
Abstract
CD20 is a receptor expressed on B cells with anonymous functions. The receptor is the target of some food and drug administration (FDA) approved monoclonal antibodies (mAb), such as Rituximab and Obinutuzumab. Blocking CD20 using the aforementioned mAbs has improved Non-Hodgkin Lymphoma (NHL) therapy. All commercial mAbs on the market were raised in non-human animal models. Antibody humanization is inevitable to mitigate immune response. In order to keep the affinity of antibody intact, humanizations are only applied to frameworks which do not eliminate immune response to foreign CDRs sequences. To address this issue, human monoclonal antibody deemed imperative. Herein, we report the isolation and characterization of a fully human single-chain variable fragment (scFv) against the large loop of CD20 from naïve human antibody library. After three rounds of phage display, a library of enriched anti-CD20 scFv was obtained. The polyclonal phage ELISA demonstrated that after each round of phage display, the population of anti-CD20 scFv became dominant. The scFv, G7, with the most robust interaction with CD20 was selected for further characterization. The specificity of G7 scFv was evaluated by ELISA, western blot, and flow cytometry. Detecting CD20 in western blot showed that G7 binds to a linear epitope on CD20 large loop. Next, G7 scFv was also bound to Raji cell (CD20+) while no interaction was recorded with K562 cell line (CD20—). This data attested that the epitope recognized by G7 scFv is accessible on the cell membrane. The affinity of G7 scFv was estimated to be 63.41 ± 3.9 nM. Next, the sensitivity was evaluated to be 2 ng/ml. Finally, G7 scFv tertiary structure was modeled using Graylab software. The 3D structure illustrated two domains of variable heavy (VH) and variable light (VL) connected through a linker. Afterward, G7 scFv and CD20 were applied to in-silico docking using ClusPro to illustrate the interaction of G7 with the large loop of CD20. As the selected scFv from the human antibody library is devoid of interspecies immunogenic amino acids sequences, no humanization or any other modifications are required prior to clinical applications.
Similar content being viewed by others
Introduction
One of the most notable markers of B cell malignancies is CD20. This receptor is non-glycosylated expressed in all stages of B cell development, except pro-B cell [1]. This receptor is a member of MS4A (membrane-spanning 4-domain family A) protein family. CD20 spans the cell membrane four times with two extracellular domains [2]. The extracellular domains are composed of one small and one large loop. Both loops can be targeted by monoclonal antibodies in oncotherapy [3, 4]. The function of CD20 is not clear yet. There are some studies indicating that CD20 is co-localized with B cell receptor (BCR) in lipid raft, hence it might play a role in BCR signaling [5, 6]. Even though the expression level of CD20 is highly variable in distinct B-cell malignancies or even between intra-clonal populations, this receptor is an FDA-approved marker for mAb therapy, which has been proved to be very effective approach in treating CD20 + Non-Hodgkin Lymphoma (NHL) [7, 8].
Currently, there are some Food and Drug Administration (FDA)-approved mAb blocking CD20, including Rituximab [9], Ofatumumab [10], Ublituximab [11], Obinutuzumab [12], and more. The majority of which bind to the large loop of CD20. MAbs utilize four mechanisms to fight cancer: neutralizing an oncoprotein, Complement-Dependent Cytotoxicity (CDC) [13, 14], Antibody-Dependent Cellular Phagocytosis (ADCP), and Antibody-Dependent Cellular Cytotoxicity (ADCC) [15, 16]. The targeted therapies against CD20 are not limited to antibodies. Recently, CD20 found to be an potential receptor in nanomedicine [17] and cancer immunotherapy [18]. For instance, there are some CAR-T and NK cells under investigation for CD20+ lymphoma (7, 8). All of which, make a most of antibody fragment, called scFv, as a targeting moiety [19]. In order to abrogate any possible immune response against scFv, this antibody fragment must be either fully human or humanized [20, 21].
