
Abstract
A number of molecular biology techniques are available to generate variants from a particular start gene for eventual protein expression. We discuss the basic principles of these methods in a repertoire that may be used to achieve the elemental steps in protein engineering. These include site-directed, deletion and insertion mutagenesis. We provide detailed case studies, drawn from our own experiences, packaged together with conceptual discussions and include an analysis of the techniques presented with regards to their uses in protein engineering.






Similar content being viewed by others



Abbreviations
- PCR:
-
Polymerase Chain Reaction
References
Paez, J. G., Janne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., et al. (2004). EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science, 304, 1497–1500.
Kobayashi, S., Boggon, T. J., Dayaram, T., Janne, P. A., Kocher, O., Meyerson, M., et al. (2005). EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. New England Journal of Medicine, 352, 786–792.
Yun, C. H., Boggon, T. J., Li, Y., Woo, M. S., Greulich, H., Meyerson, M., et al. (2007). Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell, 11, 217–227.
Neagu, M. R., Ziegler, P., Pertel, T., Strambio-De-Castillia, C., Grutter, C., Martinetti, G., et al. (2009). Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. Journal of Clinical Investigation, 119, 3035–3047.
Lu, Q. (2005). Seamless cloning and gene fusion. Trends in Biotechnology, 23, 199–207.
Higuchi, R., Krummel, B., & Saiki, R. K. (1988). A general method of in vitro preparation and specific mutagenesis of DNA fragments: Study of protein and DNA interactions. Nucleic Acids Research, 16, 7351–7367.
Horton, R. M., Hunt, H. D., Ho, S. N., Pullen, J. K., & Pease, L. R. (1989). Engineering hybrid genes without the use of restriction enzymes: Gene splicing by overlap extension. Gene, 77, 61–68.
Kammann, M., Laufs, J., Schell, J., & Gronenborn, B. (1989). Rapid insertional mutagenesis of DNA by polymerase chain reaction (PCR). Nucleic Acids Research, 17, 5404.
Sarkar, G., & Sommer, S. S. (1990). The “megaprimer” method of site-directed mutagenesis. Biotechniques, 8, 404–407.
Lai, R., Bekessy, A., Chen, C.C., Walsh, T., & Barnard, R. (2003) Megaprimer mutagenesis using very long primers. Biotechniques, 34, 52–54, 56.
Tyagi, R., Lai, R., & Duggleby, R. G. (2004). A new approach to ‘megaprimer’ polymerase chain reaction mutagenesis without an intermediate gel purification step. BMC Biotechnology, 4, 2.
Ochman, H., Gerber, A. S., & Hartl, D. L. (1988). Genetic applications of an inverse polymerase chain reaction. Genetics, 120, 621–623.
Triglia, T., Peterson, M. G., & Kemp, D. J. (1988). A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Research, 16, 8186.
Braman, J., Papworth, C., & Greener, A. (1996). Site-directed mutagenesis using double-stranded plasmid DNA templates. Methods in Molecular Biology, 57, 31–44.
Wang, W., & Malcolm, B. A. (1999). Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques, 26, 680–682.
Geiser, M., Cebe, R., Drewello, D. & Schmitz, R. (2001) Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques, 31, 88–90, 92.
Clark, J. M. (1988). Novel non-templated nucleotide addition reactions catalyzed by procaryotic and eucaryotic DNA polymerases. Nucleic Acids Research, 16, 9677–9686.
Mole, S. E., Iggo, R. D., & Lane, D. P. (1989). Using the polymerase chain reaction to modify expression plasmids for epitope mapping. Nucleic Acids Research, 17, 3319.
Adereth, Y., Champion, K.J., Hsu, T. & Dammai, V. (2005) Site-directed mutagenesis using Pfu DNA polymerase and T4 DNA ligase. Biotechniques, 38, 864, 866, 868.
Aslanidis, C., & de Jong, P. J. (1990). Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Research, 18, 6069–6074.
Cabrita, L. D., Dai, W., & Bottomley, S. P. (2006). A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC Biotechnology, 6, 12.
Yang, Y. S., Watson, W. J., Tucker, P. W., & Capra, J. D. (1993). Construction of recombinant DNA by exonuclease recession. Nucleic Acids Research, 21(8), 1889–1893.
Donahue, W. F., Turczyk, B. M., & Jarrell, K. A. (2002). Rapid gene cloning using terminator primers and modular vectors. Nucleic Acids Research, 30, e95.
Bracchi-Ricard, V., Barik, S., Delvecchio, C., Doerig, C., Chakrabarti, R., & Chakrabarti, D. (2000). PfPK6, a novel cyclin-dependent kinase/mitogen-activated protein kinase-related protein kinase from Plasmodium falciparum. Biochemical Journal, 347(Pt 1), 255–263.
Atamas, S. P., Choi, J., Yurovsky, V. V., & White, B. (1996). An alternative splice variant of human IL-4, IL-4 delta 2, inhibits IL-4-stimulated T cell proliferation. Journal of Immunology, 156, 435–441.
Ledru, E., Fevrier, M., Lecoeur, H., Garcia, S., Boullier, S., Gougeon, M. L. (2003). A nonsecreted variant of interleukin-4 is associated with apoptosis: Implication for the T helper-2 polarization in HIV infection. Blood, 101, 3102–3105.
Goulder, P. J., Phillips, R. E., Colbert, R. A., McAdam, S., Ogg, G., Nowak, M. A., et al. (1997). Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nature Medicine, 3, 212–217.
Martinez-Picado, J., Prado, J. G., Fry, E. E., Pfafferott, K., Leslie, A., Chetty, S., et al. (2006). Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. Journal of Virology, 80, 3617–3623.
Leslie, A. J., Pfafferott, K. J., Chetty, P., Draenert, R., Addo, M. M., Feeney, M., et al. (2004). HIV evolution: CTL escape mutation and reversion after transmission. Nature Medicine, 10, 282–289.
Chopera, D. R., Woodman, Z., Mlisana, K., Mlotshwa, M., Martin, D. P., Seoighe, C., et al. (2008). Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog, 4, e1000033.
Brockman, M. A., Schneidewind, A., Lahaie, M., Schmidt, A., Miura, T., Desouza, I., et al. (2007). Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. Journal of Virology, 81, 12608–12618.
Acknowledgements
S. Barichievy is supported by a Sydney Brenner Fellowship.
Conflict of Interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendices
Appendices
Box 1: The Generation of the EGFR Kinase Domain Deletion Mutant Del 746-750
Introduction
The EGFR kinase domain deletion mutation, in which residues with numbers ranging from 746 to 750 (residues ‘ELREA’) are deleted, is one of a number of mutations that have been identified in patients of non-small-cell lung cancer (NSCLC) and has been associated with the greatest rate of patient survival of all the known EGFR kinase domain mutations [2, 3]. We shall refer to this deletion mutant as Del746-750.
Primer Design
The double-stranded sequence of the EGFR kinase domain used in our studies to generate the Del746-750 mutant using overlap extension is shown below (rendered using DNAMAN software). The colour-code and design of the primers are identical to that in Fig. 1a. The specific primer sequences are as follows:


