
Abstract
The use of high stringency selection systems often results in the induction of very few recombinant mammalian cell lines, which limits the ability to isolate a cell line with favorable characteristics. The employment of for instance STAR elements in DNA constructs elevates the induced number of colonies and also the protein expression levels in these colonies. Here, we describe a method to systematically identify genomic DNA elements that are able to induce many stably transfected mammalian cell lines. We isolated genomic DNA fragments upstream from the human Rb1 and p73 gene loci and cloned them around an expression cassette that contains a very stringent selection marker. Due to the stringency of the selection marker, hardly any colony survives without flanking DNA elements. We tested fourteen ~3500 bp DNA stretches from the Rb1 and p73 loci. Only two ~3500 bp long DNA fragments, called Rb1E and Rb1F, induced many colonies in the context of the stringent selection system and these colonies displayed high protein expression levels. Functional analysis showed that the Rb1 DNA fragments contained no enhancer, promoter, or STAR activity. Our data show the potential of a methodology to identify novel gene expression augmenting DNA elements in an unbiased manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mammalian cell cultures are a predominant vehicle for the production of proteins at an industrial scale. Production methods range from transient, but large-scale, high efficiency transfections of cell cultures [1–3] to the establishment of stable cell lines that are subsequently grown in large-scale reactors [4, 5]. In the last case, identification of clones that couple high protein expression to good growth conditions is pivotal. There are many ways to achieve these characteristics, ranging from optimization of cell culture conditions [6, 7] to the employment of DNA elements that augment gene expression [8–17]. Here, we describe a method to identify DNA elements that positively influence gene expression levels, resulting in a high number of stably transfected colonies that display high protein expression levels.
Many of the known DNA elements that augment exogenous (trans)gene expression levels have been found on a premeditated basis. For instance, Matrix Attachment Regions (MARs) are thought to be involved in the large-scale organization of chromatin loops. As such they are envisioned to protect exogenous genes against all sorts of neighboring influences from the surrounding chromatin [18, 19]. Likewise, insulators that block the interaction between a gene and enhancers are also envisioned to shield an exogenous (trans)gene against unwanted influences from the surrounding chromatin [11]. Finally, we previously identified genomic sequences that are thought to block the activity of chromatin repressors such as Polycomb group proteins and heterochromatin-associated repressors such as HP1. We called these genomic sequences STAR elements and application of these elements in gene expression constructs resulted in increased protein expression levels [9].
In contrast, not many DNA elements that augment gene expression levels have been identified on a non-premeditated basis. In one approach, the surrounding DNA of a high protein expressing CHO cell clone has been analyzed for DNA elements that convey the increases in gene expression levels. Two DNA elements were identified in this fashion and they were called EASE (for expression augmenting sequence element). The two elements shared a large array of HMG (High Mobility Group) protein binding sites [20, 21]. Although this methodology is in potency very powerful, the identification of such DNA elements in the chromatin that surrounds the randomly inserted (trans)gene can still be tedious. Furthermore, the question remains how such DNA elements will behave when applied to a gene expression construct that is subsequently transfected and inserted into another, random site of the genome. In another approach, the Chinese hamster elongation factor 1α (EF-1α) promoter, together with 5′ and 3′ flanking sequences of the EF-1α gene was used to drive expression of a gene of interest, resulting in high expression levels [22].
Here, we followed a different approach. Previously, we created a very stringent selection system. This system is based on the hampered translation efficiency of a selection marker, such as the Zeocin resistance gene [23–25]. In short, when the translation efficiency of the Zeocin marker mRNA is low, high mRNA levels are needed to obtain sufficient Zeocin resistance protein levels for a cell to survive. Since the Zeocin resistance gene is coupled to a gene of interest in such a way that a bicistronic mRNA is created, high levels of this mRNA also imply high translation of the protein of interest. In order to achieve such high bicistronic mRNA expression levels, we need to flank the constructs with STAR elements. Without such elements, the mRNA expression levels are simply too low to obtain sufficient Zeocin resistance protein and as a consequence, cells die. In fact, when used in a specific consuration; a TTG Zeocin selection marker, driven by the human β-actin promoter, will hardly induce any colony when transfected to CHO-DG44 cells.
Since the background level of colony numbers in CHO-DG44 is almost zero, we reasoned that this could be the basis of an unbiased screen to identify DNA elements that positively influence the establishment of colony formation and concomitantly gene expression. That is, without a premeditated thought on how such DNA elements might function. In principle, it could be a screen with randomly cloned genomic DNA. Since this might be an inefficient methodology, we pre-selected the non-coding regions surrounding the promoters of two genes that are essential for the homeostasis of mammalian cells. We analyzed two genomic loci, retinoblastoma 1 (Rb1) and p73, both being involved in growth regulation of the cell. Out of ~50,000 bp tested genomic DNA, we identified two stretches of ~3500 bp genomic Rb1 DNA that induce more colonies than STAR elements in the context of the same stringent selection system. Furthermore, these colonies displayed at least as high protein expression levels as colonies induced by STAR elements. Analyzes showed that the Rb1 DNA fragments are not a STAR element, enhancer, or likewise. The DNA elements retained their positive effects in the context of multiple reporter proteins, as well as in serum-free suspension cell cultures. Overall, the results demonstrate the feasibility of a methodology to identify DNA elements that positively influence protein expression levels on a non-premeditated basis.
