
Abstract
Presently, the world needs safe and effective vaccines to overcome the COVID-19 pandemic. Our work has focused on formulating two types of mRNA vaccines that differ in capacity to copy themselves inside the cell. These are non-amplifying mRNA (NRM) and self-amplifying mRNA (SAM) vaccines. Both the vaccine candidates encode an engineered viral replicon which can provoke an immune response. Hence we predicted and screened twelve epitopes from the spike glycoprotein of SARS-CoV-2. We used five CTL, four HTL, and three B-cell-activating epitopes to formulate each mRNA vaccine. Molecular docking revealed that these epitopes could combine with HLA molecules that are important for boosting immunogenicity. The B-cell epitopes were adjoined with GPGPG linkers, while CTL and HTL epitopes were linked with KK linkers. The entire protein chain was reverse translated to develop a specific NRM-based vaccine. We incorporate gene encoding replicase in the upstream region of CDS encoding antigen to design the SAM vaccine. Subsequently, signal sequences were added to human mRNA to formulate vaccines. Both vaccine formulations translated to produce the epitopes in host cells, initiate a protective immune cascade, and generate immunogenic memory, which can counter future SARS-CoV-2 viral exposures before the onset of infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
SARS-CoV-2 is responsible for a dreadful disease (COVID-19) that poses a serious threat to public health worldwide [1]. The virus was first detected in Wuhan city of Central China’s Hubei province. Subsequently, it spread at an impetuous speed to become a worldwide pandemic [2]. At the onset of the pandemic, 88 countries enforced lockdown mandates to eliminate community transmission and lower the rates of new infections [3]. More than 70 million people have been infected and around 1.7 million casualties have been declared by the World Health Organization (WHO).
Coronaviruses belong to the kingdom Riboviria, phylum Pisuviricota, class Pisoniviricetes, order Nidovirales, family Coronaviridae, and subfamily Orthocoronavirideae. There are four genera under Orthocoronavirideae: alpha, beta, gamma, and delta coronaviruses [4, 5]. Among them, alpha and beta coronaviruses mainly infect humans, while gamma and delta coronaviruses cause diseases in birds and non-human mammalian species [6]. The spike protein on the surface of this virus mimics the sun’s corona; thus, the virus name was derived. The SARS-CoV-2 belongs to the beta coronavirus family and possesses a single-stranded RNA genome. It has a nucleocapsid composed of viral spikes, glycoproteins, and envelope proteins [7,8,9].
At present, there is no specified clinical therapy for COVID-19. Application of hand sanitizer, washing of hand, use of mask-wearing, and maintaining physical distance can lower the risk of infection. Although some drugs, including remdesivir, favipiravir, and hydroxychloroquine, are being repurposed for treatment and their efficacy remains unclear [10, 11]. The development of therapeutics in the near future to combat COVID-19 will be beneficial but will not be sufficient to end the pandemic. Therapeutics can cure infected patients but cannot limit infection. It will be necessary to contain the infection to cure the patients. An effective vaccine can provide a permanent solution to the COVID-19 pandemic [12]. Scientists from many parts of the world are rigorously investing tireless efforts to develop an effective vaccine that can bring an end to the pandemic. As a result, nearly 30 RNA, 19 DNA, 29 non-replicating viral vectors, 21 replicating viral vectors, 14 inactivated virus, four live-attenuated virus, 71 protein, 13 nanoparticles, four whole cells, and three viral vector-based vaccines are under development in pre-clinical trials, clinical trials, or approved [13]. Covaxin, Covishield, Moderna, Oxford AstraZeneca, Janssen COVID-19, Sputnik V, and Pfizer-BioNTech vaccines have been approved for public use in several countries [14, 15]. Many vaccines are undergoing phase III clinical trials to establish their safety and efficacy [15].
Our current work aims to develop a suitable mRNA-based vaccine expressing antigenic CD4+ and CD8+ T cell and B-cell-activating sequences. The novel goal of mRNA vaccine modeling is to deliver the transcript of interest, encoding one or more antigens or immunogens, into the host cell cytoplasm to generate a sufficient quantity of the encoded immunogen to boost the immune system [16]. Herein, two types of mRNA vaccine constructs are dynamically considered: (a) self-amplifying mRNA (SAM) and (b) non-replicating mRNA (NRM) (Fig. 1) [17, 18]. Both mRNA vaccine constructs have common features, including a 5′ cap sequence, 5′ and 3′ translated regions (UTRs), an open reading frame (ORF) carrying coding sequence (CDS), and a 3′ poly(A) tail, as shown in Fig. 1 [16, 19]. We focused on a comprehensive NRM-mediated immune activation mechanism and a SAM vaccine construct that carries an insertion of genetic replication machinery (replicase) obtained from a positive-stranded mRNA virus. The ORF encoding the viral structural proteins is replaced with the antigen-expressing transcript and the viral RNA-dependent RNA polymerase (RdRp) is retained for direct cytoplasmic amplification of the replicon construct.