Experimental Protocol
Titration of Template Plasmid (Step 1 in Fig. 1a)
The PCR reactions were set up as shown in Table 2. Template/parental plasmid dilutions were prepared and amplified at 0.01, 0.1, 0.5, 1, 10 and 50 ng/μL.
The PCR cycle was as follows:
-
95 °C for 2 min
-
25 cycles of the following steps:
-
95 °C for 1 min 30 s
-
50 °C for 1 min
-
72 °C for 1 min
-
-
72 °C for 10 min
Agarose analysis of the 12 PCR reactions is shown Fig. 6.

Titration of parental plasmid in Step 1 PCR reactions
Lanes 1 and 16 contain the O’GeneRulerTM 1 kb DNA ladder from Fermentas (www.fermentas.com) with molecular weight sizes labelled in base pairs. Lanes 3–8 are PCR reactions with primers: Fow_outer and Rev_inner. Lanes 9–14 are PCR reactions with primers: Fow_inner and Rev_outer. Lanes 3–9 used 0.01 ng of parental plasmid DNA, lanes 4 and 10 used 0.1 ng, lanes 5 and 11 used 0.5 ng, lanes 6 and 12 used 1 ng, lanes 7 and 13 used 10 ng and lanes 8 and 14 used 50 ng of parental plasmid DNA. Reactions in lanes 5 and 11 (0.5 ng of template) were carried forward to Step 2 (overlap extension reaction).
Overlap Extension Reaction (Step 2 of Fig. 1a) and Rescue PCR (Step 3 of Fig. 1a)
The overlap extension PCR was set up as shown in Table 3. A 10-μL aliquot (20% of reaction volume) of lanes 5 and 11 was carried over to the overlap extension reaction.
The annealing temperature of the overlap extension reaction has to be lowered sufficiently to allow hybridisation of the overhangs. The temperature range 30–37 °C is usually sufficient, but melting temperatures of the overlap regions can be calculated and employed to select the annealing temperatures.
The PCR cycle was as follows:
-
95 °C for 2 min
-
10 cycles of the following steps:
-
95 °C for 1 min 30 s
-
30 °C for 1 min
-
72 °C for 2 min
-
-
72 °C for 10 min
The rescue PCR is set up as follows: a 20-μL aliquot of the 100-μL overlap extension reaction (from Step 2 above) is set aside for Agarose electrophoresis analysis. The remaining 80 μL is divided into two aliquots and each added separately to a 60-μL PCR reaction mix using the set up shown in Table 4. Importantly, division of the previous PCR reaction ensures dilution of the PCR primers used in the previous step.
The PCR cycle used was as follows:
-
95 °C for 2 min
-
25 cycles of the following steps:
-
95 °C for 1 min 30 s
-
50 °C for 1 min
-
72 °C for 1 min
-
-
72 °C for 10 min
Agarose gel electrophoresis analyses of 5 μL aliquots from Step 1 PCRs, the 20 μL aliquot from Step 2 (overlap extension) and 5 μL aliquot from Step 3 (rescue) are shown in Fig. 7.