Materials and Methods
Vector Constructions
For cloning the stretches of DNA, we used our STAR-Select vector [25] and replaced the CMV promoter by the human β-actin promoter. STAR elements 7 and 67 (5′) and 7 (3′) were replaced by stretches of DNA originating from the Rb1 and p73 loci of approximately the same length (~3500 bp). The elements were isolated by PCR using BAC clones as template. The numbers of these BAC clones were, respectively, RP11-136N2 and RP5-1092A11 for Rb1 and p73. Primer sequences used for these PCRs (in 5′ → 3′ orientation; F: forward; R: reverse):
RB1 Z | F | ggagcgtctgcagaatggtgacagg | R | agactctcgctctgttgccaggctg |
RB1 A | F | ctgaaggagtctcaaactgaagagag | R | acaaagagtctggtgggtgactgtg |
RB1 B | F | tgtttgcattcctgtagcccacaag | R | cgttctaaaaagccttccttcaaag |
RB1 C | F | gtgatgtaaatctttgcaattcttc | R | tcttaatggcttgatgagccacac |
RB1 D | F | tagtcttttgtatgtgataaatctc | R | taccattcaattctcccgtctgac |
RB1 E | F | gcccaccctaaatacttatacaggc | R | acaccccaggaacagaatcagtgc |
RB1 F | F | actatgtcatttttgctaacatgtaatgg | R | gctattcactcattcctgtagctgtctaat |
E (1450–3500) | F | aaagggcccaatgaatgtccaatattcc | R | acaccccaggaacagaatcagtgc |
E (1–2000) | F | gcccaccctaaatacttatacaggc | R | gcaagagcctgacagctaagcatcag |
E (1–1500) | F | gcccaccctaaatacttatacaggc | R | caaaaggaatattggacattcattgggcc |
E (1–1000) | F | gcccaccctaaatacttatacaggc | R | ccacatcccactcagacctaccttt |
E (1–500) | F | gcccaccctaaatacttatacaggc | R | gagtcatggaaactgcaggctgtga |
E (500–2000) | F | tcacagcctgcagtttccatgactc | R | gcaagagcctgacagctaagcatcag |
E (1000–2000) | F | aaaggtaggtctgagtgggatgtgg | R | gcaagagcctgacagctaagcatcag |
E (1500–2000) | F | ggcccaatgaatgtccaatattccttttg | R | gcaagagcctgacagctaagcatcag |
F (1–2500) | F | actatgtcatttttgctaacatgtaatgg | R | ggaaacacaatggctaagaccaaatggctag |
F (2500–3500) | F | ctagccatttggtcttagccattgtgtttcc | R | gctattcactcattcctgtagctgtctaat |
F (2500–3500) | F | ctagccatttggtcttagccattgtgtttcc | R | gcaagagcctgacagctaagcatcag |
/E (1–2000) | ||||
p73 Z | F | gcaccacgtttgagcacctctggag | R | cagttttccagggggcactcagagc |
p73 A | F | tgtgatttggaataaaacctccctgaagagg | R | gcgggcgttagcgcctttttag |
p73 B | F | ccagacagctatgagcactcagtggact | R | cagggaggttttattccaaatcaca |
p73 C | F | aaatacatttaaaaatctggcagagccggg | R | tgatggagttggatcccagtgtttgg |
p73 D | F | atcaacgccaccgttcttccatgtc | R | cagtgccacctttctcttggttaggatttt |
p73 E | F | tactatcttgggatcattaatggctgcagg | R | caggcatccagttctgagctttctctct |
p73 F | F | cgcgaacagcctcagcttctgaatg | R | ggtgggaaactgctccttcactttgct |
p73 Ai | F | cactgcccacatctcaggcaaacag | R | cgctctgcaggaaggaaggaatga |
p73 Bi | F | ttggaatcgttgatctgctgaaataacagg | R | gatgcatcttccaggacacccctcc |
p73 Ci | F | tcacagggctgagcggctgactccagaaca | R | gaagtggcaaagaaacctggggcc |
A short DNA sequence run was performed on the isolated DNA sequences to verify that we indeed isolated the intended sequences.
For analyzing STAR activity, the selection vector for antirepressor elements with four LexA operator sites [9] was used to clone the Rb1E/F fragments between the LexA operators and the SV40 promoter-driven Zeocin gene. For testing enhancer function, the minimal SV40 promoter was combined with either STAR 7 or Rb1E/F or the SV40 enhancer and placed upstream of the d2EGFP reporter gene. For testing promoter function, STAR 7 and Rb1E/F were placed upstream of the d2EGFP reporter gene.
Cell Culture, Transfection, and Analysis of Clones
CHO-DG44 [26] cells were grown in HamF12:DMEM medium (Invitrogen), supplemented with 4.6% fetal bovine serum (FBS) (Invitrogen), 2 mM glutamine (Invitrogen), 100 U/ml penicillin (Invitrogen),100 μg/ml streptomycin (Invitrogen), 100 μM sodium hypoxanthine (Invitrogen), 16 μM thymidine (Invitrogen), and 10 mM MgCl2 at 37°C/5% CO2.
For transfections, 0.4 × 106 CHO-DG44 cells were seeded in 6-well culture plates 24 h prior to transfection. Cells were transfected with 3 μg of plasmid DNA using LipofectamineTM 2000 (Invitrogen) as described by the manufacturer. In brief, LipofectamineTM 2000 was combined with plasmid DNA at 4 μl/μg DNA. The mixture was added to the cells, which had grown to 70–90% confluence. After 5 h, the transfection mixture was replaced by fresh medium. The following day, cells were seeded in serial dilutions into medium containing Zeocin (Invitrogen) at a concentration of 400 μg/ml. Approximately 12 days after transfection, individual colonies became visible and these were isolated and propagated in 24-well plates in medium containing Zeocin. When grown to ~70% confluence, cells were transferred to 6-well plates. Cells were continued to grow in 6-well plates for another 1–2 weeks before FACS analysis or ELISA was performed. The d2EGFP expression levels were determined on an Epics XL Beckman Coulter flowcytometer. In the case of transient transfections, d2EGFP expression levels were determined 24 h following transfection. Values were visualized using Graphpad Prism 5 for Windows.