Schematic illustration of two different mRNA vaccine constructions and their cellular interaction
The nucleocapsid spike (S) protein is the preferred candidate antigen, as it is expressed on the virus’s outer surface mediates viral entry into the host and is specific to the SARS-CoV-2 virus [20,21,22]. mRNA vaccines are better and safer than conventional vaccines, such as killed virus and attenuated live virus vaccines [19, 23]. It is also better and safer than modern viral vector-based vaccines [24, 25]. The expressivity of mRNA vaccines is higher than that of DNA vaccines because they do not need to cross the nuclear membrane for translation [26, 27]. All these attributes of mRNA vaccines prompted us to develop mRNA-based vaccines against SARS-CoV-2. An immunoinformatic strategy is required for the successful selection of effective antigens. This strategy recruits online databases and software to map the immunogenic epitope fragments within the target sequence [28,29,30]. This method is easy, reliable, cost effective, and widely accepted [31, 32]. These epitopes are the building blocks of candidate vaccines. We subsequently formulated an mRNA vaccine coding sequence and performed qualitative assessments through molecular docking. This contemporary approach might support safe and effective COVID-19 vaccine development in the near future. The final NRM-based mRNA vaccine construct was cloned in silico into pET28a (+) to propagate the vaccine, whereas the SAM-based mRNA vaccine is self-replicating and needs only to be delivered into the host cell cytoplasm for translation. It does not require cloning [17]. In this work, SARS-CoV-2 S-protein sequence were used to develop the two mRNA vaccines. We have used different antigenic parts using MHC-I and MHC-II for their high antigenicity. We have calculated the antigenicity, allergenicity, and physiochemical properties of the vaccine candidates. Finally, we have performed the molecular docking of Cytotoxic T lymphocytes (CTLs) and Helper T lymphocytes (HTLs) epitopes with HLA alleles to understand the suitability of vaccine candidates.
Materials and Methods
Briefings of Research Design
The methods and strategies used in this research are summarized in a flow diagram. From SARS-CoV-2 S-protein sequence retrieval to molecular docking between predicted epitopes and their corresponding receptors are shown in Fig. 2.

The flowchart describes the mRNA vaccine construct design against the spike protein of SARS-CoV-2
Retrieval of S-Protein Sequence
Protein sequence data are the primary prerequisite for the formulation of a peptide-based mRNA vaccine. The necessary data about the spike glycoprotein of SARS-CoV-2 were acquired from GenBank (NCBI accession no. 6VYB_C) and PDB (ID. 6VYB) databases.
Prediction of CTL Epitopes
Cytotoxic T lymphocytes (CTLs) are the key immune system component for maintaining adaptive immunity against virus-infected cells. It recognizes viral epitopes presented to surface receptors by the MHC-I complex [33]. Therefore, the prediction and selection of CTL epitopes are pivotal for developing peptide-based mRNA vaccines. CTL epitopes were predicted by the NetCTL 1.2 Server [34]. The server deploys an artificial neural network (ANN)-based methodology to execute epitope prediction [35]. At the default threshold of 0.75, a transporter associated with antigen presentation (TAP) transport efficiency of 0.05, and C-terminal cleavage weighting of 0.15, we predicted the CTL epitopes among all 12 A and B supertypes of the HLA allele. Predicted CTL epitopes were then selected for vaccine formulation after antigenicity and allergenicity evaluation. Antigenic properties of epitopes were evaluated using the VaxiJen server. Allergenic characteristics were evaluated using the AllerTOP v2.0 server [36, 37].
Prediction of HTL Epitopes
Helper T lymphocytes (HTL) play critical roles in both cell mediated and humoral immunity upon viral infection. MHC-II-mediated cell surface receptors on HTLs recognize viral epitopes and initiate a responding immune cascade [38]. MHC-II binding and HTL epitopes were predicted using the immune epitope database (IEDB) analysis resource of MHC-II-binding prediction module v2.22 [39]. To predict epitopes, the whole HLA reference set was used, maintaining other parameters at default settings. After epitope prediction, functional epitopes were chosen based on the antigenic and allergenic evaluations of VaxiJen and AllerTOP servers, respectively.
Prediction of B-Cell Epitopes
B-cell epitopes play a major role in computational vaccine design [40]. In this work, we used the BCPREDS server to identify B-cell epitopes within the target protein [41]. The BCPred prediction method was applied to predict fixed-length linear epitopes. The length of the predicted epitopes was selected as 20 amino acids. The B-cell epitopes were also screened for antigenic and allergenic properties in VaxiJen and AllerTOP server, correspondingly.
Population Coverage Calculation
The MHC-I and MHC-II epitopes of SARS-CoV-2 S-glycoprotein, filtered for high antigenicity and absence of allergenicity, were assessed for percent coverage of the world population. The selected MHC-I and MHC-II interactive alleles were evaluated using the IEDB population coverage calculation tool [42].
Designing NRM and SAM Vaccine Constructs
The expression of mRNA vaccines is much higher than that of DNA vaccines [17]. Non-Replicating mRNA molecules used in mRNA vaccines contain five key elements critical to the lifespan and expression of an mRNA: a Kozak sequence, poly-A tail, 5′ cap (m7Gp3N), and 5′ and 3′ untranslated regions must be incorporated within an mRNA vaccine. By contrast, generating a self-amplifying mRNA molecule-based vaccine candidate requires only the insertion of a replicase-coding gene sequence and the five key elements flanking a CDS. This provides an mRNA vaccine the ability to self-replicate within the host’s body. The previously selected epitopes were the desired products of translation of the mRNA vaccine. To link the epitopes together, we used a ‘GPGPG’ linker between the B-cell epitopes and a ‘KK’ linker between the CTL epitopes and the HTL epitopes. The complete epitope-linker sequence was reverse translated using the EMBOSS Backtranseq tool of EMBL-ESI [43]. The reverse-translated DNA sequence was converted to mRNA sequence by applying the Bio model transcription and translation tool to obtain the vaccine construct.
Characterization of Vaccine Constructs
We obtained the translated primary amino acid sequences of mRNA vaccines and these are characterized for different physicochemical properties, which are also crucial for SAM and NRM molecule-based model vaccines. Physicochemical properties including molecular weight, molecular formula, theoretical isoelectric point (pI), instability index (II), aliphatic index (AI), estimated half-life, and grand average of hydrophobicity (GRAVY) were obtained using the ExPASy ProtParam online web server. Simultaneously, the antigenicity and allergenicity of these vaccine constructs were evaluated using VaxiJen and AllerTOP servers, respectively. Finally, Protein-Sol, an online server, was used to evaluate the expressed vaccine protein’s solubility [44].