Agarose analyses of all the overlap extension reactions (Step 1–Step 3)
Lanes 1 and 8 contain the O’GeneRulerTM 1 kb DNA ladder. Lane 3 is the PCR reaction using primers: Fow_outer and Rev_inner. Lane 4 is the PCR reaction using primers: Fow_inner and Rev_outer (Step 1 Fig. 1a). Lane 5 is the overlap extension PCR reaction (Step 2), and Lane 6 is the rescue PCR reaction (Step 3).
Notably, while an amplicon was detected in lane 5 (indicating the desired deletion mutant had been generated), the rescue PCR (Lane 6) did not show an expected increase in product yield.
TOP TIP: When the expected amplicon from the overlap extension reaction has been confirmed, the remaining sample should be similarly electrophoresed, purified from the gel and T-A cloned into a highly efficient system (such as the pGEM-T Easy kit from Promega). We followed this method to confirm the generation of Del746-750.
Variations in the above protocol can be included to limit ‘carry-over contamination’ from Step 1 PCRs. We explored one variation in which the Step 1 PCRs were purified, treated with Exonuclease I (NEB, www.neb.com) to degrade primers, heat-treated to inactivate the Exonuclease I enzyme (80 °C, 20 min) and purified and aliquots used in the overlap extension reaction. Rescue PCR can also be subsequently performed. We have found that degradation of primers from Step 1 PCR reactions by Exonuclease I treatment improves the yield and quality of Step 3 rescue PCR reactions (Fig. 8). However, the inclusion of Exonuclease I treatment can form part of trouble-shooting exercises for difficult overlap extension reactions and not routinely included.

The impact of Exonuclease I treatment on Step 3 (rescue) PCR
Lane 1 contains the O’GeneRulerTM 1 kb DNA ladder, while Lane 2 is empty. Lanes 3 and 4 contain the overlap extension PCR amplicon (Step 2 Fig. 1a). Lane 5 contains the PCR reaction using primers: Fow_inner & Rev_outer (Step 1 Fig. 1a). Lane 6 contains the PCR reaction using primers Fow_outer & Rev_inner. In this protocol, Step 1 PCR reactions were purified, treated with Exonuclease I and aliquots used in the overlap extension PCR reactions (Step 2).
The prominent points from the example described above are as follows:
-
(i)
When using this method, it is good practice to titrate the amount of template plasmid in each of the PCR reactions in Step 1. Titration of plasmid involves the identification of the lowest concentration of template that generates a visible band when 10% of the reaction is analysed by agarose electrophoresis. If the PCR conditions do not yield a single target band, gradient PCR using 50 ng of template to determine the optimal annealing temperature can be performed. Thereafter, the concentration of template plasmid in each of the PCR reactions in Step 1 can be optimised. This information is also useful for rescue PCR reactions or if low yields in the overlap extension PCR are obtained, or if one opts not to use the T-A-cloning system, but clone directly into the expression vector. Sub-cloning into pGEM-T Easy has the advantage that it quickly yields the clone of interest although this introduces further cloning to move the gene variant of interest to the target expression vector or shuttle vector (in the case of insect cell expression).
-
(ii)
Typically, 20 % by volume (e.g. 10 μL from a 50 μL PCR reaction) of each of the two PCR reactions in Step 1 is carried over to the Step 2 overlap extension leading to carry-over contamination. This is particularly relevant with inner primers as they are unwanted and deleterious in Step 3 PCR. One strategy that is commonly employed to minimise the effect of carry-over contamination (with Step 3 PCR in-mind) is to carry over smaller volumes of PCR reactions into subsequent steps and/or to dilute carry-over volumes by performing PCR in larger volumes. Another approach is to clean-up the reaction after the overlap extension in Step 2 and digest the carry-over primers by treatment with Exonuclease I. The exonuclease enzyme is heat inactivated before aliquots are cleaned-up and used in Step 3 rescue PCR. We generally advise against performing Step 3 in cases where the overlap extension gives the desired product visible by Agarose analysis and recommend the use of pGEM-T Easy cloning thereafter. However, it is not uncommon to perform ‘blind-cloning’—whereby the entire Step 2 reaction is loaded on a gel and the gel is cut in the area where a product of the expected size would migrate, purified and then cloned using an efficient vector such as the pGEM-T Easy system. This option can be pursued with or without the rescue PCRs of Step 3.
-
(iii)
Further minimisation of parental gene contamination can be achieved by digestion of the parental DNA by Dpn I enzyme before Step 3 PCR reactions. This requires the parental plasmid to be propagated in E. coli cell lines that constitutively express the enzyme deacetyl-methyltransferase (dam+ cell lines). Complete digestion of parental plasmids with Dpn I is a recurrent requirement in this review and is further described in Box 7.
Box 2: The Generation of a Deletion Variant of PfPK6, a Serine–Threonine Kinase from Plasmodium falciparum
Introduction
PfPK6 is a single-domain serine/threonine kinase from Plasmodium falciparum that bears sequence resemblance to both mitogen-activated protein kinases (MAPK) and cyclin-dependent kinases (CDK) [24]. We identified and targeted for deletion a 13-residue charged loop in the PfPK6 sequence that was likely to be mediating aggregation of purified PfPK6 protein. Homology modelling studies identified this stretch of 13 residues as a surface-exposed loop. We deleted these residues (DIEDKLKGENLEN) and designated the variant ∆PfPK6.
Primer Design
Using DNAMAN software, we identified the relevant double-stranded sequence of PfPK6 and designed mega-primers to generate ∆PfPK6. The colour-code and design of the primers are identical to that discussed in Fig. 1b.
-
(i)
The strategy in Fig. 1b of the main text has been slightly modified in that the outer reverse primer, Rev_outer, has been moved further upstream in the gene. That is, Fow_outer and Rev_outer do not span the entire length of the target gene.
-
(ii)
There is a SacI restriction site (GAGCTC) upstream of Rev_outer that is employed to achieve cloning of ∆PfPK6 into pGEX-6P-1 via digestion of the parental pGEX-PfPK6 construct with the same set of enzymes (BamHI and Sac I, the BamHI site is introduced as an overhang in the Fow_outer primer).
-
(iii)
Fow_outer:
 The BamHI site is highlighted in bold.
The BamHI site is highlighted in bold. -
(iv)
Rev_inner:

-
(v)
Rev_outer:

-
(vi)
There is a BstNI site (CCWGG—underlined in red in the sequence below) in the DNA sequence targeted for deletion. This forms the basis for a strategy to screen for clones in which deletion of the target sequence has been achieved. However, discussion of this topic is beyond the scope of this review.
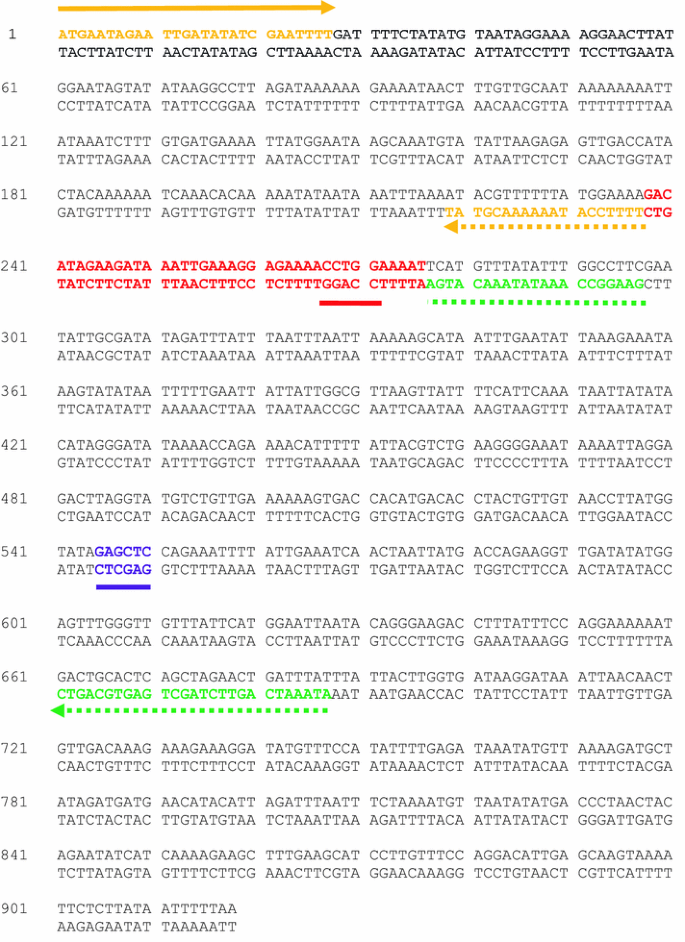
 The BamHI site is highlighted in bold.
The BamHI site is highlighted in bold.


Experimental Protocol
Production of the mega-primer was set up as shown in Table 5.
The PCR cycle was as follows:
-
95 °C for 2 min
-
30 cycles of the following steps:
-
95 °C for 1 min 30 s
-
50 °C for 1 min
-
72 °C for 1 min
-
-
72 °C for 10 min
Agarose gel electrophoresis analysis of the PCR reactions is shown in Fig. 9.

Generation of mega-primers in Step 1 PCR reaction
Lane 1 contains DNA ladder with molecular weight sizes labelled in base pairs. Lanes 2–8 are PCR reactions with primers: Fow_outer and Rev_inner (mega-primer generation reactions). The mega-primer corresponds to the bottom band (~200 bp) in Lanes 2–8.
Production of the Quasi Full-Length ∆PfPK6 (Step 2 & 3 Reactions in Fig. 1b)
As noted previously, Rev_outer has been shifted further upstream into the PfPK6 sequence. The reactions using Rev_outer therefore generate a quasi full-length version that can be cloned into appropriately digested parental plasmid using restriction sites located within or upstream of the Rev_outer primer-binding site (such as SacI).
Following the Step 1 PCR reaction, the mega-primer is purified from the gel and mixed with Rev_outer in a subsequent PCR as follows (Table 6):
The PCR cycle was as follows:
-
95 °C for 2 min
-
30 cycles of the following steps:
-
95 °C for 1 min 30 s
-
50 °C for 1 min
-
72 °C for 1 min
-
-
72 °C for 10 min
Agarose gel electrophoresis analysis of the PCR reactions is shown in Fig. 10.

Generation of the quasi full-length ∆PfPK6 construct in Step 2 & 3 PCR reactions
Lane 1 contains DNA ladder with molecular weight sizes labelled in base pairs. Lanes 2–8 are PCR reactions with the mega-primer and Rev_outer. The quasi full-length ∆PfPK6 corresponds to the bottom band (~650 bp) in Lanes 2–8.
The lower band of the two bright bands between 500 and 1000 bp in Fig. 10 was purified, and the recovered DNA was further analysed by restriction analysis. Double digestion of the quasi full-length ∆PfPK6 with BamHI & Sac I should generate an ~545-bp fragment for cloning into BamHI & Sac I-digested parental plasmid (pGEX-PfPK6).
The reaction in Step 3 is a rescue PCR reaction performed using Fow_outer and Rev_outer. It was not necessary to perform rescue PCR in this study.
Box 3: The Generation of an IL4∆2 Deletion Mutant by Inverse PCR of the IL4 Gene
Introduction
Interleukin-4 (IL-4) is an anti-inflammatory cytokine normally associated with the T-helper type 2 (Th2) immune response. Recently, an IL4∆2 (IL4 delta 2) splice variant of, and natural antagonist to IL4 was identified [25, 26]. IL4∆2 is a deletion mutant of IL4 in which residues at position number 46–61 inclusive (residues ‘TLCTELTVTDIFAASK’) are deleted. This stretch of residues is encoded by exon 2 in the mature wild-type IL4 mRNA.
Primer Design
Below is the double-stranded sequence of the entire IL4 cDNA sequence (rendered in DNAMAN software) with the inverse PCR primers included. Note that these have to be phosphorylated (P) at the 5′ end to facilitate efficient blunt-end ligation of the plasmid after deletion of the unwanted sequence (exon 2—shown in red). The colour-code and design of the primers are identical to that discussed in Fig. 1c of the main text.