Traditionally, mAb used to be generated in mice, such as anti-CD3 Muromomab. However, murine-derived mAbs raised human anti-mouse antibody (HAMA) responses in some patients [17]. To address this issue, mAbs were engineered to incorporate more human sequences to alleviate side effects, which resulted in Chimeric and Humanized mAb. Nevertheless, none of the approaches eradicated immune response against the foreign amino acid sequences. Therefore, the fully human antibody has recently been developed to root out any possible moderate to severe side effects in patients [22]. There are three approaches to generate human antibodies [23]. First, the antigen of interest is injected into a humanized murine, a genetically modified mice that carry human antibody gene. This approach was utilized to generate panitumumab [24]. Second, the phage display is applied to a human CDRs library in order to isolate the strongest binders. Next, the optimal CDRs are grafted into human antibody scaffold. This technique was applied to generate adalimumab. Finally, the phage display can be applied to human naïve antibody library to find the best binders. Fully human antibodies have no immunogenicity due to lack of foreign sequence [25].
In this study, we isolated and characterized fully human anti-CD20 scFv from naive human antibody repertoire (Fig. 1). Generation of antibodies using a naive antibody library is an approach when the classical immunization is not applicable. After rounds of phage display, a library of highly specific scFv binding to CD20 was procured. The specificity, sensitivity, and affinity of selected scFv were evaluated. Next, the isolated scFv was characterized using ELISA and flow cytometry. The selected antibody presented very tight and specific binding to CD20 receptor.

A schematic figure of all steps to isolate fully human scFv that specifically bind to CD20 large loop. The library of human naïve antibody library was kindly provided by Dr. Yamabhai (School of Biotechnology, Suranaree University of Technology, Thailand). Briefly, PMBCs were isolated from human blood using Ficoll gradient of density method. Next, VH and VL genes were amplified by PCR and assembled through SOE PCR using G4S linker. Afterward, the scFv genes were cloned into pMOD1 phagemid using SfiI and NotI restriction sites. Here in this manuscript, we used the Yama I scFv library to isolate a high affinity and specific scFv targeting CD20 large loop. Next, the isolated scFv were subjected to characterization using ELISA, western blot, and flow cytometry. Affinity estimation unveiled the affinity in nanomolar range. Finally, the 3D structure of top candidate was modeled using GrayLab server and docking was performed, using ClusPro server, to illustrate the interaction of G7 scFv to CD20. scFv, single-chain variable fragment; PMBC, peripheral blood mononuclear cell; SOE, splicing by overlapping extension; PCR, polymerase chain reaction; ELISA; enzyme-linked immunosorbent assay
Materials and methods
Production and characterization of CD20
In this manuscript, the phage display was performed using commercial CD20-GST protein purchased from Abnova. Next, in order to find the binders with the highest binding, monoclonal phage ELISA was performed using lab-made CD20, hereof called rCD20.
Expression and purification of rCD20
The extracellular domain of CD20 is composed of two loops, the small and large loop. Most commercial antibodies, such as Rituximab, interact with the large loop. Hence, this domain of CD20 was selected for protein purification to be used in panning.
For protein expression, first, residues between 141 and 188 of CD20 gene (NM_021950) were selected for gene synthesis. The nucleotide sequence was codon-optimized, synthesized, and cloned into PGH, a cloning vector, by Genray Company. For further sub-cloning to the expression vector, the gene was flanked by BamHI and XhoI restriction sites. The gene was inserted into pET28a (Novagen, USA) between the restriction sites, as mentioned earlier. Finally, the recombinant CD20 sequence was corroborated by sequencing.
The recombinant CD20-pET28a plasmid was chemically transformed to BL21 Star™ (DE3) (MilliporeSigma, USA). Briefly, BL21 Star™ (DE3) was culture in 20 ml of antibiotic free LB. Next day, 250 ml of fresh LB was inoculated 1:100 using pre-culture. After optical density of 600 nm (OD 600 nm) was reach between 0.5 and 0.8, bacteria were harvested using centrifugation at 5000 xg, 10 min, 4 °C. The bacteria were washed twice with 50 mM CaCl2. Afterward, 1 ng purified plasmid was added to the competent bacteria and incubated on ice for 30 min. Heat shock treatment (45 s at 42 °C and 5 min on ice) was performed, and then 800 µl of fresh LB wash added and incubated at 37 °C while shaking. Finally, 100 µl of bacterial culture was streaked on a pre-warmed LB agar supplemented with 50 µg/ml Kanamycin (MilliporeSigma, USA).