Molecular Docking Between Epitopes and HLA Alleles
Selected CTL and HTL epitopes were studied for structural compatibility with corresponding MHC alleles and binding interactions were evaluated via molecular docking studies. MHC alleles were retrieved from the RCSB PDB database and refined using PyMOL software to remove unnecessary ligands [45]. For molecular docking, epitopes need a 3D conformation to bind with MHC alleles. We used the DISTILL 2.0 server to predict the 3D structures of epitopes [46]. AutoDock Vina software was used for docking of epitopes and MHC molecules [47]. We set default parameters at the time of grid box preparation using AutoDock 4 Tool [48]. Finally, the molecular docking complex visualization and preparation were performed using PyMOL software [49].
Molecular Docking of mRNA Vaccine with Human TLR-7
As rationalized by molecular docking studies, we designed a novel and effective mRNA-based vaccine that supports antigen expression and immune stimulation and is facilitated by Toll-like receptor 7 (TLR-7). We employed the HDOCK server to perform protein docking against mRNA molecules [50]. This server provides protein–DNA/RNA docking based on a hybrid algorithm of template-based modeling and ab initio free docking. However, a cryo-EM structure of TLR-7 is not currently available in the RCSB protein data bank database. Therefore, we obtained this structure by performing homology modeling using the SWISS-MODEL server at ExPASy and protein sequences acquired from GenBank (NCBI accession no. AAZ99026).
Immune Simulation of Vaccine Construct
Immune responses of the two selected mRNA vaccine constructs (NRM- and SAM-based) were evaluated using in silico immune simulation. An agent-based online immune simulator server, known as C-ImmSim, was employed to perform immune response simulations of our designed mRNA-based vaccine constructs. This server furnished machine learning techniques and a position-specific scoring matrix (PSSM) to predict immune interactions and immune epitopes [51]. With a gap of four weeks, three injections were administered for targeted immune profiling against newly emerging novel coronaviruses. Expression of conserved HLA alleles in the host body was used for immune profiling. Immune analyses were computed at time steps 1, 84, and 168 post-injection (each time step is equal to 8 h and the time at first injection is step 0) [52]. The volume of the immune run simulation was set at 25 µm and the number of simulation steps was 1050.
In Silico Cloning to Understand Translation Efficiency of the Vaccine Candidate
To understand the translation efficiency of our mRNA vaccines candidate, we used the JCat server, which optimizes codon sequence based on default parameters that are crucial to successful recombinant DNA cloning [53]. For cloning, we ligated the proposed NRM vaccine candidate into an appropriate E. coli K12 expression vector by reverse transcribing the construct mRNA sequence into the corresponding complementary DNA (cDNA) sequence using the EMBOSS Backtranseq tool, with codon usage optimized for the E. coli K12 (high) strain. The construct DNA sequence was cloned in silico into the expression vector pET28a (+) (retrieved from the Addgene vector database) using SnapGene software [54].
Results
Retrieval of the S-Protein Sequence
The amino acid sequence of the targeted surface spike glycoprotein of SARS-CoV-2 (PDB ID. 6VYB_C) was retrieved from the NCBI database. This S-protein is 1281 amino acids long and forms a homotrimeric structure. It has two subunits: subunit s1 interacts with the host receptor, while subunit s2 mediates cell membrane fusion for SARS-CoV-2 viral entry. As this protein mediates the fusion and internalization of the virus into host cells, it is a crucial component of viral pathogenicity.
Prediction of CTL Epitopes
CTL epitopes were predicted by screening for antigenicity scores > 1 using the VaxiJen v2.0 server. A total of seven CTL epitopes meeting this criterion, with a length of nine amino acids, were obtained from screening. We next evaluated these epitopes’ allergenicity. Two of the seven epitopes displayed allergenic properties. Thus remaining five CTL epitopes were selected for vaccine incorporation. These CTL epitopes are displayed in Table 1 with their exact antigenic scores and allergenic properties.
Prediction of HTL Epitopes
The IEDB MHC-II-binding server predicted a total of 15 HTL epitopes. Five of these 15 epitopes were deemed non-antigenic by VaxiJen v2.0 analysis. Because of the safety measures, we further screened these 10 antigenic epitopes for allergenicity. Six epitopes emerged as allergenic. Thus, we chose the four remaining HTL epitopes to incorporate into our mRNA vaccine candidate, as these are antigenic and non-allergenic (Table 2).
Prediction of B-Cell Epitopes
The BCPred prediction module identified 21 linear epitopes within the SARS-CoV-2 S-protein [20] amino acids each. Among these, 12 epitopes were found to be antigenic. Three of these 12 B-cell epitopes were found to be both antigenic and non-allergenic. Thus, these three epitopes were selected for vaccine incorporation based on their highest antigenicity (Table 3).
Population Coverage Calculation
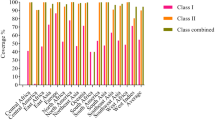
Population coverage assessment was performed to determine the efficacy of epitope recognition by known MHC-I and MHC-II alleles among the world population.
Population coverage for MHC-I was 75.04%. Population coverage results for all peptides binding to MHC-I alleles and their corresponding alleles are shown in Fig. 3. Population coverage for MHC-II alleles was 45.70%, as shown in Fig. 4.

