 promoter we cloned could regulate the expression of luciferase (Fig. 4). It was consistent with the result that the alternative 5-splicing and usage of multiple promoters may contribute regulatory mechanisms for tissue- and context-specific expression of the mGluR5 gene [30]. The results of luciferase assays (Fig. 4) and Western blot (Fig. 5) suggested that pu-erh tea contains active compounds that could inhibit the transcription and the translation of mGluR5 compared to black tea and green tea. It may be during the fermentation, the types, activity and content of the components in pu-erh tea are different from those of black and green teas [31]. Owing to the complexity of the composition of pu-erh tea, the active ingredients that protect neural cells via reducing the expression of mGluR5 await further study. The mGluR5 agonist CHPG increased the expression of mGluR5 and an mGluR5 antagonist MPEP reduced the expression of mGluR5 (Fig. 5). These results suggested that pu-erh tea was a candidate as mGluR5 antagonist.
Importantly, the activity of mGluR5 is tightly regulated by its association with scaffolding protein, Homer [32]. In Western blot, we found that pu-erh tea could downregulate the expression of Homer significantly compared to black or green teas (Fig. 5). Homer proteins share a commom Ena/Vasp homology protein (EVH1) domain at the N-terminus that binds to the intracellular C-terminal tail of mGluR5 and localises mGluR5 to the postsynaptic density (PSD) [6, 33, 34]. In addition, it has been shown that disruption of mGluR5-Homer interactions selectively blocks mGluR5 activation [35]. These results indicated that the inhibition of mGluR5 by pu-erh tea disrupted mGluR5–Homer scaffolds, resulting in decreasing Homer.
A recent study suggests that enhanced mGluR5-Homer binding leads to a reduction in mGluR5-induced cytosolic Ca2+ increase [36]. However, in our study, both L-Glu and mGluR5 agonist CHPG induced nerve cell necrosis and increased intracellular Ca2+ response. These findings are in agreement with the report that Homer proteins bind IP3 receptor [37]. IP3 induces calcium release from intracellular stores, potentially leading to cell death [38]. Pretreatment with pu-erh tea or an mGluR5 antagonist MPEP could protect cells from necrosis and decrease intracellular Ca2+ response. These results suggested that pu-erh tea had the role of mGluR5 antagonist that prevented Homer binding IP3 receptor, which contributed to blocking the glutamate stimulation and inhibiting the release of intracellular Ca2+ in response to neuronal injury.
The mGluR5-induced Ca2+ response could also be negatively regulated by Homer 1a under some condition [39]. Given that group I mGluRs are positively coupled to PLC, leading to PKC activation and phosphorylation of serine 839 mediated by PKC activation is involved in the regulation of mGlu5 receptor-mediated Ca2+ influx [40, 41]. Therefore, we wondered whether the ability of pu-erh tea to attenuate the release of intracellular was dependent mGluR5-PKC. We tested the activation of PKCα and found that both L-Glu and the mGluR5 agonist CHPG stimulated PKC activation, whereas pu-erh tea or an mGluR5 antagonist MPEP inhibited PKCα activation. Thus, we theorise that pu-erh tea protected neuronal injury by inhibiting PKC activation which resulted from inhibiting the release of intracellular Ca2+. However, PKC isoenzymes have been classified into two subfamilies: the conventional subtypes (cPKC: α, βI, βII, γ) are Ca2+ and DAG-dependent and novel subtypes (nPKC: δ, γ, η, θ,) are DAG-dependent, but Ca2+-independent [42]. Group I mGluRs coupled to Gαq/11 proteins stimulate the activation of PLC, resulting in DAG formation. Pu-erh tea decreased the contents of IP3 and DAG in the hippocampus (Fig. 7a, b). Accordingly, whether pu-erh tea influences the novel subtypes of PKC awaits further investigation.