Wild-type CHO-DG44-S suspension cells (Invitrogen) were grown in serum-free CD-DG44 medium (Invitrogen) supplemented with 8 mM glutamine (Invitrogen), pluronic acid (Invitrogen), and anti-clumping agent (Invitrogen) at 37°C/8% CO2 on a shaker (130 rpm). Cells were passaged every 2–3 days. Cells were transfected (nucleofected) with an Amaxa Nucleofector, using the Nucleofection-kit Amaxa V, as described by the manufacturer. In brief, culture medium was supplemented with ITS (Invitrogen) and medium was equilibrated in the incubator to adjust pH. For each nucleofection, 1 × 106 wild-type CHO-DG44-S cells, grown to a density between 0.7 × 106 and 1 × 106/ml and with a viability of >90%, were centrifuged in a swing out centrifuge (900 rpm, 5 min). Cell pellets were dissolved in 100 μl nucleofector solution and 5 μg DNA (in a volume of 5 μl) was added. Samples were transferred to a cuvette and electroporation in the Amaxa Nucleofector was performed (using program U-30), after which the samples were transferred to the equilibrated culture medium (in 6-well culture plates). After 5–6 h, the cells were transferred to T25 (suspension) culture flasks, in a total volume of 5 ml. After 48 h, selection was started by adding 50 μg/ml Zeocin to the culture medium. Medium was refreshed every 2–3 days. During the next 3 weeks, the viability of the cells was monitored and, if applicable, d2EGFP expression levels were determined. Three weeks after nucleofection, 5000–10000 viable cells/ml were poured in Semi Solid medium (Genetix), to form subclones. After 10 days, colonies were isolated and transferred to 96-wells culture plates in 100 μl culture medium. After another week, cells could be transferred to 24-wells plates (in 0.5 ml medium). Selected subclones were propagated to grow in T25 culture flasks (in 5 ml medium). After another 2–3 weeks, d2EGFP or hEPO expression levels were determined.
To analyze STAR activity, the RB1E/F fragments, negative control (no DNA fragment), and positive control (STAR 7 fragment) in pSelect were transfected into the U2-OS/Tet-off/LexA-repressor cell line [9], and transfected cells were cultured under hygromycin selection (25 μg/ml) and tetracycline repression (doxycycline, 10 ng/ml) for 1 week (50% confluence). The doxycycline concentration was reduced to 0.1 ng/ml to induce the LexA-repressor HP1 gene, and after 2 days Zeocin was added to 250 μg/ml. The cells were cultured for an additional 3–4 weeks, until the negative control cultures were killed by the Zeocin.
ELISA
For hEPO measurements, equal numbers of CHO-DG44 cells (0.1 × 106) were seeded in 6-well plates 3 days prior to cell counting and collection of the medium. The amount of secreted hEPO was determined using an ELISA kit (Quantikine IVD kit; R&D systems). The hEPO concentration was determined by comparing optical density at 450 nm with that of the known hEPO standard as supplied in the ELISA kit.
Determination of Copy Numbers
Individual CHO-DG44 clones transfected with TTG Zeo d2EGFP containing plasmids were isolated and assayed for d2EGFP expression. Genomic DNA was purified and the copy number of both the d2EGFP gene and the β-actin gene were determined using realtime PCR. The ratio between the d2EGFP and actin signal was taken as the relative copy number.
Results
Identification of Genomic Loci that Induce Many Colonies Under Stringent Selection Criteria
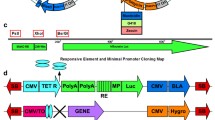
When CHO-DG44 cells are transfected with a plasmid that harbors a stringent selection marker such as the Zeocin resistance marker that is modified at its translation initiation codon, little or no colonies will emerge. This is specifically the case with a Zeocin resistance marker that has a TTG translation initiation codon and that is placed under the control of the human β-actin promoter (Fig. 2). However, when STAR elements are placed to flank the entire expression cassette, many more colonies will emerge, typically in the range of 50–100 CHO-DG44 colonies per transfection, applying the conditions described in experimental protocol (Fig. 2). In general, the resulting clones convey high protein expression levels. Here, we attempted to identify genomic sequences that are able to induce at least as many CHO-DG44 colonies as STAR elements under the same selection conditions. We therefore used the same attenuated Zeocin resistance marker as is used with STAR elements. The expression cassette was placed under control of the human β-actin promoter (Fig. 2). Genomic loci of two human genes were chosen: Rb1 and p73. Stretches of DNA of approximately the same length (~3500 bp) were isolated by PCR using human BAC clones as template. For each locus, we isolated and analyzed six (A–F) ~3500 bp long adjacent DNA stretches upstream from the transcription start site, as well as a ~3500 bp DNA stretch coding region of the genes, encompassing the start of translation in the corresponding mRNA (dubbed Z). Furthermore, we isolated three ~3500 bp long DNA stretches from the intron, downstream of the transcription start site, and direct upstream of the translation start of p73, which we dubbed Ai, Bi, and Ci. A length of 3500 bp was taken to create constructs of similar length as the construct with the combined STAR 7/67/7 elements. The upstream DNA stretches, containing only non-coding DNA, were dubbed A to F (Fig. 1). The particular stretches of DNA were cloned to flank a construct encompassing the human β-actin promoter, the TTG Zeo resistance gene and the d2EGFP reporter gene. A short DNA sequence run was performed on the isolated DNA sequences to verify that we indeed isolated the intended sequence. As control constructs, we took the same construct without any flanking DNA element and the same construct flanked with STARs 7 and 67 upstream of the expression cassette and STAR 7 downstream of the expression cassette (Fig. 2).