Population coverage of the most promising five epitopes binding with MHC-I alleles of SARS-CoV-2

Population coverage of the most promising four epitopes binding with MHC-II alleles of SARS-CoV-2
Designing of NRM and SAM Vaccine Constructs
Epitopes and their linkers were reverse translated to obtain NRM and SAM vaccine sequences. Their total lengths were 588 and 7098 nucleotides, respectively. We used beta-globin 5′ UTR and the alpha globin 3′ UTR to stabilize our mRNA vaccines [55]. We placed a 5′ m7G cap at the start and a 150 nucleotide poly-A tail at the 3′ end of the mRNA vaccine. The poly-A tail has 120–150 nucleotides in the NRM vaccine construct (Fig. 5) [56,57,58]. SAM-type mRNA vaccines require replicase for self-amplification within the host body. We inserted a codon-optimized replicase gene within the vaccine structure with an EAAAK peptide linker flanking the CDS sequences, as shown in Fig. 6.

Graphical representation of the formulated NRM-based mRNA vaccine construct

Graphical representation of the formulated SAM-based mRNA vaccine construct
Characterization of the Vaccine Construct
Physicochemical characterization of the translated mRNA vaccine constructs was performed as shown in Tables 4 and 5. The molecular weights of the primary peptide sequences encoded by the NRM and SAM molecules were 21.36868 and 272.207 kDa, respectively. The calculated pI’s of NRM- and SAM-encoded proteins were 10.23 and 9.14, respectively. The chemical formulae of the respective NRM- and SAM-encoded primary amino acid chains were C959H1548N272O274S3 and C12330H19448N3246O3508S91. The instability indices of NRM and SAM-encoded proteins were 26.23 and 34.53, respectively. The aliphatic indices of NRM- and SAM-encoded proteins were 78.01 and 99.57, respectively. The GRAVY scores for NRM- and SAM-encoded proteins were observed as − 0.470 and − 0.213, respectively.
The antigenicity features and allergenic potencies of each construct were evaluated using another online server. The antigenicities of NRM- and SAM-encoded proteins were 0.6661 and 0.4684, respectively, and both vaccine constructs were non-allergenic. Both vaccine candidates were water soluble, with scores of 0.850 and 0.120 out of 1 (Table 4).
Molecular Docking of Epitopes to HLA Alleles
Using AutoDock 4, both CTL and HTL epitopes were docked with their corresponding HLA alleles. Binding affinities of epitopes with HLA alleles are shown in Table 6. The docked complex of QIAPGQTGK with the HLA*C-06 molecule is shown in Fig. 7. Molecular docking of other CTL and HTL epitopes with their respective HLA molecules is depicted in Supplementary figure S1.

Molecular docking showing the CTL epitope QIAPGQTGK with HLA*C-06 molecule
Molecular Docking of each mRNA Vaccine with Human TLR-7
Molecular interactions between intracellular TLR-7 and the vaccine mRNAs can potentially be characterized by accurately defining the binding grooves most likely to be activated in bound conformation. The HDOCK program uses a fast and flexible Fourier transform (FFT) search strategy to identify the best binding conformations for RNA binding to the protein, focusing on cavity prediction and shape complementarity. Server-generated output model quality is associated with negative score amplitude. We selected the most significant negative docking score (− 399.51) with 204.64 A0 ligand RMSD, signifying high-affinity binding between the two macromolecules. The complex of the mRNA vaccine docked with TLR-7 is shown in Fig. 8.

Molecular docking of mRNA vaccines with human TLR-7
Immune Response Simulation
We examined simulated immune responses of our mRNA-based vaccine constructs, similar to real immunological phenomena provoked by specific pathogens. The results of these simulations are presented in Figs. 9 and 10. These simulations indicate that the secondary and tertiary responses are much higher than the primary responses. The IgG + IgM antibody concentrations in response to different antigen concentrations were higher than those of other immunoglobins (Figs. 9a, 10a). This server also quantitated the long-lasting B-cell population (cells per mm3) to assess its potential for the development of memory cells to mediate future responses (Figs. 9b, 10b). Elevated levels of active TH (helper) and TC (cytotoxic) cell populations were also observed, together with strong memory development (Figs. 9c–f, 10c–f, respectively). The elevation in cytotoxic T cells reached maxima of 1130 cells per mm3 after 10 days (approx.) of NRM vaccine administration and 1150 cells per mm3 after 8–10 days (approx.) of SAM vaccine administration (Figs. 9e, 10e, respectively). During NRM and SAM mRNA vaccine exposure, steady proliferation was observed among natural killer (NK) cells, together with higher macrophage and dendritic cell activity, as shown in Figs. 9g–i and 10g–i, respectively. Lastly, increased levels of cytokines (IFN-γ and IL-2) were found, as an innate immune response, with lower Simpson index (D), indicating greater diversity of immune response (Figs. 9j, 10j).

