
Abstract
The orphan nuclear receptor TLX, also called NR2E1, is a factor important in the regulation of neural stem cell (NSC) self-renewal, neurogenesis, and maintenance. As a transcription factor, TLX is vital for the expression of genes implicated in neurogenesis, such as DNA replication, cell cycle, adhesion and migration. It acts by way of repressing or activating target genes, as well as controlling protein-protein interactions. Growing evidence suggests that dysregulated TLX acts in the initiation and progression of human disorders of the nervous system. This review describes recent knowledge about TLX expression, structure, targets, and biological functions, relevant to maintaining adult neural stem cells related to both neuropsychiatric conditions and certain nervous system tumours.
Similar content being viewed by others
Introduction
Drosophila tailless (Tll) gene and its homolog Tlx in vertebrates belong to the NR2E subclass of orphan nuclear receptors. Tll was discovered more than two decades ago as a molecule able to specify cell fate during the embryogenesis of Drosophila. Recent studies on Tll and Tlx in both Drosophila and mouse have revealed critical roles in the developing nervous and visual systems, as well as in maintaining neural stem cells (NSC). Tll was identified as a gene needed for terminal structures in Drosophila embryonic development [1–3]. The Tll mutant not only lacks the posterior to the eighth abdominal segment structures (tail), but also those of the head and the brain [2, 4–6]. Similar to its phenotype, the Tll transcript expresses as symmetrical caps at both poles of the embryo [7].
In mammals, four distinct areas of the postnatal brain harbour cells with stem cell properties: (i) the subventricular zone (SVZ) [8], (ii) the subgranular zone (SGZ) [9], (iii) the subcallosal zone (SCZ) [9], and (iv) the cerebellum [10]. Neural stem cells keep the capacity to proliferate and self-renew, as well as the ability to differentiate into neuronal and glial lineages [11–14]. The NR2E1 derives from the evolutionary preserved nuclear receptor superfamily member of transcription factors (with at least 89–97 % homology). It is found in both vertebrates and invertebrates, as mentioned above [3, 15–18].
Tlx is a key regulator of NSC maintenance and self-renewal in the adult brain [19]. Expression of Tlx is specific to the neurogenic regions of the developing forebrain in several species, including the frog [16], zebra fish [17], and mouse [3]. In early neural development, NSCs increase in the neural tube over a limited number of cell cycles, expanding the size of the NSC pool by symmetric division [70]. When cells are about to determine their fate, a subset of NSCs becomes neuroprogenitors producing specialised cell types i.e. neurones, oligodendrocytes, and astrocytes. Neuroprogenitor cells are thought to have a decreased potential for self-renewal and pluripotency.
In the developing embryo and adult mouse, the protein TLX is localised to the neurogenic regions of the telencephalon, diencephalon, nasal placode, and retina [3, 18]. In the adult brain, TLX is expressed in the neural stem cells of the two neurogenetic zones—strong expression in the subventricular zone (SVZ) of the lateral ventricle and in the subgranular zone (SGZ) of the hippocampal dentate gyrus [19]. TLX has been found in proliferating neuroprogenitors of the adult SVZ, although most Tlx-expressing cells in the SVZ are quiescent [20, 21]. Tlx regulates transcriptionally the expression of multiple genes by repressing or activating target genes. Dysregulation of Tlx appears to affect the initiation and progression of human neurological disorders [22, 23], including various nervous system tumours [24–27], making TLX an interesting therapeutic target.
Structure and Regulation of TLX
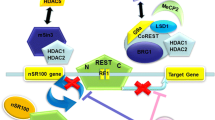
Tlx contains two structural subunits—the highly conserved DNA-binding domain (DBD), and the less conserved ligand-binding domain (LBD) [18, 28]. The TLL and TLX proteins are identical at the levels of 81 and 41 % in the DNA-binding and the ligand-binding domains, respectively [3, 18]. TLX and TLL were validated as functional homologs [18]. TLX has been recognised to act as a transcriptional repressor [29]. In the LBD region, TLX interacts with its cofactors, such as atrophin [30–32], BCL11A [33], LSD1 [34–36], histone deacetylases (HDACs) [30–32, 34, 35], and the von Hippel-Lindau suppressor protein (VHL) [25].
Analysis of the X-ray structure of human TLX-LBD [106] unveiled that TLX-LBD does not have a canonical NR structure. The LBD of the NR superfamily is composed of 12 α-helices (H) and a β-sheet forming an anti-parallel “α-helical sandwich” [107]. The human TLX-LBD lacks two LBD helices H1 and H2 that could potentially form an open LB pocket, and it folds into an auto-repressed ligand-free confirmation [105]. By using homology models of TLX-LBD, Benod and her group suggested that TLX could keep a large LB pocket, which would enable adaptation to ligands [105]. In screening for small molecules that directly bind TLX, three synthetic ligands with sufficient affinity and specificity were detected for TLX, but not for other NR2 subgroup members [105, 108]. However, the possibility remains that TLX functions independently of the ligand, regardless of whether endogenous ligands are discovered, since the structure of TLX LB pocket is similar to Nurr1 (NR4A2) which functions without ligands [108].
The functions of Tll and Tlx are affected by cofactors [14], such as transcriptional repression, being partly mediated by associations with atrophin family proteins [14, 30–32]. Yokoyama et al. [36] demonstrated that TLX binds the promoter of Pten, which is also bound by LSD1. Since Tlx-silencing hampers NSC renewal caused by LSD1 inhibitors, the LSD1-regulated self-renewal of NSCs might depend on Tlx [34]. Recently, Notch1/RBPJ were found to directly regulate transcription factors that are critical for NSC self-renewal, including Tlx, Sox2, Pax6, and Id4 [112]. The microRNA (miR) let-7d also modulates Tlx expression and activity through a conserved binding site on the Tlx mRNA transcripts [37]. Thus, in embryonic mouse brains, over-expression of let-7d inhibits NSC proliferation, promotes neuronal differentiation, and induces neuronal migration, being similar to Tlx knockdown models. Interleukin-1 beta (IL-1β) has been shown to be a negative regulator of embryonic and adult hippocampal neurogenesis, and the expression of Tlx is repressed in neuroprogenitors [6]. Conversely, IL-1β represses Tlx in differentiating newborn and mature neurones and astrocytes [38, 39]. Recently, it was demonstrated that this repression of Tlx and neuroprogenitor proliferation is mediated through the IL-1 receptor type I [40]. Another transcription factor regulating adult NSC proliferation is the sex-determining region-box 2 (SOX2), which has been shown to bind the promoter region of Tlx and activate proliferation of adult mouse NSCs [41].