Genomic structure of the Rb1 and p73 loci that are screened for fragments that elevate the formation of colonies in the context of a stringent selection system. Shown are the six ~3500 bp DNA stretches upstream from the transcription start site, as well as a ~3500 bp DNA stretch coding region of the genes, encompassing the start of translation in the corresponding mRNA (dubbed Z) for each locus. The six upstream DNA stretches, containing only non-coding DNA, were dubbed A to F. For the p73 locus, also three fragments (Ai to Ci) that are located in the indicated intron are shown

Genomic sequences that induce more colonies than STAR elements in the context of a stringent selection system. a CHO-DG44 cells were transfected with DNA of constructs as shown, using TTG Zeo as selectable marker. For the negative control, there was no sequence introduced as Element X. For the positive control, STAR 7/67 was used as Element X at the 5′ end and STAR 7 is used as Element X at the 3′ end. The different stretches of DNA of Fig. 1 were used as element X as indicated. Approximately 2 weeks after transfection, the number of stably established colonies were counted. b Rb1E and Rb1F fragments induce equal or higher GFP expression levels than STAR elements. d2EGFP expression levels were determined in stable colonies comprising DNA constructs that were flanked with Rb1E, Rb1F, or STAR elements. The relative fluorescence levels were taken as arbitrary units. The average d2EGFP expression levels for each construct are indicated with a line
We transfected equal amounts of the plasmids containing the isolated DNA stretches from the Rb1 and p73 loci to CHO-DG44 cells. Selection was performed with 400 μg/ml Zeocin in the culture medium, which was added 24 h after transfection. After approximately 2 weeks, the number of stably established colonies were counted. As shown in Fig. 2a, transfection of the construct encompassing STAR 7/67/7 resulted in 105 stable colonies. Transfection of seven constructs containing DNA sequences from the p73 locus gave rise to hardly any colonies (Fig. 2a). In contrast, transfection of the constructs containing the DNA sequences from the Rb1 locus gave a significant number of colonies. Specifically, the Rb1E and Rb1F sequences induced 217 and 113 colonies, respectively (Fig. 2a). Since the Rb1E and Rb1F sequences induced ~2.5 and ~1.1 more colonies than STAR 7/67/7 elements, respectively, we decided to focus on these sequences. Analysis of the sequences in databases such as blast revealed that Rb1E and F are intergenic non-coding fragments located between the integral membrane protein 2B (ITM2B) gene and the Rb1 gene on the human chromosome 13. We found no known sequence motifs, promoter regions, or repeats. No duplications of the sequences in the human genome were found either.
The Rb1E and Rb1F Sequences Induce Equal or Higher d2EGFP Expression Values than STAR Elements
Since the constructs that contain the Rb1E and Rb1F sequences also harbor the d2EGFP reporter gene, we were able to analyze the influence of the Rb1E and Rb1F DNA sequences on the d2EGFP expression levels. Up to 24 independent colonies induced by the indicated constructs were isolated. Colonies were propagated before analysis by flow cytometry (FACS), approximately 6 weeks after transfection. The fluorescence signal derived from d2EGFP (destabilized) is linear with d2EGFP expression levels in the cell and is thus a reliable indicator for promoter activity. In a single FACS analysis, fluorescence signals from a sample that contain up to 4000 cells are analyzed. One such sample of cells is taken from an independent, stably transfected cell colony. Since the signal will vary among the individual cells in the colony, the mean fluorescence level of the ~4000 cells in the sample is taken as a measure for the d2EGFP expression level in the stably transfected cell colony. As shown in Fig. 2b, incorporation of the Rb1E and Rb1F sequences induced equal or higher d2EGFP expression levels, as compared to the control construct with the STAR 7/67/7 elements. The average d2EGFP expression level in 24 independent colonies transfected with the Rb1E or Rb1F were 835 and 725, respectively, as compared to the average d2EGFP expression of 615 induced by STARs 7/67/7 (Fig. 2b).
Above-described experiments were performed with the human β-actin promoter that drives the expression cassettes. We tested whether the Rb1E/F elements also operate in a similar way in the context of the CMV promoter. We placed the Rb1E and Rb1F sequences around an expression cassette harboring the CMV promoter, the TTG Zeo selection marker and the d2EGFP reporter gene. The constructs containing Rb1E or Rb1F induced 176 and 107 colonies, as compared to the 152 colonies induced by the STAR 7/67/7 combination (Fig. 2b). Up to 24 independent colonies were isolated, propagated and d2EGFP was analyzed. As shown in Fig. 2b, the Rb1E and Rb1F sequences induced average d2EGFP expression levels of 585 and 478, respectively, as compared to the average d2EGFP expression of 483 induced by STARs 7/67/7 (Fig. 2b).
We conclude that flanking an expression cassette with Rb1E or Rb1F sequences not only induces more colonies, but these colonies also display equal or slightly higher d2EGFP expression level than STAR-induced colonies, either in the context of the human β-actin or the CMV promoter.
We also placed the Rb1E/F element upstream of the β-actin promoter, driving the human erythropoietin (EPO) reporter gene. As shown in Fig. 3, we found that the Rb1E/F element was able to induce large numbers of stable EPO producing colonies (252), as compared to the 124 colonies induced by the STAR 7/67/7 combination. When specific EPO production levels were analyzed in the clones, we found that the Rb1E/F element induced similar EPO expression levels as the STAR 7/67/7 combination (Fig. 3). We conclude that the Rb1E/F element is able to induce a higher number of EPO producing colonies with similar EPO expression. This is the same conclusion as with d2EFP as reporter gene.