In silico immune response simulation of the mRNA vaccine construct. a Elevation of immunoglobulins at different concentrations of antigen, b Population of B lymphocytes (IgM, IgG1, and IgG2) after three injections, c Amount of helper T-lymphocytes population, d Helper T-cell count in the resting and active states, e Amount of Cytotoxic T-lymphocytes population, f Cytotoxic T-lymphocytes population in different states; resting and active, g Population of natural killer cells, h Population of macrophages, i Population of dendritic cells in the active and resting states, j Concentration of cytokines and interleukins with Simpson index

In silico immune response simulation of the SAM-based mRNA vaccine construct. a Elevation of immunoglobulins at different concentrations of antigen, b Population of B lymphocytes (IgM, IgG1, and IgG2) after three injections, c Amount of helper T-lymphocytes population, d Helper T-cell count in the resting and active states, e Amount of Cytotoxic T-lymphocytes population, f Cytotoxic T-lymphocytes population in different states; resting and active, g Population of Natural Killer cells, h Population of Macrophages, i Population of Dendritic cells in the active and resting states, j Concentration of cytokines and interleukins with Simpson index
In Silico Cloning to Understand Translation Efficiency of the Vaccine Candidate
After the vaccination of the mRNA vaccine candidate, we studied the translation process efficiency for expressing the protein. To examine the expression of the mRNA vaccine constructs within a vector-based expression model, we performed in silico cloning. The total length of the cDNA sequence after codon optimization was 594 nts. We inserted XbaI and XhoI restriction sites upstream and downstream, respectively, of the CDS to permit cloning. The recombinant plasmid was constructed by inserting the optimized codon sequence vaccine between XbaI (5026) and XhoI (6318) restriction sites in the pET28a (+) vector, as shown in figure S2. The recombinant vector had a length of 5.728 kbp. This in silico cloning practice substitutes an artificial technique to amplify the proposed mRNA vaccine candidate for its accurate application. In this way, the constructed vaccine can be marked for its probable effectiveness in protecting from SARS-CoV-2 infection.
Discussion
The world is currently facing the continued threat of the COVID-19 pandemic caused by SARS-CoV-2 infection [7, 59, 60]. The disease has spread across 218 nations and territories, reaching every corner globally [59]. Millions of infections and casualties are a major public health concern. The face-to-face transmission mode of the disease has made people socially disconnected. Lockdown in many countries has resulted in the strongest interruption [61]. The spread of this infection must be rendered manageable as early as possible. Only an effective vaccine has the power to eradicate an infectious disease [62].
Vaccines represent the most promising therapeutic modality to control the outbreak and spread of infectious diseases [63, 64]. A vaccine exerts its full potential when it can excite both humoral and cell-mediated immunity. Both memory B and T cells help maintain a defense against future infections by the pathogen [65, 66]. Currently, there are many available routes to developing vaccines against certain diseases [67]. These routes can be broadly classified into two categories: (i) classical and (ii) non-classical. Classical strategies include the preparation of killed or inactivated pathogens, live-attenuated pathogens, and whole-cell vaccines [68]. Non-conventional strategies include the preparation of DNA vaccines, RNA vaccines, peptide vaccines, replicating viral vectors, and non-replicating viral vectors [69]. All these practices have their own pros and cons. We pursued two types of mRNA vaccine formulation strategy in this work: an NRM vaccine candidate lacking a replicase-coding gene sequence and a SAM vaccine candidate encoding a replicase gene upstream of the antigen CDS. These modern strategies are more safe and reliable than conventional practices. In addition, they offer a high expression rate and do not require nuclear localization for functionality, as do DNA vaccines. These vaccine strategies have some limitations, including high susceptibility to RNase and the innate immune system.
Our study adopted mRNA vaccine strategies targeting the SARS-CoV-2 S-protein. The S-protein primary sequence was obtained from NCBI and PDB databases (accession no. 6VYB_C). This protein is present on the outer surface of the virus and mediates viral entry into host cells. CTL, HTL, and B-cell epitopes present within this protein were predicted, analyzed, and selected as vaccine components. Seven highly antigenic epitopes were chosen using the NetCTL 1.2 server. Allergenic analysis eliminated two of these due to predicted allergenic properties. The IEDB server simultaneously provided 15 HTL epitopes, among which 10 were predicted to possess antigenicity. Six of the 10 antigenic epitopes were predicted to exhibit allergenicity. Therefore, only four HTL epitopes were selected for inclusion in our mRNA vaccines against SARS-CoV-2. We selected three B-cell epitopes from among 21 predicted epitopes following the previously described criteria using the BCPred method. All selected epitopes were used for mRNA vaccine formulation. The B-cell epitopes were linked using a GPGPG spacer and a KK spacer was used to link CTL and HTL epitopes. The assembly of epitopes was reverse translated to obtain an mRNA sequence. The 5′ m7G cap, 5′ UTR, 3′ UTR, and poly-A tail were added for expression in human cells. Molecular docking of CTL and HTL epitopes with their corresponding HLA molecules showed effective binding. All bonds between CTL or HTL epitopes and HLA alleles displayed negative bonding energies, indicating spontaneous interactions. Molecular docking between these epitopes and HLA molecules strongly substantiates the utility of the vaccine components.
A further molecular docking study using HDOCK provided a genuine interaction and calculated a docking score of − 399.51, indicating proper interaction with its binding pockets and stimulation of innate immunity via the TLR-7 cascade machinery. The physicochemical parameters, including solubility score and instability index, were found quite satisfactory. The assembled antigen collection encoded by the vaccine candidates is also antigenic and non-allergenic in protein form. This is very important for vaccine safety. In summary, these mRNA vaccines are reliable and safe and most importantly, they are effective against SARS-CoV-2. Our immune simulation study with the designed mRNA vaccine construct yielded promising humoral and cellular immune response outcomes. A server-based immune simulation showed an increase in long-lasting memory B and T cells, indicating the potential of both constructs to serve as increased potency vaccine candidates (Figs. 9, 10).
Furthermore, our in silico cloning model (Figure S2) also revealed the effectiveness of the mRNA vaccine construct along with the recognized epitopes against the SARS-CoV-2 virus. Even though the proposed mRNA vaccine candidate has potent efficiency against the COVID-19 infection, succeeding experimental work is required to validate the actual effectiveness in vivo and in vitro models.