Tlx Targets
Tlx regulates a broad area of cellular activities, i.e. DNA replication, mitogen-activated protein kinase (MAPK) signal, adhesion, cell cycle, and migration [24, 27, 42, 43]. Through the conserved motifs or DNA-binding assays, the TLX targets Ascl1, Pou5f1 (Oct4), miR 137, Pax2, miR 9, Pten, Cdkn1a, Wnt7a, and Sirt1 have been identified [19, 32, 44–49]. Evidence indicates Tlx to activate Ascl1 and to promote neuronal induction in adult hippocampal neuroprogenitors [45]. During hypoxic conditions, Tlx balances the NSC commitment to the neuronal lineage, maintaining neural progenitor pools through transcriptional activation of Ascl1 and Pou5f1 expression [44, 45]. Similarly, TLX induces matrix metalloproteinase (MMP)-2, which is crucial for NSC to migrate to target areas and establish a neural network [27]. TLX can also physically bind and sequester VHL in normoxia in order to create a hypoxic environment for NSCs [25]. As a result, HIF-2α will be stabilised and recruited to the promoters of angiogenic factors i.e. VEGF and erythropoietin, both being important in maintaining NSC niches [25].
TLX represses the transcription of Pax2 [49, 50]. miR137 is a TLX target and an upstream regulator of LSD1. By recruiting LSD1 to the genomic regions of miR137, TLX represses miR137 in NSCs [35]. miR9 , another direct TLX target, forms a negative-feedback loop resulting in the modulation of Tlx expression in NSCs. This event affects the status of progenitor proliferation and differentiation [51]. Tlx is also regulated by another miR, let-7b and miR378, both of them increasing NSC differentiation [52, 113]. Emerging evidence suggests that microRNAs promote factors that induce differentiation by modulating Tlx expression in neuroprogenitor cells, and by interplay with TLX, these miRs appear to be involved in neurological disorders and neural tumours, as described below.
By analysing RNAs isolated from adult brains of wild type and Tlx mutant mice with a gene profile screen, Qu et al. [46] identified Wnt7α as a downstream target of Tlx. Wnt proteins bind cell membrane-Frizzled receptors, activating Wnt signalling pathways. Furthermore, they translocate the cytoplasmic β-catenin to the nucleus, where β-catenin binds T cell factor family (TCF) transcription factors, together activating the target genes [53]. Several TLX-binding sites are contained in the promoter region of Wnt7α, suggesting that Tlx regulates neurogenesis in an autonomous manner. The Wnt pathway has been experimentally proven to control self-renewal of NSCs.
By suppressing the G0-to-G1 cell-cycle transition, Tlx controls how progenitors proliferate and differentiate by the Pten-cyclin D1 pathway [32, 54]. As a regulator of NSC proliferation, Tlx also represses Pten in both the developing retina and the adult brain [32, 47, 54–56]. Tlx also regulates the differentiation of retinal progenitors via the phospholipase C and MAPK pathways [54]. Thus, by controlling the expression of Pten, Tlx regulates proliferation of stem cell and cell cycle re-entry during retinogenesis [47, 54, 55].
Tlx is considered to regulate NSC proliferation by governing expression of the Cip/Kip family cyclin-dependent kinase inhibitors, such as Cdkn1a (p21), Cdkn1c (p57, Kip2), and several genes downstream of p53 [19, 20, 24, 32, 42, 43, 56]. In fact, p21 and p57 are frequently expressed in differentiating neuroprogenitors [57]. The NAD-dependent deacetylase Sirt1 has been shown to co-localise with TLX in neuroprogenitors [58]. In HEK293 cells, TLX enhances the expression of Sirt1 through binding to the TLX-activating element in the Sirt1 promoter. Moreover, Tlx knockdown diminishes Sirt1 protein expression in neuroprogenitors [58]. Additionally, TLX controls the timing of the postnatal genesis of astrocytes by modulating the BMP-SMAD signalling pathway [59]. TLX binds to the enhancer region of BMP4 to repress its expression in the NSCs, and BMP4 is upregulated in nestin-expressing cells from Tlx mutant mice [59].
Biological Functions
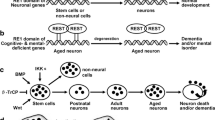
During the development of mouse brain, the expression of Tlx is restricted to the ventricular zone [20]. A detailed inspection of this region displayed a gradient of Tlx along the dorsal-to-ventral axis in the telencephalon [3]. Even though Tlx mutant mice display no abnormality at birth, the Tlx gene is required in embryonic brains for the building of superficial cortical layers [60]. In the cortex, the timing of neurogenesis is regulated by TLX [61] and the formation of lateral telencephalic progenitor regions [62]. Mature Tlx knockout mice display reduced cerebral hemispheres [63] and retinopathies due to deficient cell proliferation and decreased neuroprogenitors [32, 49, 50, 64]. Structures developing late in these mice are diminished in size, such as the upper cortical layers, the hippocampal dentate gyrus, and the olfactory bulbs—the active neurogenetic regions [19]. Transducing Tlx into Tlx-null cells will rescue their capacity to proliferate and self-renew [19]. Adult Tlx mutants exhibit enhanced aggressiveness, decreased copulation, epilepsy, and learning disabilities [43, 63, 65]. Conversely, in Tlx transgenic mice, hippocampal neurogenesis was stimulated, resulting in enhanced learning and memory [66]. This might suggest that the status of adult neuroprogenitors in the SGZ and the reduced neurogenesis precede neuropsychiatric conditions such as cognitive deficits and mood disorders [23], as described below.
Astrocyte markers are repressed by Tlx expression in NSCs [16], i.e. GFAP and aquaporin. The suppressor gene Pten is also involved, suggesting that the transcriptional repression is essential in maintaining their undifferentiated state [19, 32]. In the SGZ of Tlx-mutant mice, there is a significant decrease of stem cell proliferation and a deficiency in spatial learning, whereas no effect is seen on contextual fear conditioning, diurnal variation, or locomotion [43]. Retinal neuroprogenitors expresses Tlx in the mouse during retinal neurogenesis [49, 50]. In Tlx-mutant mice, the normal retinal cell types are specified early, but the number of cells in each layer later progressively decreases, ending as malformation of the vascular system [50]. Studies of conditional depletion of Tlx that will save visual function suggested that blindness at least partially is related to the cognitive defect and other behavioural abnormalities observed in these mice [43, 67, 68]. Additionally, Tlx-mutant animals suffer from defects in retinal vasculature, reaffirming the role for Tlx in the assembly of fibronectin matrices secreted by proangiogenic astrocytes [64].