The Rb1E fragment also induces more colonies when hEPO is used as reporter gene. CHO-DG44 cells were transfected with DNA of constructs that either contained Rb1E or STAR elements, using TTG Zeo as selectable marker and hEPO as reporter gene. Approximately 2 weeks after transfection, the number of stably established colonies were counted. After another approximately 3 weeks, specific hEPO expression levels were determined in these stable colonies. The average hEPO expression levels (pg/cell/day) for each construct are indicated with a line
The Ability of Rb1E and Rb1F Sequences to Induce High Colony Numbers is Localized
Next, we analyzed different portions of the Rb1E and Rb1F elements for their ability to induce colony formation, as well as the respective d2EGFP values in those colonies. As shown in Fig. 3a, the 1–3500 bp of the Rb1E and Rb1F elements were compared to a range of shorter Rb1E and Rb1F fragments. The Rb1E (1450–3500) fragment was by large dispensable, since it induced hardly any colony. In contrast, the 1–2000 bp portion of Rb1E induced similar colony numbers as the entire 1–3500 bp element (Fig. 4a). When we further dissected the 1–2000 bp fragment we noted a decreasing ability to induce colonies: the 1–1500 bp fragment induced 106 colonies, the 1–1000 and 1–500 bp fragment hardly any. Also, deletion of the first 500 bp (500–2000 bp) showed a decrease in induced colony number. However, this small 1–500 bp fragment itself was not able to induce colonies at all. Therefore, within the 1–2000 bp Rb1E fragment, we could not further pinpoint the exact region that is responsible for inducing colony formation. Any further shortening, 5′ or 3′, of this 1–2000 bp fragment induces fewer colonies.

Testing of regions within Rb1E and Rb1F for highest activity. a Indicated regions of Rb1E and Rb1F, as well as a specific combination of fragments were cloned to flank the TTG Zeo d2EFGP expression cassette and the constructs were transfected to CHO-DG44 cells. Approximately 2 weeks after transfection, the number of stably established, Zeocin resistant colonies were counted, as schematically indicated with bars. b As in a, indicated regions of Rb1E and Rb1F, as well as a specific combination of fragments were cloned to flank the TTG Zeo d2EFGP expression cassette and the constructs were transfected to CHO-DG44 cells. Stably transfected colonies were isolated and propagated, before d2EGFP values were determined. The average d2EGFP expression levels for each construct are indicated with a line
Similarly, we dissected the Rb1F element. As shown in Fig. 4a, the highest potential to induce colony formation resided in the 2500–3500 bp region of the Rb1F fragment. In contrast, the 1–2500 bp region was to a great extent dispensable. Since the 2500–3500 bp region is relatively small, we did not attempt to further delineate this fragment. It is noteworthy that in the endogenous Rb1 locus, the 2500–3500 bp region of Rb1F is located adjacent to the 1–2000 Rb1E region (Fig. 1). We, therefore, joined these two fragments to create Rb1F (2500–3500)/Rb1E (1–2000). This larger fragment, when placed upstream of the human β-actin promoter induced the highest number of colonies in CHO-DG44 (Fig. 4a). The Rb1E and Rb1F elements were chosen randomly to create equal length genomic fragments, but apparently, the activity within the genomic locus that harbors the ability to induce high colony numbers stretches over the border between the Rb1E and Rb1F fragments.
When we analyzed the d2EGFP expression levels in the clones that were induced by the fragments as described above, we noted a mixed picture (Fig. 4b). In all cases, the average d2EGFP values were between 500 and 1000. When, however, the average of the full length Rb1E (1–3500) was arbitrarily put at 100% (shown as line in Fig. 4b), only the Rb1E (1–2000), the Rb1F (1–3500), and the combined Rb1E/F fragments displayed a higher d2EGFP value. When combined with the above-described analysis on colony formation, we conclude that the highest activity within the Rb1E and Rb1F elements is indeed confined to the region overlapping the border of these elements. We, therefore, confined our next analyses to this combined Rb1F (2500–3500)/Rb1E (1–2000) element.
The Rb1E/F Element Is No STAR Element and Does Not Display Promoter or Enhancer Activity
A possible explanation for the ability of the Rb1E/F element to induce a high number of colonies with high d2EGFP expression levels could be that these elements are STAR elements. We directly tested this possibility by placing the element between targeted LexA-HP1 repressors and the SV40 driven Zeocin selection gene. When a genomic element has no STAR activity, the HP1-mediated gene repression will silence the Zeocin selection marker gene [9]. Subsequent addition of Zeocin to the culture medium will result in cell death. On the other hand, when an element does contain STAR activity, the HP1-mediated gene repression is blocked and silencing of the Zeocin selection marker does not occur. Subsequent addition of Zeocin to the culture medium will result in survival of these cells. These experiments were performed in the human U2-OS cells, expressing the LexA-HP1 fusion protein under control of a DOX-off switch, as was the original screen to identify and isolate STAR elements [9]. As shown in Fig. 5, placing STAR 7 between the LexA-HP1 binding sites and the Zeocin marker gene does indeed result in cell survival and fast growing colonies. Placing the Rb1E (1–2000), Rb1F (2500–3500), or the combined Rb1E/F element between the LexA-HP1 binding sites and the Zeocin selection gene did not, however, result in the emergence of viable colonies. We, therefore, conclude that neither the entire Rb1E/F nor the two smaller fragments harbor STAR activity, as is found in the STAR 7 element.