Conclusion
With rigorous and extensive formulation, prediction, and validation, we have developed two different mRNA vaccines against SARS-CoV-2. We developed two mRNA-based vaccine candidates (both NRM and SAM) using the immunoinformatics approach in this work. Further researchers or developers can choose any vaccine candidate from these two options to develop an mRNA-based vaccine. These mRNA vaccines produce a protein chain containing CTL, HTL, and B-cell epitopes linked with flexible peptide spacers. Molecular docking of these epitopes with corresponding HLA molecules suggests good efficacy, prompt recognition, and high binding affinity. These epitopes are antigenic and immune cells can be easily detected, leading to immune system activation. This immune response produces memory B cells and T cells, which may provide prolonged immunity against SARS-CoV-2. Although the vaccine shows promising results via computational analysis, it has yet to be assessed in vitro and in vivo trials prior to human administration.
References
Salgotra, R., Gandomi, M., & Gandomi, A. H. (2020). Evolutionary modelling of the COVID-19 pandemic in fifteen most affected countries. Chaos, Solitons & Fractals, 140, 110118.
Hall, D. C., Jr., & Ji, H.-F. (2020). A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Medicine and Infectious Disease, 35, 101646.
Rahman, M., Thill, J.-C., & Paul, K. C. (2020). COVID-19 pandemic severity, lockdown regimes, and people’s mobility: Early evidence from 88 countries. Sustainability, 12(21), 9101.
Burrell, C. J., Howard, C. R., & Murphy, F. A. (2016). Fenner and White’s medical virology (pp. 1–604). Academic Press Elsevier Science.
Dong, Y., Dai, T., Wei, Y., Zhang, L., Zheng, M., & Zhou, F. (2020). A systematic review of SARS-CoV-2 vaccine candidates. Signal Transduction and Targeted Therapy, 5(1), 1–14.
Harapan, H., Itoh, N., Yufika, A., Winardi, W., Keam, S., Te, H., Megawati, D., Hayati, Z., Wagner, A. L., & Mudatsir, M. (2020). Coronavirus disease 2019 (COVID-19): A literature review. Journal of Infection and Public Health, 13(5), 667–673.
Bhattacharya, M., Sharma, A. R., Ghosh, P., Lee, S.-S., & Chakraborty, C. (2021). A next-generation vaccine candidate using alternative epitopes to protect against wuhan and all significant mutant variants of SARS-CoV-2: An immunoinformatics approach. Aging and Disease. https://doi.org/10.14336/AD.2021.0518
Chakraborty, C., Sharma, A. R., Sharma, G., Bhattacharya, M., & Lee, S.-S. (2020). SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): Diagnostic and proposed therapeutic options. European Review for Medical and Pharmacological Sciences, 24(7), 4016–4026.
Savastano, A., de Opakua, A. I., Rankovic, M., & Zweckstetter, M. (2020). Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nature Communications, 11(1), 1–10.
Cusinato, J., Cau, Y., Calvani, A. M., & Mori, M. (2020). Repurposing drugs for the management of COVID-19. Expert Opinion on Therapeutic Patents, 4(31), 295–307.
Saha, A., Sharma, A. R., Bhattacharya, M., Sharma, G., Lee, S.-S., & Chakraborty, C. (2020). Probable molecular mechanism of remdesivir for the treatment of COVID-19: Need to know more. Archives of Medical Research, 51(6), 585–586.
Rehman, H. M., Mirza, M. U., Ahmad, M. A., Saleem, M., Froeyen, M., Ahmad, S., Gul, R., Alghamdi, H. A., Aslam, M. S., & Sajjad, M. (2020). A Putative prophylactic solution for COVID-19: Development of novel multiepitope vaccine candidate against SARS-COV-2 by comprehensive immunoinformatic and molecular modelling approach. Biology, 9(9), 296.
Tregoning, J. S., Brown, E. S., Cheeseman, H. M., Flight, K. E., Higham, S. L., Lemm, N. M., Pierce, B. F., Stirling, D. C., Wang, Z., & Pollock, K. M. (2020). Vaccines for COVID-19. Clinical & Experimental Immunology, 202(2), 162–192.
Shrotri, M., Swinnen, T., Kampmann, B., & Parker, E. P. (2021). An interactive website tracking COVID-19 vaccine development. The Lancet Global Health, 9(5), e590–e592.
Craven J (2021) COVID-19 vaccine tracker. Regulatory Affairs Professionals Society. Retrieved November 25, 2021, from https://www.raps.org/news-and-articles/news-articles/2020/3/covid-19-vaccine-tracker
Jackson, N. A., Kester, K. E., Casimiro, D., Gurunathan, S., & DeRosa, F. (2020). The promise of mRNA vaccines: A biotech and industrial perspective. npj Vaccines, 5(1), 1–6.
Brito, L. A., Kommareddy, S., Maione, D., Uematsu, Y., Giovani, C., Scorza, F. B., Otten, G. R., Yu, D., Mandl, C. W., Mason, P. W., & Dormitzer, P. R. (2015). Self-amplifying mRNA vaccines. Advances in Genetics, 89, 179–233.
Magini, D., Giovani, C., Mangiavacchi, S., Maccari, S., Cecchi, R., Ulmer, J. B., De Gregorio, E., Geall, A. J., Brazzoli, M., & Bertholet, S. (2016). Self-amplifying mRNA vaccines expressing multiple conserved influenza antigens confer protection against homologous and heterosubtypic viral challenge. PLoS ONE, 11(8), e0161193.
Pardi, N., Hogan, M. J., Porter, F. W., & Weissman, D. (2018). mRNA vaccines—a new era in vaccinology. Nature Reviews Drug Discovery, 17(4), 261.
Chung, M., Bernheim, A., Mei, X., Zhang, N., Huang, M., Zeng, X., Cui, J., Xu, W., Yang, Y., & Fayad, Z. A. (2020). CT imaging features of 2019 novel coronavirus (2019-nCoV). Radiology, 295(1), 202–207.