mGluR5 is well known to play important roles in neuronal function and synaptic plasticity, and defects in mGluR5 signals are thought to cause a variety of neurological disorders. Activation of mGluR5 increases epileptiform activity and an mGluR5 antagonist MPEP inhibits convulsive and non-convulsive primary generalised seizures [43]. In this study, we constructed an epilepsy model by using Licl-pilocarpin [21]. Pilocarpine activates the G-protein which mediates the receptor stimulation to PLC. PLC hydrolyses PI into IP3 and DAG. IP3 and DAG cause neuronal stimulation and when in excess, may contribute to neuronal injury [44]. In general, mGluR5 combined with glutamate activates the G-protein Gq/11, and subsequently, the activated G-protein activates PLC which hydrolyses membrane phosphoinositides to form IP3 and DAG [5]. We found that pu-erh tea could decrease the severity of seizure behaviours and the content of IP3 and DAG. Immunofluorescence and Western blot both showed that pu-erh tea reduced the expression of mGluR5 that was increased in epilepsy rats. These results coincide with that the expression of mGluR5 is higher in the hippocampus of epileptic patients, than in those of a control group [45]. The results indicated that pu-erh tea could decrease the content of IP3 and DAG through reducing the expression of mGluR5. Taken together, these results indicate that pu-erh tea played an important role in protecting neural cells from injury induced by glutamate and antiepileptic via inhibiting the activity of mGluR5 promoter and the expression of mGluR5 (Fig. 8).

The mechanism of pu-erh tea protects neural cell from injury stimulated by glutamate
Conclusion
Dysregulation of mGluR5 is implicated in multiple brain disorders. Accordingly, many selective mGluR5 antagonists have been developed for clinical therapy. However, these drugs have produced unacceptable side effects in clinical trials. The findings of this study suggested that pu-erh tea can inhibit mGluR5 activation stimulated by glutamate via inhibiting the activity of mGluR5 promoter and the expression of mGluR5, which blocks Ca2+ influx and suppresses the activity of PKC α to protect nerve cell from necrosis. Pu-erh tea also relieves rat epilepsy and reduce the contents of IP3 and DAG in epilepsy rats. These results suggest that pu-erh tea is a potential neuroprotective agent acting via regulation of mGluR5. However, the specific compounds in pu-erh tea that function in neural protection await further study.
References
Robbins TW, Murphy ER (2006) Behavioural pharmacology: 40+ years of progress, with a focus on glutamate receptors and cognition. Trends Pharmacol Sci 27(3):141–148. doi:10.1016/j.tips.2006.01.009
Moghaddam B, Javitt D (2012) From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37(1):4–15. doi:10.1038/npp.2011.181
Power EM, English NA, Empson RM (2016) Are type 1 metabotropic glutamate receptors a viable therapeutic target for the treatment of cerebellar ataxia? J Physiol. doi:10.1113/JP271153
Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322. doi:10.1146/annurev.pharmtox.011008.145533
Matosin N, Newell KA (2013) Metabotropic glutamate receptor 5 in the pathology and treatment of schizophrenia. Neurosci Biobehav Rev 37(3):256–268. doi:10.1016/j.neubiorev.2012.12.005
Shiraishi-Yamaguchi Y, Furuichi T (2007) The Homer family proteins. Genome Biol 8(2):206. doi:10.1186/gb-2007-8-2-206
Krystal JH et al. (2010) Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs 24(8):669–693. doi:10.2165/11533230-000000000-00000
Bruno V et al. (2000) Selective blockade of metabotropic glutamate receptor subtype 5 is neuroprotective. Neuropharmacology 39(12):2223–2230