Rb1E and Rb1F elements do not contain STAR activity. Schematically is shown what happens if an element has STAR activity or not. Genomic elements (X) were placed between targeted LexA-HP1 repressors and the Zeocin selection gene. When the elements have no STAR activity, the HP1-mediated gene repression will silence the Zeocin selection marker gene. Subsequent addition of Zeocin to the culture medium will result in cell death. On the other hand, when an element does contain STAR activity, the HP1-mediated gene repression is not strong enough to silence the Zeocin selection marker. Subsequent addition of Zeocin to the culture medium will result in survival of these cells. Figure 4 shows the results on survival of cells when either STAR 7 was inserted as positive control, or with the indicated Rb1E, Rb1F, and the Rb1E/F combination. “++” indicates cell survival and STAR activity, “-” indicates cell death and the lack of STAR activity
Another potential explanation for the ability of the Rb1E/F element to induce a high number of stable colonies and high d2EGFP expression values could be that they harbor a strong CHO-DG44-specific enhancer. We tested this by placing the element upstream of the SV40 minimal promoter and the d2EGFP gene. In the control construct, we placed the SV40 enhancer upstream of the SV40 minimal promoter, which is the natural occurring SV40 enhancer/promoter configuration. Only the construct with the SV40 enhancer/promoter combination induced high transient d2EGFP value (put arbitrarily at 100%, Fig. 5a). In contrast, neither the SV40 minimal promoter alone nor the Rb1E/F or STAR 7 element induced any transient d2EGFP expression (Fig. 6a). This indicates that neither the Rb1E/F element nor the STAR 7 element harbors a functional enhancer.

The Rb1E/F element does not possess enhancer or promoter activity. a Constructs as indicated were made. The Rb1E/F or STAR 7 element was placed upstream of the SV40 minimal promoter and the d2EGFP gene. As control either the SV40 minimal promoter alone, or the SV40 enhancer/minimal promoter combination was placed upstream of the d2EGFP gene. The relative transient d2EGFP values of the different constructs are shown, whereby the value obtained with the SV40 enhancer/minimal promoter combination was put at 100. b Constructs as indicated were made. Either the β-actin promoter, STAR 7 alone or Rb1E/F alone were placed upstream of the d2EGFP gene. We transfected the constructs to CHO-DG44 cells and measured the transient d2EGFP values, 24 h after transfection. The relative transient d2EGFP values of the different constructs are shown, whereby the value obtained with the β-actin promoter-d2EGFP gene combination was put at 100
Finally, we tested whether the Rb1E/F has promoter activity. The construct that contained the β-actin promoter was modified in such a way that the β-actin promoter was replaced by the Rb1E/F or STAR 7 element, while the TTG Zeo was removed. This created constructs that contained the Rb1E/F and STAR 7 elements, placed immediately upstream of the d2EGFP cassette. We transfected the constructs to CHO-DG44 cells and measured the transient d2EGFP values. As shown in Fig. 6b, the constructs with either the Rb1E/F or STAR 7 elements, but without β-actin promoter gave no d2EGFP signal at all. This indicates that the elements are no functional promoters. Instead, the control constructs with the β-actin promoter gave a transient d2EGFP signal (put arbitrarily at 100%) (Fig. 6b). We conclude that the Rb1E/F fragment contains neither STAR, enhancer nor promoter activity.
The Rb1E element induces more colonies than STAR elements and with at least equally high d2EGFP values. One possibility could be that inclusion of the Rb1 fragments might result in stable colonies that have more copies of the plasmid incorporated. We tested this by directly determining the copy numbers of the respective plasmids in independently isolated stable colonies. We isolated DNA from seven clones that were transfected with either STAR 7/67/7 elements or the Rb1E fragment. The average d2EGFP values in the seven STAR-induced colonies was 691.7, and 1056.6 in the seven Rb1E-induced colonies (Fig. 7). The average copy number in STAR-induced colonies was 36, whereas the average copy number in Rb1E-induced colonies was 27 (Fig. 7). It, therefore, appears that the high d2EGFP values induced by the Rb1E fragment are not due to an increased copy number, but that, instead more d2EGFP is produced per copy.

Inclusion of the Rb1E element does not lead to increased copy numbers. CHO-DG44 cells were transfected with DNA of constructs that either contained Rb1E or STAR elements and using TTG Zeo as selectable marker. Seven stably transfected colonies were analyzed for d2EGFP expression levels. Genomic DNA from these clones were isolated and the copy number of the integrated plasmids were determined by real time PCR
The Rb1E/F Element Operates Well in Transfected Serum-Free Suspension Cell Cultures
The above-described results were all obtained with adherent CHO-DG44 cells. Next, we tested whether the Rb1E/F element retained its positive effects when transfected to cells growing in suspension. We, therefore, transfected the construct with the d2EGFP reporter construct to CHO-DG44-S suspension cells. Cells were transfected (nucleofected) in chemically defined medium for DG44 suspension cells, with an Amaxa Nucleofector, and Zeocin selection was started. Three weeks after nucleofection, a stably transfected polyclonal population was poured in semi solid medium, for subcloning. Single colonies were isolated, transferred to 24-wells plates and finally to T25 culture flasks. After another 2–3 weeks, protein expression levels were measured.