Experimental Protocol
Inverse PCR Amplification of Template Plasmid (Step 1 of Fig. 1c)
The template plasmid we used, pPR30, is an in-house shuttle vector that targets protein expression in insect cell systems. The vector is ~6.3 kbp and contains an ampicillin resistance gene for antibiotic selection in E. coli. We used 100 ng (although 10–200 ng can be used) of template plasmid for the PCR reaction using a high fidelity DNA polymerase as shown in Table 7. The enzyme of choice here is Kapa HiFi from Kapa Biosystems (www.kapabiosystems.com):
The PCR cycle was as follows:
-
95 °C for 2 min
-
30 cycles of the following steps:
-
95 °C for 1 min 30 s
-
69 °C for 1 min
-
72 °C for 6 min
-
-
72 °C for 10 min
Agarose gel electrophoresis analysis of IL-4 and IL4∆2 is shown in Fig. 11.

Agarose analyses of inverse PCR reactions
Lane 1 is the O’GeneRulerTM 1 kb DNA ladder. Lanes 3 and 4 are duplicate inverse PCR reactions with primers Inv_PCR1 and Inv_PCR2 to generate the deletion mutant IL4Δ2.
Ligase-Mediated Circularisation of Amplicons (Step 2 of Fig. 1c)
Following electrophoresis analysis, the inverse PCR amplicon was purified using a commercial DNA purification kit (Zymogen DNA Clean and Concentrator-25 kit) according to the manufacturer’s instructions. Ligation reactions were set up using 100 ng of total DNA substrate as shown in Table 8.
The ligation reaction was incubated at 16 °C overnight and the entire aliquot was utilised to transform 50 μL of JM109 competent cells (Invitrogen). After this, 940 μL of SOC media was added, and cells were incubated with shaking at 37 °C for 90 min. The entire 1-mL culture was plated in 100 μL aliquots on ten Ampicillin-containing agar plates and incubated overnight 37 °C. A total of 70 colonies were obtained from 1 mL of culture (plated over 10 plates), six were sent for sequencing, and three contained the correct mutation deletion giving a success rate of 50%.
Additional notes
Our protocol omits DpnI digestion of the parental plasmid as we still get the desired clones with high success rates. This affords a simpler, quicker and cheaper protocol for inverse PCR-based protein engineering. However, this simpler protocol has only been tested in cases where the yields of inverse PCR reactions are high. DpnI digestion might be essential in cases where the yields of inverse PCR reaction are low and the experimenter does not wish to spend time optimising the PCR reaction. One can assign an inverse PCR reaction as high yield or otherwise on the basis of the combination of Agarose analyses, and the ratio of the amount of DNA harvested per 50 μL PCR reaction to the amount of template used in the reaction (here 100 ng). This gives an indication of the enrichment for the desired product versus the parental material. The ratio in our protocol is typically in the range from 45:1 to 55:1.
If transformation of E. coli cells with 100 ng of amplicon does not yield a sufficiently high number of colonies, then higher concentrations of PCR product may be used. To illustrate this, we ligated 2 μg of amplicon from the IL4∆2 reaction (in a 10 μL ligation reaction) and transformed the entire aliquot. A total of 476 colonies were obtained across 10 plates. Furthermore, supplementation of the plates with D-glucose (1 g/L) increases the colony yield possibly by providing a ready source of carbon to stressed cells post-heat-shock.
Box 4: The Generation of HIV-1 p24 gag T242N and H219Q/T242N Mutants Using Circular Mutagenesis
Introduction
The HIV-1 p24 capsid region is highly conserved, and immune evasion mutations in this sequence may have a fitness cost on viral replication [27, 28]. One of the well-known mutations that occurs in vivo is the T242N mutation in helix six of this protein [28–30]. This mutation is selected for in infected individuals which carry the HLA-B*57/B*5801 alleles and is not targeted by their cytotoxic T lymphocytes. Secondary mutations upstream of site 242 partially restore the fitness cost of the T242N mutation [29, 31]. To elucidate the structural and functional mechanisms associated with the T242N mutation and the H219Q compensatory mutation, there is need to introduce the mutations into HIV-1 infectious molecular clones (IMCs) and carry out in vitro analyses. In this experiment, we used circular mutagenesis to perform site-directed mutagenesis (SDM) to generate the T242N and the T242N/H219Q mutants. Two important considerations are
-
(a)
The PCR cycle number must be kept low (≤16 cycles)
-
(b)
A proof-reading polymerase that has no strand-displacement activity under reaction conditions must be used, such as Pfu polymerase.
Primer Design
The double-stranded sequence of the full-length subtype C consensus gag gene is shown below. The bold underlined (black) characters represent start-and-stop codons. The consensus sequence was synthesised by Integrated DNA Technologies (IDT) for use on SDM experiments as follows:
-
(i)
The synthetic DNA was amplified using Gag-BssHIIF and Gag-XhoIR primers and subsequently cloned into pGEM-T Easy vector (Promega).
-
(ii)
SDM to generate the T242N and T242N/H219Q mutants was carried out using primer sets T242NF/T242NR and NQF/NQR, respectively, and the Platinum Taq DNA Polymerase High Fidelity kit (Invitrogen). The mutated codons are in purple, and the mutated residues are underlined (in bold type). The primer design used here is shown in Panel B of Fig. S4 in the Supplementary Materials.