TLX and Neural Stem Cells
In the developing brain, the primary role of TLX is to prevent a precocious differentiation of NSCs [20, 61] and to maintain them in an undifferentiated state [19, 42]. These undifferentiated precursor cells are the driving force behind the formation of a complete and functional CNS [69]. TLX plays an important role in leading NSCs to the neurogenic niche [42]. The microvascular vessels of the SVZ is critical for the stem cell niche in activating and maintaining NSCs through Wnt, EGF, and other renewal and survival factors produced by the endothelial cells of blood vessels [109]. Hypoxia also plays an essential role for stem cell renewal [110], partly by enhancing the expression of Tlx [25, 27], which, in turn, binds and sequesters VHL, resulting in stabilisation of HIF-α [25]. Most of NSC expresses TLX that induces Wnt7a and increases HIF-α, resulting in stabilisation of the vasculature in the stem cell niche [111]. All these molecules synergise in maintaining the stem cell niche.
Moreover, whole-genome RNA-sequencing has revealed that Tlx coordinates many signalling pathways, regulating NSC behaviour [42]. TLX-positive cells in adult SVZ are relatively quiescent stem cells, and inactivation of TLX in these cells leads to a loss of neurogenesis in the SVZ [24, 56].
In the SVZ of the adult brain, Tlx is expressed in astrocyte-like quiescent or slowly cycling stem cells, named B cells [8, 9], being necessary for the transition of radial glia into adult NSCs [56]. Tlx is expressed both in quiescent and active transit-amplifying NSCs, C cells, in the SVZ [8, 21]. Tlx-inducible mutation eventually leads to a loss of neurogenesis in this region, whereas its overexpression leads to increased neurogenesis of NSCs. The increased neurogenesis prevents age-dependent exhaustion of NSCs. This will even make small glioma-like lesions progress to aggressive gliomas, promoted by p53 inactivation [24, 56]. This finding suggests Tlx to be a critical regulator of self-renewal in the NSCs of SVZ.
In the developing mouse embryo, Tlx emerges early at embryonic day (E) 8 in the ventricular zone, where NSCs and neuroprogenitors reside [3, 20, 61]. As cortical neurones are generated between E11 and E17, the expression of Tlx within the ventricular zone will reach its maximum at E13, diminishing by E16 [71]. In Tlx-mutant mice, neuroprogenitors undergo proliferation with shorter cell cycles from E9.5 to E12.5, resulting in precocious maturation [61]. In these mice, at E14.5, NSCs are decreased in number and undergo slower cell cycles [20]. The deficiency observed during the development of Tlx-mutant mice described above caused reduced cortical depth and size of the dentate gyrus, as well as a smaller forebrain [60]. The impaired limbic system manifests as abnormal behaviours of Tlx-mutant mice [63]. Similarly, adult NSCs, however small their population, also require Tlx in order to remain in a proliferative state. In these mice, NSCs may differentiate into glial cells expressing GFAP and aquaporin—markers of astrocytes [19].
The reduced number of cortical layer neurones and the defective limbic system found in Tlx-mutant mice are, moreover, also seen in Pax6 [72] and in T-box transcription factor (Tbr2) mutants [73]. Tbr2 mutants behave as aggressively as Tlx-mutant mice. The interaction between Tlx and Pax6 [20] facilitates formation of the pallido-subpallidal boundary, as noted above [62]. This suggests that Tlx and Pax6 cooperate to control the differentiation of radial glia into late neurogenic progenitors in the SVZ [73]. Thus, it is likely that deletion or mutation of Tlx may alter Tbr2 expression, and its deficient expression may partly lead to the defects seen in Tlx mutants.
TLX and Disorders in the Nervous System
Recently, large genome-wide studies have revealed links between neuropsychiatric diseases and gene variations. As mentioned elsewhere, loss of the Tlx (NR2E1) gene in mice leads to thinner superficial cortical layers and fewer subsets of GABAergic interneurones in the neocortex [65]. Mutant adult mice display microencephaly, involving hypoplasia in the olfactory bulbs, entorhinal cortex, amygdala, hippocampus, and part of the medial temporal lobe [65]. These animals are aggressive and exhibit decreased anxiety as well as signs of reduced memory. As expected, Tlx transgenic mice demonstrate higher hippocampal neurogenesis, resulting in better learning and memory [66]. Prior to resident-intruder challenges, the aggressive behaviour of Tlx-mutant mice was diminished by a selective 5-HT2A/C receptor antagonist. This suggests that this 5-HT2 receptor is involved in the mechanism behind aggression [68]. As for the basis of conditioned emotional responses, inactivation of the amygdala will prevent fear conditioning to both cue and context, whereas hippocampal dysfunction prevents fear conditioning to context only [74, 75]. Thus, both amygdala and hippocampus may be affected by the loss of Tlx. Dysfunctions of the limbic system are implicated in several psychiatric disorders [65].
Mutation of the NR2E1 gene has been related to microcephaly. Studies have been carried out on coding, un-translated, and regulatory sequences, as well as on evolutionary conserved non-coding regions [22]. Alterations were observed in the non-coding regulatory region, and a number of candidate mutations have been identified in patients with severe cortical disorders [22]. Interestingly, the evolutionary constraint in the coding region of NR2E1 was strong when compared to many other genes examined for gene diversity.
The NR2E1 region on chromosome 6q21-22 has been related to bipolar disorder (BP), especially in patients with manic episodes and a distinct heredity (bipolar I). This link has also been described in schizophrenia (SZ), as well as in some neurological disorders [76–78]. In addition, a meta-analysis of BP demonstrated the strongest genome-wide linkage at 6q21-22 [79]. Profiling of miR expression in SZ and BP post-mortem brains as well as genome-wide association studies (GWAS) have indicated miRs in the aetiologies of these disorders [80]. TLX is targeted by several miRs, among which miR137 displayed the highest degrees of gene variation in a GWAS study of SZ and BP [81]. Administration of lithium and other mood stabilisers seems to downregulate the miR let-7 family in both treatment responders and non-responders [81]. The let-7 family has also been suggested to affect synaptic development [82], and its expression in the brain can be modulated by sleep deprivation [83]. miR9-3p has been reported to associate with SZ as well [84]. The miR field is continuously expanding as new non-coding RNAs are being discovered. Associations between miRs and TLX are likely to be increasingly found in neuropsychiatric disorders. Moreover, these miRs might be used as therapy targets for regulation of Tlx in both positive and negative ways, dependent on the specific target and function.