Schmidt D, Schachter SC (2014) Drug treatment of epilepsy in adults. BMJ 348:g254. doi:10.1136/bmj.g254
Loscher W, Schmidt D (2011) Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 52(4):657–678. doi:10.1111/j.1528-1167.2011.03024.x
Loscher W (2002) Current status and future directions in the pharmacotherapy of epilepsy. Trends Pharmacol Sci 23(3):113–118. doi:10.1016/S0165-6147(00)01974-X
Jeng KC et al. (2007) Effect of microbial fermentation on content of statin, GABA, and polyphenols in Pu-erh tea. J Agric Food Chem 55(21):8787–8792. doi:10.1021/jf071629p
Xu Y et al. (2011) Variations of antioxidant properties and NO scavenging abilities during fermentation of tea. Int J Mol Sci 12(7):4574–4590. doi:10.3390/ijms12074574
Ding Y et al. (2015) Pu-erh tea down-regulates sterol regulatory element-binding protein and stearyol-CoA desaturase to reduce fat storage in Caenorhaditis elegans. PLoS One 10(2):e0113815. doi:10.1371/journal.pone.0113815
Lin JK, Lin-Shiau SY (2006) Mechanisms of hypolipidemic and anti-obesity effects of tea and tea polyphenols. Mol Nutr Food Res 50(2):211–217. doi:10.1002/mnfr.200500138
Su Y et al. (2012) Antibacterial property and mechanism of a novel Pu-erh tea nanofibrous membrane. Appl Microbiol Biotechnol 93(4):1663–1671. doi:10.1007/s00253-011-3501-2
Zhao H et al. (2011) Changes of constituents and activity to apoptosis and cell cycle during fermentation of tea. Int J Mol Sci 12(3):1862–1875. doi:10.3390/ijms12031862
Xu Y et al. (2012) Pu-erh tea reduces nitric oxide levels in rats by inhibiting inducible nitric oxide synthase expression through toll-like receptor 4. Int J Mol Sci 13(6):7174–7185. doi:10.3390/ijms13067174
Hou CW (2011) Pu-erh tea and GABA attenuates oxidative stress in kainic acid-induced status epilepticus. J Biomed Sci 18:75. doi:10.1186/1423-0127-18-75
Yu Y et al. (2014) Pu-erh tea extract induces the degradation of FET family proteins involved in the pathogenesis of amyotrophic lateral sclerosis. Biomed Res Int 2014:254680. doi:10.1155/2014/254680
Grabenstatter HL et al. (2014) Effect of spontaneous seizures on GABAA receptor alpha4 subunit expression in an animal model of temporal lobe epilepsy. Epilepsia 55(11):1826–1833. doi:10.1111/epi.12771
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32(3):281–294
Luo P et al. (2014) Postsynaptic scaffold protein Homer 1a protects against traumatic brain injury via regulating group I metabotropic glutamate receptors. Cell Death Dis 5:e1174. doi:10.1038/cddis.2014.116
Zhai MZ et al. (2016) Dexmedetomidine dose-dependently attenuates Ropivacaine-induced seizures and negative emotions via inhibiting phosphorylation of amygdala extracellular signal-regulated kinase in mice. Mol Neurobiol 53(4):2636–2646. doi:10.1007/s12035-015-9276-1
Mandel SA et al. (2008) Targeting multiple neurodegenerative diseases etiologies with multimodal-acting green tea catechins. J Nutr 138(8):1578S–1583S
Wang D et al. (2011) Acute and subchronic oral toxicities of Pu-erh black tea extract in Sprague-Dawley rats. J Ethnopharmacol 134(1):156–164. doi:10.1016/j.jep.2010.11.068
Dolen G et al. (2010) Mechanism-based approaches to treating fragile X. Pharmacol Ther 127(1):78–93. doi:10.1016/j.pharmthera.2010.02.008
Piers TM et al. (2012) Translational concepts of mGluR5 in synaptic diseases of the brain. Front Pharmacol 3:199. doi:10.3389/fphar.2012.00199
de Bartolomeis A et al. (2012) Targeting glutamate system for novel antipsychotic approaches: relevance for residual psychotic symptoms and treatment resistant schizophrenia. Eur J Pharmacol 682(1–3):1–11. doi:10.1016/j.ejphar.2012.02.033