Experimental Protocol (Table 9)
The PCR cycle for the amplification of the gag gene was as follows:
-
95 °C for 5 min
-
30 cycles of the following steps:
-
95 °C for 20 s
-
56 °C for 30 s
-
68 °C for 1.5 min
-
-
68 °C for 7 min
The PCR cycle for the mutagenesis reactions was as follows:
-
95 °C for 5 min
-
16 cycles of the following steps:
-
95 °C for 20 s
-
68 °C for 6 min
-
-
68 °C for 20 min
Agarose gel analyses of the mutagenesis PCR reactions is shown in Fig. 12.

PCR products from the reaction for site-directed mutagenesis of the gag gene by the circular mutagenesis method
Lane 1 contains the O’GeneRulerTM 1 kb DNA ladder (Fermentas, www.fermentas.com). Relevant molecular weight sizes are labelled in base pairs. Lanes 2 and 3 are the T242N and T242N/H219Q mutants, respectively (8 μL each). The pGEM-T Easy vector with the gag gene is ~4.5 kb.
Following the mutagenesis PCR reactions, DNA was purified using the MinElute PCR Purification kit (Qiagen) and digested using FastDigest DpnI (Fermentas) to remove template DNA. The low number of PCR cycles results in low yields. When no visible band is obtained from the PCR reactions, one may still proceed to do ‘blind transformations’ after DpnI digestion of the purified PCR reactions.
Transformation of Circular Mutagenesis PCR Products into E. coli (Step 2 of Fig. 1d)
Following removal of template DNA by DpnI digestion, DNA was purified with the MinElute PCR purification kit (Qiagen). Chemically competent JM109 E. coli cells were transformed with 5 μL of the purified DNA. Ten colonies were picked and grown in overnight cultures followed by plasmid miniprep. The plasmids were sequenced using an ABI PRISM dye terminator cycle-sequencing kit (Applied Biosysytems). Sequencing analysis revealed that seven out the 10 clones (70%) contained the desired mutations.
Notably, high success rates were achieved here without the need for commercial circular mutagenesis systems such as QuickChangeII (Stratagene). Furthermore, if the gel-purification step after the circular mutagenesis PCR is omitted, then the method yields the same number of desired mutants.
Box 5: Cloning of the CYP123 Gene from Mycobacterium tuberculosis into the Expression Vector pMD004 by Exonuclease Recession Method (i) (Operational Sequence-Dependent Exonuclease Recession, OSDER)
Introduction
CYP123 is a putative cytochrome P450 from Mycobacterium tuberculosis (Mtb). The CYP123 ORF (MTCY369.11c) was assigned as a CYP450-like sequence via electronic annotation of the sequenced Mtb genome. The ordered locus names for CYP123 have been assigned as Rv0766c and MT0790. We amplified the CYP123 gene from heat-inactivated Mtb cells and cloned it in-frame in the in-house expression vector pMD004 for the expression and characterisation of CYP123 recombinant protein.
Primer Design
The vector pMD004 is an ~5-kb in-house expression vector used for recombinant protein expression in bacterial cells. Below are the double stranded sequences of the multiple cloning site of pMD004 and of CYP123. Primers were designed to achieve the insertion of the CYP123 gene into pMD004 at the NotI site of the vector (5′-GCGGCCGC-3′ underlined in the pMD004 sequence). A HindIII site (5′-AAGCTT-3′) is also part of the pMD004 multiple cloning site and is underlined.
The CYP123 gene-specific parts of the primers are shown in green, including the second (additional) stop codon in green. The upstream stretch of sequence encodes the TEV protease recognition site (5′-GAA AAC CTG TAT TTT CAG GGC GCC-3′) which we appended to the original (wild-type) CYP123 gene by using PCR.
The primers ‘Fow_Add-Op-to-CYP123’ and ‘Rev_Add-Op-to-CYP123’ were designed to generate a version of the CYP123 gene that has the operational sequences for exonuclease recession-mediated cloning. The operational sequences are shown in red in the two primers.

The sequence flanking the pMD004 multiple cloning site is shown below with the vector-specific parts of the primers shown in purple. The primers ‘Fow_Add-Op-to-pMD004’ and ‘Rev_Add-Op-to-pMD004’ were designed to generate a linear version of the pMD004 expression vector that has the operational sequences for exonuclease recession-mediated cloning of the CYP123 gene (shown in red in the two primers).