TLX and Tumour Stem Cells
Several studies indicate that brain tumour stem cells (BTSCs) may derive from NSCs. The most striking supporting evidence is that there are numerous similarities between these cell types. Accumulating evidence also indicates that different malignant tumours, including brain cancers, contain cells that maintain features of tissue-specific stem cells and are malignant [85–90]. BTSC expresses nestin, Musashi-1, Sox2, and MELK, all of which are also expressed in adult NSC [85, 89–91]. Cancer stem cells (CSCs) are capable of self-renewal and multipotent lineage differentiation, and these cells have been isolated from numerous tumours. Particularly for brain tumours, research groups have advocated that a hierarchical concept of tumour development [87, 91] is applicable in many cases. BTSCs are at the top of the hierarchy similar to NSCs, and are able to self-renew and differentiate—which might go wrong (Fig. 1) [87].

Relationships between TLX and its upstream, downstream, and interacting molecules are illustrated. Their hierarchical relations are indicated by arrow lines (positive) and cut lines (negative). Molecules in rectangular boxes indicate repressors, and circular boxes activators
In aggressive brain tumours, the existence of BTSC was demonstrated using a nucleostemin-based murine model [92]. Further evidence has been presented describing CSCs to exist within ependymomas [91], glioblastoma multiforme [93], astrocytomas [94], and medulloblastomas [87]. What mechanisms lead to NSC transformation? For instance, ependymomas have been shown to be derived from CSC and to carry localization-specific radial glia phenotypes [91]. Additional evidence for the hypothesis that BTSCs are derived from NSCs evolves from the fact that numerous brain tumours develop in the SVZ where NSCs are located [93, 100]. Indeed, Tlx is expressed in adult NSCs [19, 21, 56, 74, 93]. The high degree of similarity between NSCs and BTSCs is a potential source of inspiration in identifying efficient targets and in designing novel treatments.
In glioblastoma, many of the signalling pathways that control stem cell development are aberrant as for the EGF receptor and the PDGF α-receptor. Other examples of stem or progenitor cell determinants being up-regulated and maybe playing a role in gliomagenesis include the transcription factors Tlx [93], Oct4 [95], Olig2 [96], and the stem cell markers, i.e. CD15, CD133, and nestin [87, 97–99], as well as markers of different neuroepithelial cells.
As for the peripheral nervous system (PNS) tumours, over-expression of TLX has been demonstrated in high-risk neuroblastoma (NB)—the childhood tumour of embryonic origin derived from the adrenosympathetic branch of neural crest cells (NCCs) [27]. When tumour-initiating cells (TICs) are enriched by tumour spheres, Tlx expression increases [27], and the side population cells are enriched (in manuscript). NCCs are highly plastic and mobile cells, being able to form both neuroepithelial and mesenchymal cells. Thus, tumours derived from NCC, such as NB and melanoma, maintain these properties, making them difficult to target.
The overexpression of Tlx alongside with common genetic lesions (e.g. mutant p53) will induce gliomas [24, 93, 101]. Tlx-GFP reporter mice overexpressing PDGFB and AKT were used to develop gliomas with differential GFP expression [26]. When compared to cells not expressing Tlx-GFP, cells doing so were largely quiescent, but they could self-renew and showed an increased sphere formation with tumour-promoting potential. Remarkably, Tlx-GFP-expressing cells were different from cells that express other putative CSC markers (i.e. Sox2 and Olig2). This may suggest that tumours may contain variable CSCs and/or stem and progenitor cells. These observations suggest that Tlx is a potential molecular target of gliomas [26, 92].
The tumour-suppressor gene Pten was identified as a TLX target during a global gene expression in a profiling study [42, 43]. Pten and p21 are likely to be the responsible genes for Tlx-induced glioma. Loss of Pten and p53 in adult NSCs leads to expansion of these cells, resulting in the formation of glioblastoma. Cell-cycle regulators and neuronal differentiation genes, such asTGFbR1 and Dlx2, were upregulated in Tlx knockout CSCs [42, 43]. These findings agree with prior reports that Tlx represses transcription in controlling CSCs. HDACs are required for this function of Tlx, which is essential for maintaining CSC self-renewal [35]. Thus, HDAC inhibitors may target CSCs to overexpress Tlx.
Since Tlx-amplified NSCs in combination with other genetic alteration contribute to tumorigenesis in the nervous system, Tlx may become a good diagnostic marker and therapeutic target for patients with malignant gliomas. In fact, Tlx is overexpressed in human glioma and neuroblastoma, suggesting that Tlx is involved in human neural tumour development. Tlx expression has furthermore been shown to correlate with poor prognosis and shorter survival of these tumours [24, 27, 56, 101]. Indeed, conditional ablation of Tlx-expressing cells will slow tumor growth and inhibit CSC self-renewal, which is associated with induction of senescence and neurogenetic differentiation [26, 102].
It is important to identify specific CSC targets, since CSCs contribute to treatment resistance [103]. A recent investigation of the full Cancer Genome Atlas dataset (that includes other prognostic factors) indicated that the prognostic importance of Tlx is related to its low expression of the glioma CpG island methylator phenotype (G-CIMP), with a single mutation of isocitrate dehydrogenase 1 (IDH1). Gliomas associated with IDH1 mutation are considered to be a genetically distinct entity predicting better survival [104]. Thus, Tlx may be a poor prognostic factor in itself. Nevertheless, its low expression in G-CIMP patients might reveal the biology in one distinct population of tumours. Future research will demonstrate the importance of CSCs, highlighting Tlx as a novel glioma CSC marker.
Conclusion
It is urgent to examine different subtypes of malignant gliomas as for other genomic alterations with amplified TLX. Are there any mutations in the TLX gene? These questions are also relevant for neuropsychiatric disorders, which are usually considered to be triggered by combinations of multiple gene variations and epigenetics—such as methylation and changes in regulatory RNAs. Moreover, nuclear receptors interact with a variety of proteins, enabling protein stabilisation, activation, or degradation. No endogenous ligands or inhibitors of TLX have as yet been identified, but TLX is indeed a druggable target since synthetic ligands bind to the TLX-LBP [105]. Understanding the mechanisms of NSC renewal will provide insights into basic science as well as offer clinically useful replacement therapies for several disorders in the nervous system, including tumours. Mapping the TLX-controlled network that regulates these outcomes will be a major step forward in our understanding of NSC self-renewal, neurogenesis, and possibility to eradicate cancer stem cells.