Corti C et al. (2003) Gene structure of the human metabotropic glutamate receptor 5 and functional analysis of its multiple promoters in neuroblastoma and astroglioma cells. J Biol Chem 278(35):33105–33119. doi:10.1074/jbc.M212380200
Wang D et al. (2010) Comparative safety evaluation of Chinese Pu-erh green tea extract and Pu-erh black tea extract in Wistar rats. J Agric Food Chem 58(2):1350–1358. doi:10.1021/jf902171h
Ronesi JA et al. (2012) Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci 15(3):431–440 . doi:10.1038/nn.3033S1
Hayashi MK, Ames HM, Hayashi Y (2006) Tetrameric hub structure of postsynaptic scaffolding protein homer. J Neurosci 26(33):8492–8501. doi:10.1523/JNEUROSCI.2731-06.2006
Hayashi MK et al. (2009) The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell 137(1):159–171. doi:10.1016/j.cell.2009.01.050
Ronesi JA, Huber KM (2008) Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J Neurosci 28(2):543–547. doi:10.1523/JNEUROSCI.5019-07.2008
Hu JH et al. (2012) Preso1 dynamically regulates group I metabotropic glutamate receptors. Nat Neurosci 15(6):836–844. doi:10.1038/nn.3103
Tu JC et al. (1998) Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21(4):717–726
Power JM, Sah P (2007) Distribution of IP3-mediated calcium responses and their role in nuclear signalling in rat basolateral amygdala neurons. J Physiol 580(Pt.3):835–857. doi:10.1113/jphysiol.2006.125062
Tappe A et al. (2006) Synaptic scaffolding protein Homer1a protects against chronic inflammatory pain. Nat Med 12(6):677–681. doi:10.1038/nm1406
Nakanishi S (1994) Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron 13(5):1031–1037
Kim CH et al. (2005) Protein kinase C phosphorylation of the metabotropic glutamate receptor mGluR5 on serine 839 regulates Ca2+ oscillations. J Biol Chem 280(27):25409–25415. doi:10.1074/jbc.M502644200
Parker PJ, Murray-Rust J (2004) PKC at a glance. J Cell Sci 117(Pt 2):131–132. doi:10.1242/jcs.00982
Chapman AG et al. (2000) Anticonvulsant activity of two metabotropic glutamate group I antagonists selective for the mGlu5 receptor: 2-methyl-6-(phenylethynyl)-pyridine (MPEP), and (E)-6-methyl-2-styryl-pyridine (SIB 1893. Neuropharmacology 39(9):1567–1574
Savolainen KM, Hirvonen MR (1992) Second messengers in cholinergic-induced convulsions and neuronal injury. Toxicol Lett 64-65 :437–445Spec No
Notenboom RG et al. (2006) Up-regulation of hippocampal metabotropic glutamate receptor 5 in temporal lobe epilepsy patients. Brain 129(Pt 1):96–107. doi:10.1093/brain/awh673
Acknowledgments
We appreciate the Pu-erh Tea Research Institute for pu-erh tea. This study was supported by grants from the National Nature Science Foundation of China (no. 51473060), Science and Technology Department of Jilin Province (no. 20130206010YY) and Pu-erh Tea Research Institute (no. 2013220101001352).
Author Contributions
The experiments were designed by Chunjie Li, Jun Sheng and Wei Shi and performed in the laboratory of Wei Shi. Pu-erh tea was supplied by Jun Sheng. The date was collected and analysed by Chunjie Li and Hang Zhao. Flow cytometry experiments were performed by Chunjie Li and Shaomeng Chai. Animal experiments were performed by Chunjie Li, Yongzhi Ju and Tian Li. The manuscript was written by Chunjie Li, Lu Hou, Wei Ma and Wei Shi. All authors read and approved the final version.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Li, C., Chai, S., Ju, Y. et al. Pu-erh Tea Protects the Nervous System by Inhibiting the Expression of Metabotropic Glutamate Receptor 5. Mol Neurobiol 54, 5286–5299 (2017). https://doi.org/10.1007/s12035-016-0064-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0064-3