Notes:
-
(i)
The bracketed operational sequence DNA residue highlighted in bold-type indicates the site at which T4 polymerase-mediated exonuclease recession stops due to inclusion of the correct (complementary) dNTP in the reaction mix.
-
(ii)
The operational sequences appended to the primers ‘Fow_Add-Op-to-pMD004’ and ‘Rev_Add-Op-to-CYP123’ are complementary to each other up to the exonuclease recession stop residue. The same relationship applies for the primers ‘Fow_Add-Op-to-CYP123’ and ‘Rev_Add-Op-to-pMD004’.
-
(iii)
The addition of two extra bases (GG) in the primer ‘Fow_Add-Op-to-CYP123’ ensures that the CYP123 gene sequence is fused in-frame into the pMD004 vector. These extra bases result in the addition of a Glycine codon (GGG) immediately upstream of the CYP123 gene.
Fow_Add-Op-to-pMD004
The primer ‘Fow_Add-Op-to-pMD004’ has the following architecture:
Substitution with the appropriate sequences gives the following:
Rev_Add-Op-to-pMD004
The primer ‘Rev_Add-Op-to-pMD004’ has the following architecture:
Substitution with the appropriate sequences gives the following:
The final primer sequence is as follows:
Fow_Add-Op-to-CYP123
The primer ‘Fow_Add-Op-to-CYP123’ has the following architecture:
Substitution with the appropriate sequences gives the following:
Rev_Add-Op-to-CYP123
The primer ‘Rev_Add-Op-to-CYP123’ has the following architecture:
Substitution with the appropriate sequences gives the following:
The final primer sequence is as follows:
Experimental Protocol
Amplification of pMD004 Vector
The PCR reactions were set up as shown in Table 10.
The PCR cycle was as follows:
-
95 °C for 2 min
-
30 cycles of the following steps:
-
95 °C for 1 min 30 s
-
69 °C for 1 min
-
72 °C for 6 min
-
-
72 °C for 10 min
Amplification of CYP123 Insert
The PCR reactions were set up as shown in Table 11.
The PCR cycle was as follows:
-
95 °C for 2 min
-
30 cycles of the following steps:
-
95 °C for 1 min 30 s
-
69 °C for 1 min
-
72 °C for 6 min
-
-
72 °C for 10 min
The PCR reactions were purified using the DNA Clean and Concentrator-25 kit from Zymo Research and subsequently DpnI-digested to remove parental plasmid. These were gel-purified using the Zymoclean Gel DNA Recovery kit from Zymo Research and the DNA concentration measured by spectrophotometry.
CYP123 recession reaction
The recession reactions were set up as shown in Table 12.
pMD004 Recession Reaction
The recession reactions were set up as shown in Table 13.
The above recession reactions were incubated at 22 °C for 60 min followed by T4 polymerase enzyme denaturation at 75 °C for 20 min.
Annealing
1 μL of 1 mM EDTA was added to the recessed vector and insert before annealing. Equal concentrations (weight/weight) of insert and vector were then added as shown in Table 14.
The annealing reactions were incubated at 22 °C for 1 h.
The entire annealing reaction (10 μL) was transformed into chemically competent E. coli cells and plated onto Ampicillin LB-Agar plates. Typically, five colonies per plate were picked, processed and screened for the relevant insert by double-digestion with NotI and HindIII (Fig. 13).

NotI/HindIII double digest screen of CYP123 insert
Lane 1 contains the O’GeneRulerTM 1 kb DNA ladder from Fermentas. Lane 2 is empty. Lanes 3–7 are NotI/HindIII double-digested plasmids with all five colonies yielding the expected insert size (~1200 bp).
Box 6: Cloning of the Glutathione-S-transferase (GST) Gene into the In-house Shuttle/Transfer Vector pPR30 by Exonuclease Recession Method (ii) (Seamless Exonuclease Recession, SER)
Introduction
Glutathione-S-transferase is commonly fused on to recombinant proteins to facilitate their purification by affinity chromatography. This exploits the highly specific affinity interaction between GST and the glutathione ligand. In commercial affinity purification systems, glutathione is immobilised on inert resin such as Sepharose and packed into columns. GST-fusion proteins bound to these columns after application of clarified cell extract are released by elution with free glutathione. In this study, we created a GST-fusion shuttle/transfer vector to enable the expression of GST-fusion proteins in insect cells using the in-house shuttle vector pPR30.
Primer Design
The pPR30 in-house shuttle/transfer vector is ~6 kb. Below is a portion of the double-stranded sequence where the GST gene is to be inserted. The inverse PCR primers designed to remove the unwanted BCCP (Biotin Carboxy Carrier Protein) sequence are highlighted in blue. Notably, the inverse PCR primers are not 5′-phosphorylated as blunt-end ligation is not used. Instead, a recombinant plasmid is built via ligation-independent cloning using exonuclease recession. The colour-scheme and design of the primers is identical to that discussed in Step 1a of Fig. 3 in the main text.

Below is the double-stranded GST gene sequence in the expression vector pGEX-6P-1 (GE Healthcare). The sequences highlighted in yellow correspond to GST gene-specific parts of the primer. The colour-code and design of the primers is identical to that discussed in Step 1b of Fig. 3 in the main text.