References
Jürgens G, Wieschaus E, Nüsslein-Volhard C, Kluding H (1984) Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. Wilhelm Roux’s Arch Dev Biol 193(5):283–295
Pignoni F, Baldarelli RM, Steingrimsson E, Diaz RJ, Patapoutian A, Merriam JR, Lengyel JA (1990) The Drosophila gene tailless is expressed at the embryonic termini and is a member of the steroid receptor superfamily. Cell 62(1):151–163
Monaghan AP, Grau E, Bock D, Schutz G (1995) The mouse homolog of the orphan nuclear receptor tailless is expressed in the developing forebrain. Development 121(3):839–853
Casanova J (1990) Pattern formation under the control of the terminal system in the Drosophila embryo. Development 110(2):621–628
Strecker TR, Merriam JR, Lengyel JA (1988) Graded requirement for the zygotic terminal gene, tailless, in the brain and tail region of the Drosophila embryo. Development 102(4):721–734
Younossi-Hartenstein A, Green P, Liaw GJ, Rudolph K, Lengyel J, Hartenstein V (1997) Control of early neurogenesis of the Drosophila brain by the head gap genes tll, otd, ems, and btd. Dev Biol 182(2):270–283. doi:10.1006/dbio.1996.8475
Rudolph KM, Liaw GJ, Daniel A, Green P, Courey AJ, Hartenstein V, Lengyel JA (1997) Complex regulatory region mediating tailless expression in early embryonic patterning and brain development. Development 124(21):4297–4308
Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97(6):703–716
Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurones in the adult mammalian hippocampus. J Neurosci 21(18):7153–7160
Lee A, Kessler JD, Read TA, Kaiser C, Corbeil D, Huttner WB, Johnson JE, Wechsler-Reya RJ (2005) Isolation of neural stem cells from the postnatal cerebellum. Nature Neurosci 8(6):723–729. doi:10.1038/nn1473
Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn A-M, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4(11):1313–1317
Gage FH (2000) Mammalian neural stem cells. Science 287(5457):1433–1438
Kornblum HI (2007) Introduction to neural stem cells. Stroke 38(2 Suppl):810–816. doi:10.1161/01.STR.0000255757.12198.0f
Temple S (2001) The development of neural stem cells. Nature 414(6859):112–117. doi:10.1038/35102174
Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P et al (1995) The nuclear receptor superfamily: the second decade. Cell 83(6):835–839
Hollemann T, Bellefroid E, Pieler T (1998) The Xenopus homologue of the Drosophila gene tailless has a function in early eye development. Development 125(13):2425–2432
Kitambi SS, Hauptmann G (2007) The zebrafish orphan nuclear receptor genes nr2e1 and nr2e3 are expressed in developing eye and forebrain. Gene Expr Patterns 7(4):521–528. doi:10.1016/j.modgep.2006.10.006
Yu RT, McKeown M, Evans RM, Umesono K (1994) Relationship between Drosophila gap gene tailless and a vertebrate nuclear receptor Tlx. Nature 370(6488):375–379. doi:10.1038/370375a0
Shi Y, Chichung Lie D, Taupin P, Nakashima K, Ray J, Yu RT, Gage FH, Evans RM (2004) Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature 427(6969):78–83. doi:10.1038/nature02211
Li W, Sun G, Yang S, Qu Q, Nakashima K, Shi Y (2008) Nuclear receptor TLX regulates cell cycle progression in neural stem cells of the developing brain. Mol Endocrinol 22(1):56–64. doi:10.1210/me.2007-0290
Li S, Sun G, Murai K, Ye P, Shi Y (2012) Characterization of TLX expression in neural stem cells and progenitor cells in adult brains. PLoS One 7(8):e43324. doi:10.1371/journal.pone.0043324
Kumar RA, Leach S, Bonaguro R, Chen J, Yokom DW, Abrahams BS, Seaver L, Schwartz CE et al (2007) Mutation and evolutionary analyses identify NR2E1-candidate-regulatory mutations in humans with severe cortical malformations. Genes Brain Behav 6(6):503–516. doi:10.1111/j.1601-183X.2006.00277.x
Kempermann G, Krebs J, Fabel K (2008) The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr Opin Psychiatry 21(3):290–295. doi:10.1097/YCO.0b013e3282fad375
Liu HK, Wang Y, Belz T, Bock D, Takacs A, Radlwimmer B, Barbus S, Reifenberger G et al (2010) The nuclear receptor tailless induces long-term neural stem cell expansion and brain tumour initiation. Genes Dev 24(7):683–695. doi:10.1101/gad.560310
Zeng ZJ, Johansson E, Hayashi A, Chavali PL, Akrap N, Yoshida T, Kohno K, Izumi H et al (2012) TLX controls angiogenesis through interaction with the von Hippel-Lindau protein. Biology Open 1(6):527–535. doi:10.1242/bio.2012893
Zhu Z, Khan MA, Weiler M, Blaes J, Jestaedt L, Geibert M, Zou P, Gronych J et al (2014) Targeting self-renewal in high-grade brain tumours leads to loss of brain tumour stem cells and prolonged survival. Cell Stem Cell 15(2):185–198
Chavali P, Saini R, Zhai Q, Vizlin-Hodzic D, Venkatabalasubramanian S, Hayashi A, Johansson E, Zeng Z et al (2014) TLX activates MMP-2, promotes self-renewal of tumour spheres in neuroblastoma and correlates with poor patient survival. Cell Death & Dis 5(10):e1502
Bain DL, Heneghan AF, Connaghan-Jones KD, Miura MT (2007) Nuclear receptor structure: implications for function. Annu Rev Physiol 69:201–220. doi:10.1146/annurev.physiol.69.031905.160308
Moran E, Jimenez G (2006) The tailless nuclear receptor acts as a dedicated repressor in the early Drosophila embryo. Mol Cell Biol 26(9):3446–3454. doi:10.1128/mcb.26.9.3446-3454.2006
Haecker A, Qi D, Lilja T, Moussian B, Andrioli LP, Luschnig S, Mannervik M (2007) Drosophila brakeless interacts with atrophin and is required for tailless-mediated transcriptional repression in early embryos. PLoS Biol 5(6):e145. doi:10.1371/journal.pbio.0050145