Walls, A. C., Park, Y.-J., Tortorici, M. A., Wall, A., McGuire, A. T., & Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell, 181(2), 281–292.
Bhattacharya, M., Sharma, A. R., Mallick, B., Sharma, G., Lee, S.-S., & Chakraborty, C. (2020). Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infection, Genetics and Evolution, 85, 104587.
Maruggi, G., Zhang, C., Li, J., Ulmer, J. B., & Yu, D. (2019). mRNA as a transformative technology for vaccine development to control infectious diseases. Molecular Therapy, 27(4), 757–772.
Chetverin, A. B. (2004). Replicable and recombinogenic RNAs. FEBS Letters, 567(1), 35–41.
Probst, J., Weide, B., Scheel, B., Pichler, B., Hoerr, I., Rammensee, H., & Pascolo, S. (2009). Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Therapy, 16(5), 706.
Pascolo, S. (2006). Vaccination with messenger RNA. In W. M. Saltzman, H. Shen, & J. L. Brandsma (Eds.), DNA vaccines. Methods in molecular medicine™. (Vol. 127). Humana Press.
Probst, J., Weide, B., Scheel, B., Pichler, B., Hoerr, I., Rammensee, H., & Pascolo, S. (2007). Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Therapy, 14(15), 1175–1180.
Adhikari, U. K., Tayebi, M., & Rahman, M. M. (2018). Immunoinformatics approach for epitope-based peptide vaccine design and active site prediction against polyprotein of emerging oropouche virus. Journal of Immunology Research, 6718083, 1–22.
Kazi, A., Chuah, C., Majeed, A. B. A., Leow, C. H., Lim, B. H., & Leow, C. Y. (2018). Current progress of immunoinformatics approach harnessed for cellular-and antibody-dependent vaccine design. Pathogens and Global Health, 112(3), 123–131.
Dhal, A. K., Pani, A., Mahapatra, R. K., & Yun, S.-I. (2019). An immunoinformatics approach for design and validation of multi-subunit vaccine against Cryptosporidium parvum. Immunobiology, 224(6), 747–757.
Khan, M., Khan, S., Ali, A., Akbar, H., Sayaf, A. M., Khan, A., & Wei, D.-Q. (2019). Immunoinformatics approaches to explore Helicobacter Pylori proteome (virulence factors) to design B and T cell multi-epitope subunit vaccine. Scientific Reports, 9(1), 1–13.
Chakraborty, C., Sharma, A. R., Bhattacharya, M., Sharma, G., & Lee, S.-S. (2021). Immunoinformatics approach for the identification and characterization of T cell and B cell epitopes towards the peptide-based vaccine against SARS-CoV-2. Archives of Medical Research, 52(4), 362–370.
Kumar Pandey, R., Ojha, R., Mishra, A., & Kumar Prajapati, V. (2018). Designing B-and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. Journal of Cellular Biochemistry, 119(9), 7631–7642.
Larsen, M. V., Lundegaard, C., Lamberth, K., Buus, S., Lund, O., & Nielsen, M. (2007). Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics, 8(1), 424.
Baruah, V., & Bose, S. (2020). Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. Journal of Medical Virology, 92(5), 495–500.
Doytchinova, I. A., & Flower, D. R. (2007). VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics, 8(1), 4.
Dimitrov, I., Bangov, I., Flower, D. R., & Doytchinova, I. (2014). AllerTOP v. 2—a server for in silico prediction of allergens. Journal of Molecular Modeling, 20(6), 2278.
Scholar, E. (2015). Xpharm: The comprehensive pharmacology reference (pp. 1–10). Elsevier Inc.
Vita, R., Mahajan, S., Overton, J. A., Dhanda, S. K., Martini, S., Cantrell, J. R., Wheeler, D. K., Sette, A., & Peters, B. (2019). The immune epitope database (IEDB): 2018 update. Nucleic Acids Research, 47(D1), D339–D343.
Saha, S., & Raghava, G. P. S. (2004). BcePred: Prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties. In G. Nicosia, V. Cutello, P. J. Bentley, & J. Timmis (Eds.), Artificial immune systems. (Vol. 3239). Springer.
El-Manzalawy, Y., Dobbs, D., & Honavar, V. (2008). Predicting linear B-cell epitopes using string kernels. Journal of Molecular Recognition, 21(4), 243–255.
Zhang, Q., Wang, P., Kim, Y., Haste-Andersen, P., Beaver, J., Bourne, P. E., Bui, H.-H., Buus, S., Frankild, S., Greenbaum, J., & Lund, O. (2008). Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Research, 36(suppl_2), W513–W518.
McWilliam, H., Li, W., Uludag, M., Squizzato, S., Park, Y. M., Buso, N., Cowley, A. P., & Lopez, R. (2013). Analysis tool web services from the EMBL-EBI. Nucleic Acids Research, 41(W1), W597–W600.
Hebditch, M., Carballo-Amador, M. A., Charonis, S., Curtis, R., & Warwicker, J. (2017). Protein–sol: A web tool for predicting protein solubility from sequence. Bioinformatics, 33(19), 3098–3100.
Kouranov, A., Xie, L., de la Cruz, J., Chen, L., Westbrook, J., Bourne, P. E., & Berman, H. M. (2006). The RCSB PDB information portal for structural genomics. Nucleic Acids Research, 34(suppl_1), D302–D305.
Baú, D., Martin, A. J., Mooney, C., Vullo, A., Walsh, I., & Pollastri, G. (2006). Distill: A suite of web servers for the prediction of one-, two-and three-dimensional structural features of proteins. BMC Bioinformatics, 7(1), 402–450.
Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461.
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791.
DeLano, W. L. (2002). PyMOL: An open-source molecular graphics tool. CCP4 Newsletter on Protein Crystallography, 40(1), 82–92.