pPR30_inv_1

pPR30_inv_2

GST_fuse_Fow
The primer ‘GST_fuse_Fow’ has the following architecture:
Substitution with the appropriate DNA sequences gives the following:
GST_fuse_Rev
The primer ‘GST_fuse_Fow’ has the following architecture:
Substitution with the appropriate DNA sequences gives the following:
The final primer sequence is as follows:
Experimental Protocol
The experimental methods below were adapted from Yang et al. [22] although our protocol omits the ‘gap-filling’ polymerisation reaction.
Amplification and Linearisation of pPR30 (Step 1a Fig. 3)
The inverse PCR reaction for linearisation and amplification of pPR30 is shown in Table 15.
Amplification of GST (Step 1b of Fig. 3)
The PCR reaction for amplification of the GST gene from the vector pGEX-6P-1 is shown in Table 16.
The PCR cycles for both GST and pPR30 amplifications were as follows:
-
95 °C for 2 min
-
30 cycles of the following steps:
-
95 °C for 1 min 30 s
-
63 °C for 1 min
-
72 °C for 5 min
-
-
72 °C for 10 min
The PCR reactions were purified using the DNA Clean and Concentrator-25 kit from Zymo Research and parental plasmid removed by DpnI digestion. Following this, DpnI-digested PCR reactions were gel-purified to remove dNTPs and primers, and subjected to exonuclease recession.
Exonuclease Recession and Annealing Reactions (Step 2 of Fig. 3)
Unlike in exonuclease recession method (i) (OSDER), the vector and insert are added to the same reaction mix during SER. This means that extra liquid-handling is not required after the recession reaction.
The experimental recession reaction is shown in Table 17.
The control reaction (no insert) is shown in Table 18.
The recession and annealing reaction conditions were as follows:
-
Recession reaction step: 25 °C for 5 min
-
Denaturation step (T4 polymerase heat inactivation): 70 °C for 10 min
-
Annealing step: 30 °C for 30 min
Half of the experimental reaction (10 μL) was transformed into chemically competent E. coli cells and plated onto Ampicillin LB-Agar plates. Colonies were analysed for the correct insert by PCR and sequencing.
Box 7: Tips on Executing Efficient DpnI Digestion
Introduction
Efficient DpnI digestion to remove parental plasmid DNA, assuming the parental plasmid has been propagated in E. coli strains that constitutively express the enzyme deacetyl-methyltransferase (assigned as dam+ E. coli cells), is an important step in most of the protocols described here. We have dedicated this Box to the description of an optimal DpnI digestion protocol that we have successfully used for both family I and II methods.
Experimental Protocol
-
(a)
We have compared DpnI digestion systems from various manufacturers and found the FastDigest DpnI digestion system from Fermentas (www.fermentas.com) to be the most efficient and reproducible.
-
(b)
We digest a maximum of 2 μg of total DNA in 50 μL reactions only. Reduced DpnI digestion performance with larger amounts of total DNA occurs, yielding higher colony backgrounds in negative controls. Spectrophotometric quantification of DNA before and after DpnI digestion helps us to monitor the quality of the digestion reaction—for example, it can easily indicate if non-specific digestion (star activity) has occurred.
-
(c)
DpnI digestions are incubated for a maximum of 60 min as longer periods result in non-specific degradation of DNA. Conversely, premature termination of digestion (incubation for less than 30 min) results in incomplete digestion of parental plasmid and higher backgrounds in negative controls after transformation into E. coli cells.
A typical DpnI digestion reaction is shown in Table 19.
The above reaction is incubated for 60 min at 37 °C followed by enzyme denaturation at 80 °C for 5 min. The heat-denatured reactions are cooled and purified using a commercial DNA purification kit (e.g. Zymogen DNA Clean and Concentrator-25 kit) according to the manufacturer’s instructions.
Box 8: Terminator Primer Design for the Cloning of the IL-4 Gene into the Expression Vector pGEX-6P-1
Introduction
IL-4 is an anti-inflammatory cytokine normally associated with T-helper type 2 (Th2) immune responses [25, 26]. We describe the design of terminator primers for the rapid gene cloning of the IL-4 gene into the expression vector pGEX-6P-1.
Primer Design
We describe the design of RNA/DNA chimeric primers having three consecutive ribonucleotide bases that terminate complementary-strand DNA synthesis to generate the single-stranded overhangs that permit ligation-independent cloning. The plasmid pGEX-6P-1 is an ~5 kb expression vector used for recombinant protein expression in E. coli. The section below is a double-stranded portion of the pGEX-6P-1 multiple cloning site. We used this sequence of the vector to design chimeric RNA/DNA amplification terminator primers. Following amplification with these primers, a linear version of the pGEX-6P-1 vector was generated with long single-stranded overhangs.
Note: the primer ‘Rev_Term_pGEX’ is shown in red. It is designed based on the bottom strand of pGEX-6P-1. The primer ‘Fow_Term_pGEX’ is shown in green. It is designed based on the top strand of pGEX-6P-1. The bases indicated in purple are ribonucleotides, and those in red are deoxy-ribonucleotides.

Rev_Term_pGEX

The 5’ to 3’ orientation of the primer is as follows:
Fow_Term_pGEX

The double-stranded IL-4 gene sequence is shown below. Note that in the design of the IL-4 terminator amplification primers, the pGEX-6P-1-specific regions (5′ to 3′ highlighted in blue and 3′ to 5′ highlighted in turquoise above) are appended to the corresponding IL4-specific sequences (highlighted in yellow). These form part of the overhangs in the forward (blue) and reverse (turquoise) primer for amplification of IL4 to permit seamless fusion of the IL-4 gene to the pGEX-6P-1 vector.

Fow_Term_IL4
The ‘Fow_Term_IL4’ primer for amplification of IL-4 has the following architecture:
Substitution with the appropriate DNA sequences gives the following:
The final primer sequence is as follows:
Note that the rNTP terminator codon translates to alanine and maintains the pGEX-6P-1 reading frame.
Rev_Term_IL4
The primer ‘Rev_Term_IL4’ for the amplification of IL-4 has the following architecture:
Substitution with the appropriate DNA sequences gives the following:
The final primer sequence is as follows:
Rights and permissions
About this article
Cite this article
Zawaira, A., Pooran, A., Barichievy, S. et al. A Discussion of Molecular Biology Methods for Protein Engineering. Mol Biotechnol 51, 67–102 (2012). https://doi.org/10.1007/s12033-011-9448-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-011-9448-9