Wang L, Rajan H, Pitman JL, McKeown M, Tsai CC (2006) Histone deacetylase-associating atrophin proteins are nuclear receptor corepressors. Genes Dev 20(5):525–530. doi:10.1101/gad.1393506
Zhang CL, Zou Y, Yu RT, Gage FH, Evans RM (2006) Nuclear receptor TLX prevents retinal dystrophy and recruits the corepressor atrophin1. Genes Dev 20(10):1308–1320. doi:10.1101/gad.1413606
Estruch SB, Buzon V, Carbo LR, Schorova L, Luders J, Estebanez-Perpina E (2012) The oncoprotein BCL11A binds to orphan nuclear receptor TLX and potentiates its transrepressive function. PLoS One 7(6):e37963. doi:10.1371/journal.pone.0037963
Sun G, Alzayady K, Stewart R, Ye P, Yang S, Li W, Shi Y (2010) Histone demethylase LSD1 regulates neural stem cell proliferation. Mol Cell Biol 30(8):1997–2005. doi:10.1128/mcb.01116-09
Sun G, Ye P, Murai K, Lang MF, Li S, Zhang H, Li W, Fu C et al (2011) miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat Commun 2:529. doi:10.1038/ncomms1532
Yokoyama A, Takezawa S, Schule R, Kitagawa H, Kato S (2008) Transrepressive function of TLX requires the histone demethylase LSD1. Mol Cell Biol 28(12):3995–4003. doi:10.1128/mcb.02030-07
Zhao C, Sun G, Ye P, Li S, Shi Y (2013) MicroRNA let-7d regulates the TLX/microRNA-9 cascade to control neural cell fate and neurogenesis. Sci Rep 3:1329. doi:10.1038/srep01329
Green HF, Nolan YM (2012) Unlocking mechanisms in interleukin-1beta-induced changes in hippocampal neurogenesis—a role for GSK-3beta and TLX. Transcult Psychiatry 2:e194. doi:10.1038/tp.2012.117
Koo JW, Duman RS (2008) IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. PNAS USA 105(2):751–756. doi:10.1073/pnas.0708092105
Ryan SM, O'Keeffe GW, O'Connor C, Keeshan K, Nolan YM (2013) Negative regulation of TLX by IL-1beta correlates with an inhibition of adult hippocampal neural precursor cell proliferation. Brain Behav Immun 33:7–13. doi:10.1016/j.bbi.2013.03.005
Shimozaki K, Zhang CL, Suh H, Denli AM, Evans RM, Gage FH (2012) SRY-box-containing gene 2 regulation of nuclear receptor tailless (Tlx) transcription in adult neural stem cells. J Biol Chem 287(8):5969–5978. doi:10.1074/jbc.M111.290403
Niu W, Zou Y, Shen C, Zhang CL (2011) Activation of postnatal neural stem cells requires nuclear receptor TLX. J Neurosci 31(39):13816–13828. doi:10.1523/jneurosci.1038-11.2011
Zhang CL, Zou Y, He W, Gage FH, Evans RM (2008) A role for adult TLX-positive neural stem cells in learning and behaviour. Nature 451(7181):1004–1007. doi:10.1038/nature06562
Chavali PL, Saini RK, Matsumoto Y, Agren H, Funa K (2011) Nuclear orphan receptor TLX induces Oct-3/4 for the survival and maintenance of adult hippocampal progenitors upon hypoxia. J Biol Chem 286(11):9393–9404. doi:10.1074/jbc.M110.167445
Elmi M, Matsumoto Y, Zeng ZJ, Lakshminarasimhan P, Yang W, Uemura A, Nishikawa S, Moshiri A et al (2010) TLX activates MASH1 for induction of neuronal lineage commitment of adult hippocampal neuroprogenitors. Mol Cell Neurosci 45(2):121–131. doi:10.1016/j.mcn.2010.06.003
Qu Q, Sun G, Li W, Yang S, Ye P, Zhao C, Yu RT, Gage FH et al (2010) Orphan nuclear receptor TLX activates Wnt/beta-catenin signalling to stimulate neural stem cell proliferation and self-renewal. Nat Cell Biol 12(1):31–40. doi:10.1038/ncb2001
Sun G, Yu RT, Evans RM, Shi Y (2007) Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. PNAS USA 104(39):15282–15287. doi:10.1073/pnas.0704089104
Wang Y, Liu HK, Schutz G (2013) Role of the nuclear receptor tailless in adult neural stem cells. Mech Dev 130(6-8):388–390. doi:10.1016/j.mod.2013.02.001
Yu RT, Chiang MY, Tanabe T, Kobayashi M, Yasuda K, Evans RM, Umesono K (2000) The orphan nuclear receptor Tlx regulates Pax2 and is essential for vision. PNAS USA 97(6):2621–2625. doi:10.1073/pnas.050566897
Miyawaki T, Uemura A, Dezawa M, Yu RT, Ide C, Nishikawa S, Honda Y, Tanabe Y et al (2004) Tlx, an orphan nuclear receptor, regulates cell numbers and astrocyte development in the developing retina. J Neurosci 24(37):8124–8134. doi:10.1523/jneurosci.2235-04.2004
Zhao C, Sun G, Li S, Shi Y (2009) A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat Struct Mol Biol 16(4):365–371. doi:10.1038/nsmb.1576
Zhao C, Sun G, Li S, Lang MF, Yang S, Li W, Shi Y (2010) MicroRNA let-7b regulates neural stem cell proliferation and differentiation by targeting nuclear receptor TLX signaling. PNAS USA 107(5):1876–1881. doi:10.1073/pnas.0908750107
Nusse R, Fuerer C, Ching W, Harnish K, Logan C, Zeng A, ten Berge D, Kalani Y (2008) Wnt signaling and stem cell control. Cold Spring Harb Symp Quant Biol 73:59–66. doi:10.1101/sqb.2008.73.035
Groszer M, Erickson R, Scripture-Adams DD, Dougherty JD, Le Belle J, Zack JA, Geschwind DH, Liu X et al (2006) PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. PNAS USA 103(1):111–116. doi:10.1073/pnas.0509939103
Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X et al (2001) Negative regulation of neural stem/progenitor cell proliferation by the Pten tumour suppressor gene in vivo. Science 294(5549):2186–2189. doi:10.1126/science.1065518
Liu HK, Belz T, Bock D, Takacs A, Wu H, Lichter P, Chai M, Schutz G (2008) The nuclear receptor tailless is required for neurogenesis in the adult subventricular zone. Genes Dev 22(18):2473–2478. doi:10.1101/gad.479308
Gui H, Li S, Matise MP (2007) A cell-autonomous requirement for Cip/Kip cyclin-kinase inhibitors in regulating neuronal cell cycle exit but not differentiation in the developing spinal cord. Dev Biol 301(1):14–26. doi:10.1016/j.ydbio.2006.10.035