The β-actin promoter drove the expression units, either alone, or flanked by the STAR 7/67/7 combination (Fig. 8). Since it is not possible to count independently established stable colonies in suspension cultures, we determined the percentage of green fluorescent cells within the stably transfected polyclonal population as a measure for the effectiveness of the different constructs. As shown in Fig. 8, 2 weeks after transfection the cell culture transfected with the β-actin promoter alone contained only 5% green cells. When the constructs encompassed STAR elements, this percentage of stably transfected green cells increased to 14%, 2 weeks after nucleofection. However, with the Rb1E/F containing construct, the percentage green cells rose to 41%, 2 weeks after nucleofection. This result showed the beneficial effect of the Rb1E/F element on the growth of stably transfected cells. The d2EGFP values were also determined in isolated subclones, which were cultured in T25 flasks for 2–3 weeks (Fig. 8). The average d2EGFP expression values in the three cell lines that survived after transfection with the construct without flanking DNA elements remained very low. In contrast, the average d2EGFP values induced by STAR elements or Rb1E/F were 476 and 974, respectively (Fig. 8). These results show that also in suspension growing CHO-DG44-S cells, the Rb1E/F element is an effective tool to increase the number of gene expressing cells as well as the d2EGFP expression values. This is in particular apparent when compared to the construct without DNA elements, but also when compared to STAR elements, the beneficial effects were significant.

The Rb1E/F element induces high protein expression levels in serum-free suspension medium. Constructs in which the d2EGFP expression cassette was flanked by either STAR 7/67/7 elements or the Rb1E/F combination were transfected to serum-free CHO-DG44-S cells. Transfection and isolation of independent clones are described in the text. Shown are the d2EGFP value in independent subclones. Also the percentage of green cells, 2 weeks after transfection are indicated
Discussion
We have used a very stringent selection system as means to isolate genomic DNA fragments that induce a high number of stable CHO-DG44 colonies. These colonies further display high protein expression levels. Previously, we isolated so-called STAR elements that also augmented gene expression levels. This allowed the use of a high selection stringency for the establishment of stably transfected cell lines. The advantage of selecting cells with high stringency is that most cells that would establish a colony with low to intermediate protein expression levels will not survive. This helps to simplify the laborious and time-consuming procedure for the isolation of high protein expressing cell lines. Unfortunately, the selection system we devised has such high stringency that only very few colonies can survive at all. For instance, when the human β-actin promoter drives the expression of the most stringent TTG Zeo selection marker we used, hardly any colony will form in CHO-DG44 cells. Obviously, this is at the edge of what can be considered useful. However, when STAR elements were included to flank the expression cassette, significantly more colonies were formed. This is due to the ability of STAR elements to elevate gene expression, in this case the expression of the Zeocin selection protein. Although this was an improvement, using this expression platform in serum-free suspension cells still turned out to be on the edge what cells can take. It is very difficult to obtain viable, well growing and high protein expressing suspension cell cultures with the system. Therefore, we were interested in identifying DNA elements that could induce more colonies in the context of the same stringent selection system.
In order to do that, we did not follow an approach based on prior presumptions. Our previous approach to identify STAR elements rested heavily on the assumption that such DNA element would restrict or block heterochromatin-mediated gene repression. Flanking an expression cassette with such elements would have beneficial effects in turns of shielding the reporter gene from gene repression and this would, in turn result in augmented gene expression levels. Although this turned out to be the case, the approach contained many unproven assumptions. To avoid this, we performed a screen that would immediately identify DNA elements that augmented gene expression levels in CHO-DG44 cells. As shown here, we identified a DNA stretch in the genomic region upstream of the Rb1 locus that induced more CHO-DG44 colonies than STAR elements and with equal to higher protein expression levels. The screen was performed in adherent CHO-DG44 cells. The particular ~3000 bp long region that we called Rb1E/F was able to induce more CHO-DG44-S suspension cell lines that also displayed higher protein expression values.
The Rb1E/F fragment was found to be only one among many genomic fragments to display beneficial characteristics. Besides the p73 locus, we screened in a similar way two other genomic loci, without positive results (data not shown). It does not appear that the genome contains a high density of such gene expression augmenting elements. The approach we devised was unbiased in the sense that we did not make any prior assumptions about the nature of potential DNA elements or whether we could identify any DNA element at all. The read-out of the screen was more stably transfected CHO-DG44 colonies in the context of a stringent selection system. As drawback of this approach, we have as yet no idea which mechanism underlies the action of the Rb1E/F fragment and about its function in the endogenous Rb1 locus. We excluded some possibilities, such as that the DNA fragment is no enhancer, promoter or STAR element. Besides that, the DNA fragment is simply operationally defined by its ability to induce many colonies and high protein expression levels.
In principle, the method can also be used for a systematic screen of many more randomly chosen DNA fragments. This would imply the making of a library of plasmids containing random DNA fragments and the large-scale screening and testing in CHO-DG44 cells. Although we attempted this, this approach is less efficient than it would appear. First of all, it is not easy to establish a plasmid library that can efficiently be transfected to CHO-DG44 cells. Given the complexity of the library, the introduced copies per specific DNA fragment will be low and chances are that these are insufficient to induce a large number of colonies. Furthermore, as we also observed with the screen for STAR elements, a surviving colony may contain multiple DNA fragments. Isolation of such DNA fragments from the mammalian cells, re-cloning and testing to search for the responsible DNA fragments was very tedious and inefficient. Although it may appear that isolation by PCR and cloning of >20 DNA fragments that are ~3500 bp long is tedious too, in comparison this was a very straightforward approach. Once colonies were established, there was no doubt which DNA fragment was responsible for the effects. We, therefore, prefer the methodology as we described.
In conclusion, we have presented a method to screen for DNA elements that are able to induce a high number of colonies in CHO-DG44 cells, in the context of a stringent selection system. Both the Rb1E/F fragment we identified and the screening methodology may be useful tools for high-level expression of recombinant proteins in mammalian cells.
References
Chu, L., & Robinson, D. K. (2001). Industrial choices for protein production by large-scale cell culture. Current Opinion in Biotechnology, 12, 180–187.
Wurm, F. M. (2004). Production of recombinant protein therapeutics in cultivated mammalian cells. Nature Biotechnology, 22, 1393–1398.