Yan, Y., Tao, H., He, J., & Huang, S.-Y. (2020). The HDOCK server for integrated protein–protein docking. Nature Protocols, 15(5), 1829–1852.
Rapin, N., Lund, O., Bernaschi, M., & Castiglione, F. (2010). Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE, 5(4), e9862.
Saha, R., Ghosh, P., & Burra, V. P. (2021). Designing a next generation multi-epitope based peptide vaccine candidate against SARS-CoV-2 using computational approaches. 3 Biotech, 11(2), 1–14.
Grote, A., Hiller, K., Scheer, M., Münch, R., Nörtemann, B., Hempel, D. C., & Jahn, D. (2005). JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Research, 33(suppl_2), W526–W531.
Biotech GJUsc (2015) SnapGene software (from Insightful Science; available at snapgene.com)
Wang, Z., Day, N., Trifillis, P., & Kiledjian, M. (1999). An mRNA stability complex functions with poly (A)-binding protein to stabilize mRNA in vitro. Molecular and Cellular Biology, 19(7), 4552–4560.
Bernstein, P., Peltz, S., & Ross, J. (1989). The poly (A)-poly (A)-binding protein complex is a major determinant of mRNA stability in vitro. Molecular and Cellular Biology, 9(2), 659–670.
Holtkamp, S., Kreiter, S., Selmi, A., Simon, P., Koslowski, M., Huber, C., Oz, T., & Sahin, U. (2006). Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood, 108(13), 4009–4017.
Mockey, M., Gonçalves, C., Dupuy, F. P., Lemoine, F. M., Pichon, C., & Midoux, P. (2006). mRNA transfection of dendritic cells: Synergistic effect of ARCA mRNA capping with poly (A) chains in cis and in trans for a high protein expression level. Biochemical and Biophysical Research Communications, 340(4), 1062–1068.
Sarkar, B., Ullah, M. A., Araf, Y., & Rahman, M. S. (2020). Engineering a novel subunit vaccine against SARS-CoV-2 by exploring immunoinformatics approach. Informatics in Medicine Unlocked, 21, 100478.
Bhattacharya, M., Sharma, A. R., Patra, P., Ghosh, P., Sharma, G., Patra, B. C., Saha, R. P., Lee, S.-S., & Chakraborty, C. (2020). A SARS-CoV-2 vaccine candidate: In-silico cloning and validation. Informatics in Medicine Unlocked, 20, 100394.
Elfiky, A. A. (2020). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure and Dynamics. https://doi.org/10.1080/07391102.2020.1761882
Samad, A., Ahammad, F., Nain, Z., Alam, R., Imon, R. R., Hasan, M., & Rahman, M. S. (2020). Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach. Journal of Biomolecular Structure and Dynamics. https://doi.org/10.1080/07391102.2020.1792347
Dhama, K., Sharun, K., Tiwari, R., Dadar, M., Malik, Y. S., Singh, K. P., & Chaicumpa, W. (2020). COVID-19, an emerging coronavirus infection: Advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Human Vaccines & Immunotherapeutics, 16(6), 1232–1238.
Zhang, C., Maruggi, G., Shan, H., & Li, J. (2019). Advances in mRNA vaccines for infectious diseases. Frontiers in Immunology, 10, 594. https://doi.org/10.3389/fimmu.2019.00594
Porto, P. S., Anjos, D., Dábilla, N., da Fonseca, S. G., & Souza, M. (2020). Immunoinformatic construction of an adenovirus-based modular vaccine platform and its application in the design of a SARS-CoV-2 vaccine. Infection, Genetics and Evolution, 85, 104489.
Bhattacharya, M., Sharma, A. R., Patra, P., Ghosh, P., Sharma, G., Patra, B. C., Lee, S. S., & Chakraborty, C. (2020). Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. Jounal of Medical Virology, 92(6), 618–631.
Solanki, V., Tiwari, M., & Tiwari, V. (2019). Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Scientific Reports, 9(1), 1–19.
van Riel, D., & de Wit, E. (2020). Next-generation vaccine platforms for COVID-19. Nature Materials, 19(8), 810–812.
de Queiroz, N. M. G., Marinho, F. V., Chagas, M. A., Leite, L. C., Homan, E. J., de Magalhães, M. T., & Oliveira, S. C. (2020). Vaccines for COVID-19: Perspectives from nucleic acid vaccines to BCG as delivery vector system. Microbes and Infection, 22(10), 515–524.
Acknowledgements
This research was supported by the Hallym University Research Fund and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1C1C1008694 & NRF-2020R1I1A3074575).
Author information
Authors and Affiliations
Contributions
MB, ARS, and CC performed the data collection, formal analysis, and manuscript draft writing; ARS, PG, PP, and BCP drafted the final version of manuscript; SS and CC supervised the whole work. All authors contributed substantially to the manuscript and approved the final manuscript for submission. All authors are responsible for the integrity, accuracy, and presentation of the data.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Informed Consent
Not applicable.
Research Involving Human and Animal Participants
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12033_2021_432_MOESM1_ESM.tif
Supplementary file1 (TIF 836 KB)—Molecular docking showing the CTL and HTL epitopes with corresponding HLA molecules (A–H for different epitopes)
Rights and permissions
About this article

Cite this article
Bhattacharya, M., Sharma, A.R., Ghosh, P. et al. Bioengineering of Novel Non-Replicating mRNA (NRM) and Self-Amplifying mRNA (SAM) Vaccine Candidates Against SARS-CoV-2 Using Immunoinformatics Approach. Mol Biotechnol 64, 510–525 (2022). https://doi.org/10.1007/s12033-021-00432-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-021-00432-6