Iwahara N, Hisahara S, Hayashi T, Horio Y (2009) Transcriptional activation of NAD+-dependent protein deacetylase SIRT1 by nuclear receptor TLX. Biochem Biophys Res Commun 386(4):671–675. doi:10.1016/j.bbrc.2009.06.103
Qin S, Niu W, Iqbal N, Smith DK, Zhang CL (2014) Orphan nuclear receptor TLX regulates astrogenesis by modulating BMP signaling. Front Neurosci 8:74. doi:10.3389/fnins.2014.00074
Land PW, Monaghan AP (2005) Abnormal development of zinc-containing cortical circuits in the absence of the transcription factor tailless. Brain Res Dev Brain Res 158(1-2):97–101. doi:10.1016/j.devbrainres.2005.04.006
Roy K, Kuznicki K, Wu Q, Sun Z, Bock D, Schutz G, Vranich N, Monaghan AP (2004) The Tlx gene regulates the timing of neurogenesis in the cortex. J Neurosci 24(38):8333–8345. doi:10.1523/jneurosci.1148-04.2004
Stenman J, Yu RT, Evans RM, Campbell K (2003) Tlx and Pax6 co-operate genetically to establish the pallido-subpallidal boundary in the embryonic mouse telencephalon. Development 130(6):1113–1122
Monaghan AP, Bock D, Gass P, Schwager A, Wolfer DP, Lipp HP, Schutz G (1997) Defective limbic system in mice lacking the tailless gene. Nature 390(6659):515–517. doi:10.1038/37364
Uemura A, Kusuhara S, Wiegand SJ, Yu RT, Nishikawa S (2006) Tlx acts as a proangiogenic switch by regulating extracellular assembly of fibronectin matrices in retinal astrocytes. J Clin Invest 116(2):369–377. doi:10.1172/jci25964
Roy K, Thiels E, Monaghan AP (2002) Loss of the tailless gene affects forebrain development and emotional behavior. Physiol Behav 77(4-5):595–600
Murai K, Qu Q, Sun G, Ye P, Li W, Asuelime G, Sun E, Tsai GE et al (2014) Nuclear receptor TLX stimulates hippocampal neurogenesis and enhances learning and memory in a transgenic mouse model. PNAS USA 111(25):9115–9120. doi:10.1073/pnas.1406779111
Belz T, Liu HK, Bock D, Takacs A, Vogt M, Wintermantel T, Brandwein C, Gass P et al (2007) Inactivation of the gene for the nuclear receptor tailless in the brain preserving its function in the eye. Eur J Neurosci 26(8):2222–2227. doi:10.1111/j.1460-9568.2007.05841.x
Juarez P, Valdovinos MG, May ME, Lloyd BP, Couppis MH, Kennedy CH (2013) Serotonin(2)A/C receptors mediate the aggressive phenotype of TLX gene knockout mice. Behav Brain Res 256:354–361. doi:10.1016/j.bbr.2013.07.044
Alvarez‐Buylla A, Temple S (1998) Stem cells in the developing and adult nervous system. Neurobiol 36(2):105–110
Rakic P (1995) A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci 18(9):383–388
Takahashi T, Nowakowski RS, Caviness VS Jr (1995) Early ontogeny of the secondary proliferative population of the embryonic murine cerebral wall. J Neurosci 15(9):6058–6068
Quinn JC, Molinek M, Martynoga BS, Zaki PA, Faedo A, Bulfone A, Hevner RF, West JD et al (2007) Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev Biol 302(1):50–65. doi:10.1016/j.ydbio.2006.08.035
Arnold SJ, Huang GJ, Cheung AF, Era T, Nishikawa S, Bikoff EK, Molnar Z, Robertson EJ et al (2008) The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev 22(18):2479–2484. doi:10.1101/gad.475408
Phillips RG, LeDoux JE (1992) Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106(2):274–285
Logue SF, Paylor R, Wehner JM (1997) Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci 111(1):104–113
Dick DM, Foroud T, Flury L, Bowman ES, Miller MJ, Rau NL, Moe PR, Samavedy N et al (2003) Genomewide linkage analyses of bipolar disorder: a new sample of 250 pedigrees from the National Institute of Mental Health Genetics Initiative. Am J Hum Genet 73(1):107–114. doi:10.1086/376562
Middleton FA, Pato MT, Gentile KL, Morley CP, Zhao X, Eisener AF, Brown A, Petryshen TL et al (2004) Genome-wide linkage analysis of bipolar disorder by use of a high-density single-nucleotide-polymorphism (SNP) genotyping assay: a comparison with microsatellite marker assays and finding of significant linkage to chromosome 6q22. Am J Hum Genet 74(5):886–897. doi:10.1086/420775
Wong EH, Tarazi FI, Shahid M (2010) The effectiveness of multi-target agents in schizophrenia and mood disorders: relevance of receptor signature to clinical action. Pharmacol Ther 126(2):173–185. doi:10.1016/j.pharmthera.2010.02.001
McQueen MB, Devlin B, Faraone SV, Nimgaonkar VL, Sklar P, Smoller JW, Abou Jamra R, Albus M et al (2005) Combined analysis from eleven linkage studies of bipolar disorder provides strong evidence of susceptibility loci on chromosomes 6q and 8q. Am J Hum Genet 77(4):582–595. doi:10.1086/491603
Schizophrenia Psychiatric Genome-Wide Association Study Consortium (2011) Genome-wide association study identifies five new schizophrenia loci. Nat Genet 43(10):969–976. doi:10.1038/ng.940
Hunsberger JG, Chibane FL, Elkahloun AG, Henderson R, Singh R, Lawson J, Cruceanu C, Nagarajan V et al (2015) Novel integrative genomic tool for interrogating lithium response in bipolar disorder. Transcult Psychiatry 5:e504. doi:10.1038/tp.2014.139
Corbin R, Olsson-Carter K, Slack F (2009) The role of microRNAs in synaptic development and function. BMB Rep 42(3):131–135
Davis CJ, Bohnet SG, Meyerson JM, Krueger JM (2007) Sleep loss changes microRNA levels in the brain: a possible mechanism for state-dependent translational regulation. Neurosci Lett 422(1):68–73. doi:10.1016/j.neulet.2007.06.005
Geaghan M, Cairns MJ (2015) MicroRNA and posttranscriptional dysregulation in psychiatry. Biol Psychiatry 78(4):231–239. doi:10.1016/j.biopsych.2014.12.009
Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI (2003) Cancerous stem cells can arise from pediatric brain tumours. PNAS USA 100(25):15178–15183. doi:10.1073/pnas.2036535100
Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK (2004) A distinct "side population" of cells with high drug efflux capacity in human tumour cells. PNAS USA 101(39):14228–14233. doi:10.1073/pnas.0400067101
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumours. Cancer Res 63(18):5821–5828
Tu SM, Lin SH, Logothetis CJ (2002) Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol 3(8):508–513
Cardona AF, Balana C, Torres D, Becerra H, Ortiz LD, Fadul CE (2011) Stem cells in brain tumorigenesis and their impact on therapy. Curr Stem Cell Res Ther 6(4):339–349
Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C et al (2004) Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 64(19):7011–7021. doi:10.1158/0008-5472.can-04-1364
Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J et al (2005) Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8(4):323–335. doi:10.1016/j.ccr.2005.09.001
Tamase A, Muraguchi T, Naka K, Tanaka S, Kinoshita M, Hoshii T, Ohmura M, Shugo H et al (2009) Identification of tumour-initiating cells in a highly aggressive brain tumour using promoter activity of nucleostemin. PNAS USA 106(40):17163–17168. doi:10.1073/pnas.0905016106
Zou Y, Niu W, Qin S, Downes M, Burns DK, Zhang CL (2012) The nuclear receptor TLX is required for gliomagenesis within the adult neurogenic niche. Mol Cell Biol 32(23):4811–4820. doi:10.1128/mcb.01122-12
Sharma MK, Mansur DB, Reifenberger G, Perry A, Leonard JR, Aldape KD, Albin MG, Emnett RJ et al (2007) Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res 67(3):890–900. doi:10.1158/0008-5472.can-06-0973
Holmberg J, He X, Peredo I, Orrego A, Hesselager G, Ericsson C, Hovatta O, Oba-Shinjo SM et al (2011) Activation of neural and pluripotent stem cell signatures correlates with increased malignancy in human glioma. PLoS One 6(3):e18454
Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD et al (2004) The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol 63(5):499–509
Lu WJ, Lan F, He Q, Lee A, Tang CZ, Dong L, Lan B, Ma X et al (2011) Inducible expression of stem cell associated intermediate filament nestin reveals an important role in glioblastoma carcinogenesis. Int J Cancer 128(2):343–351. doi:10.1002/ijc.25586
Son MJ, Woolard K, Nam DH, Lee J, Fine HA (2009) SSEA-1 is an enrichment marker for tumour-initiating cells in human glioblastoma. Cell Stem Cell 4(5):440–452. doi:10.1016/j.stem.2009.03.003
Rebetz J, Tian D, Persson A, Widegren B, Salford LG, Englund E, Gisselsson D, Fan X (2008) Glial progenitor-like phenotype in low-grade glioma and enhanced CD133-expression and neuronal lineage differentiation potential in high-grade glioma. PLoS One 3(4):e1936. doi:10.1371/journal.pone.0001936
Lim DA, Cha S, Mayo MC, Chen MH, Keles E, VandenBerg S, Berger MS (2007) Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumour phenotype. Neuro Oncol 9(4):424–429. doi:10.1215/15228517-2007-023
Park HJ, Kim JK, Jeon HM, Oh SY, Kim SH, Nam DH, Kim H (2010) The neural stem cell fate determinant TLX promotes tumorigenesis and genesis of cells resembling glioma stem cells. Mol Cells 30(5):403–408. doi:10.1007/s10059-010-0122-z
O’Loghlen A, Martin N, Krusche B, Pemberton H, Alonso MM, Chandler H, Brookes S, Parrinello S et al (2014) The nuclear receptor NR2E1/TLX controls senescence. Oncogene. doi:10.1038/onc.2014.335
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444(7120):756–760. doi:10.1038/nature05236
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW et al (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483(7390):479–483. doi:10.1038/nature10866
Benod C, Villagomez R, Filgueira CS, Hwang PK, Leonard PG, Poncet-Montange G, Rajagopalan S, Fletterick RJ et al (2014) The human orphan nuclear receptor tailless (TLX, NR2E1) is druggable. PLoS One 9(6):e99440. doi:10.1371/journal.pone.0099440
Zhi X, Zhou XE, He Y, Searose-Xu K, Zhang CL, Tsai CC, Melcher K, Xu HE (2015) Structural basis for corepressor assembly by the orphan nuclear receptor TLX. Genes Dev 29:440–450. doi:10.1101/gad.254904.114
Moras D, Gronemeyer H (1998) The nuclear receptor ligand-binding domain: structure and function. Curr Opin Cell Biol 10(3):384–391
Benod C, Villagomez R, Webb P (2015) TLX: An elusive receptor. J Steroid Biochem Mol Biol. doi:10.1016/j.jsbmb.2015.11.001 [Epub ahead of print]
Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, Roysam B, Temple S (2008) Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell 3(3):289–300. doi:10.1016/j.stem.2008.07.026
Panchision DM (2009) The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol 220:562–568
Li L, Candelario KM, Thomas K, Wang R, Wright K, Messier A, Cunningham LA (2014) Hypoxia inducible factor-1α (HIF-1α) is required for neural stem cell maintenance and vascular stability in the adult mouse SVZ. J Neurosci 34(50):16713–16719. doi:10.1523/jneurosci.4590-13
Li Y, Hibbs MA, Gard AL, Shylo NA, Yun K (2012) Genome-wide analysis of N1ID/RBJ targets in vivo reveals direct transcriptional regulation of Wnt, SHH, and hippo pathway effects by Notch1. Stem Cells 30(4):741–752. doi:10.1002/stem.1030
Huang Y, Liu X, Wang Y (2015) MicroRNA-378 regulates neural stem cell proliferation and differentiation in vitro by modulating tailless expression. Biochem Biophys Res Commun 466(2):214–220. doi:10.1016/j.bbrc.2015.09.011
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sobhan, P.K., Funa, K. TLX—Its Emerging Role for Neurogenesis in Health and Disease. Mol Neurobiol 54, 272–280 (2017). https://doi.org/10.1007/s12035-015-9608-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9608-1