Birch, J. R., & Racher, A. J. (2006). Antibody production. Advanced Drug Delivery Reviews, 58, 671–685.
Dekker, A. (1990). Continuous cell substrate considerations. In Lubiniecki A. S. (Ed.), Large-scale mammalian cell culture technology. New York: Marcel Dekker.
Xing, Z., Kenty, B. M., Li, Z. J., & Lee, S. S. (2009). Scale-up analysis for a CHO cell culture process in large-scale bioreactors. Biotechnology and Bioengineering, 103, 733–746.
Trummer, E., Fauland, K., Seidinger, S., Schriebl, K., Lattenmayer, C., Kunert, R., et al. (2006). Process parameter shifting: Part I. Effect of DOT, pH, and temperature on the performance of Epo-Fc expressing CHO cells cultivated in controlled batch bioreactors. Biotechnology and Bioengineering, 94, 1033–1044.
Nam, J. H., Zhang, F., Ermonval, M., Linhardt, R. J., & Sharfstein, S. T. (2008). The effects of culture conditions on the glycosylation of secreted human placental alkaline phosphatase produced in Chinese hamster ovary cells. Biotechnology and Bioengineering, 100, 1178–1192.
Kwaks, T. H., & Otte, A. P. (2006). Employing epigenetics to augment the expression of therapeutic proteins in mammalian cells. Trends in Biotechnology, 24, 137–142.
Kwaks, T. H., Barnett, P., Hemrika, W., Siersma, T., Sewalt, R. G. A. B., Satijn, D. P. E., et al. (2003). Identification of anti-repressor elements that confer high and stable protein production in mammalian cells. Nature Biotechnology, 21, 553–558.
Li, Q., Peterson, K. R., Fang, X., & Stamatoyannopoulos, G. (2002). Locus control regions. Blood, 100, 3077–3086.
Gerasimova, T. I., & Corces, V. G. (2001). Chromatin insulators and boundaries: Effects on transcription and nuclear organization. Annual Review of Genetics, 35, 193–208.
Mutskov, V. J., Farrell, C. M., Wade, P. A., Wolffe, A. P., & Felsenfeld, G. (2002). The barrier function of an insulator couples high histone acetylation levels with specific protection of promoter DNA from methylation. Genes and Development, 16, 1540–1554.
Antoniou, M., Harland, L., Mustoe, T., Williams, S., Holdstoeck, J., Yague, E., et al. (2003). Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing. Genomics, 82, 269–279.
Williams, S., Mustoe, T., Mulcahy, T., Griffiths, M., Simpson, D., Antoniou, M., et al. (2005). CpG-island fragments from the HNRPA2B1/CBX3 genomic locus reduce silencing and enhance transgene expression from the hCMV promoter/enhancer in mammalian cells. BMC Biotechnology, 5, 17.
Zahn-Zabal, M., Kobr, M., Girod, P. A., Imhof, M., Chatellard, P., De Jesus, M., et al. (2001). Development of stable cell lines for production or regulated expression using matrix attachment regions. Journal of Biotechnology, 87, 29–42.
Kim, J. M., Kim, J. S., Park, D. H., Kang, H. S., Yoon, J., Baek, K., et al. (2004). Improved recombinant gene expression in CHO cells using matrix attachment regions. Journal of Biotechnology, 107, 95–105.
Girod, P. A., Zahn-Zabal, M., & Mermod, N. (2005). Use of the chicken lysozyme 5′ matrix attachment region to generate high producer CHO cell lines. Biotechnology and Bioengineering, 91, 1–11.
Hancock, R. (2000). A new look at the nuclear matrix. Chromosoma, 109, 219–225.
Heng, H. H., Goetze, S., Ye, C. J., Liu, G., Stevens, J. B., Bremer, S. W., et al. (2004). Chromatin loops are selectively anchored using scaffold/matrix-attachment regions. Journal of Cell Science, 117, 999–1008.
Morris, A. E., Lee, C. C., Hodges, K., Aldrich, T. L., Krantz, C., Smidt, P. S., et al. (1997). Expression augmenting sequence element (EASE) isolated from Chinese hamster ovary cells. In M. J. T. Carrondo, B. Griffiths, & J. L. P. Moreira (Eds.), Animal cell technology (pp. 529–534). Amsterdam: Kluwer Academic Publishers.
Aldrich, T. L., Thomas, J. N., & Morris, A. E. (1998). Improved bicistronic mammalian expression vectors using expression augmenting sequence element (EASE). Cytotechnology, 28, 9–17.
Running Deer, J., & Allison, D. S. (2004). High-level expression of proteins in mammalian cells using transcription regulatory sequences from the Chinese hamster EF-1α gene. Biotechnology Progress, 20, 880–889.
Kozak, M. (1989). Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Molecular and Cellular Biology, 9, 5073–5080.
Kozak, M. (1990). Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proceedings of the National Academy of Sciences of the United States of America, 87, 8301–8305.
Van Blokland, H. J. M., Kwaks, T. H. J., Sewalt, R. G. A. B., Verhees, J. A., Klaren, V. N. A., Siersma, T. K., et al. (2007). A novel, high stringency selection system allows screening of few clones for high protein expression. Journal of Biotechnology, 128, 237–248.
Urlaub, G., Käs, E., Carothers, A. M., & Chasin, L. A. (1983). Deletion of the diploid dihydrofolate reductase locus from cultured mammalian cells. Cell, 33, 405–412.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hoeksema, F., van Blokland, R., Siep, M. et al. The Use of a Stringent Selection System Allows the Identification of DNA Elements that Augment Gene Expression. Mol Biotechnol 48, 19–29 (2011). https://doi.org/10.1007/s12033-010-9344-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-010-9344-8