Next, transformants were selected for colony selection to obtain the highest expressing colony. First, each colony was inoculated into 5 ml Luria–Bertani (LB) supplemented with 50 µg/ml Kanamycin and cultured at 37 °C overnight. The next day, a fresh 5 ml LB was inoculated with a 1:100 ratio of pre-culture and incubated at 37 °C until OD600nm of 0.6 was obtained. The expression was induced by 0.5 mM Isopropyl β- d-1-thiogalactopyranoside (IPTG) (MilliporeSigma, USA) and continued for 5 h at 37 °C. To run SDS-PAGE, 1 ml of each culture was collected and centrifuged at 10,000 xg for 10 min. Afterward, the pellet was lysed with 100 µl of 8 M urea (MilliporeSigma, USA) for 30 min at room temperature (RT). After adding 100 µl of 2 × protein sample buffer (Bio-Rad, USA) and heating for 20 min at 95 °C, 10 µl of each sample was loaded onto 15% SDS-PAGE gel. The colony with the highest level of expression was selected for large-scale protein production.
Large-scale expression and purification
First, 500 ml of LB supplemented with 50 µg/ml Kanamycin was inoculated 1:100 with pre-culture. After OD600nm of 0.6 was reached, 0.5 mM IPTG was added to induce protein expression, which continued for 18 h at 30 °C. Following harvesting the bacterial by centrifugation, 10 ml lysis buffer (500 mM NaCl, 20 mM NaH2PO4, 8 M Urea, and pH 8) was added to lyse the cells. After 2 h at RT, cell lysate was sonicated for 20 min on ice. To remove cell debris, cell lysate was centrifuged at 20,000 xg for 1 h at 4 °C. Then the supernatant was loaded onto Ni–NTA column (Qiagen, Hilden, Germany). To remove impurities, the column was washed using 5 ml of lysis buffer. Finally, protein was eluted using 5 ml of pH gradient (500 mM NaCl, 20 mM NaH2PO4, 6 M Urea, and pH 8, 6.5, 5.2, 4.5, 3.5). All the eluates were loaded onto 15% gel SDS-PAGE to find the best one.
Characterization of CD20
After purification of rCD20, the protein was validated by ELISA using a commercial anti-CD20 antibody, Obinutuzumab (Kindly provided by Tehran University Hospital). First, 200 ng of rCD20 was coated into a 96-well plate overnight at 4 °C. The next day, after washing once, the plate was blocked using 200 µl of 4% skimmed milk. Next, 100 µl of different Obinutuzumab dilutions 1:50, 1:125, 1:250, 1:500, 1:1000, and 1:2000, was added and incubated at 37 °C for 1 h. Then, after washing trice with PBS-T (PBS, 0.05% Tween 20), 100 µl of Goat anti-human IgG HRP conjugated (1:3000) (SouthernBiotech, AL, USA) was added and incubated at 37 °C for 1 h. Finally, after washing six times, 100 µl of 3,3′,5,5′-Tetramethylbenzidine (TMB) (MilliporeSigma, USA) solution was added and incubated in darkness for 10–20 min. Following stopping the reaction by 100 µl of 2 N HCl, the plate was read by Labsystems multiskan RS-232C (Finland, Vantaa) at 405/650 nm.
Phage display and panning
Propagation of the recombinant phages
In this step, the anti-CD20 scFvs were enriched using three steps of phage display. A naïve human library used in this study (hereof called Yama I library) was kindly provided by Dr. Yamabhai (School of Biotechnology, Suranaree University of Technology, Thailand) [26]. Briefly, the variable domains of light and heavy chains were fused through a (G4S)3 linker using splicing by overhang extension polymerase chain reaction (SOE PCR) and then cloned into pMOD1 phagemid using SfiI and NotI restriction sites upstream of GIII gene (Fig. 1). First, the naive human antibody library was propagated in ER2738 (New England Biolab, MA, USA) E. coli. For pre-culture, 5 ml 2xYT media supplemented with 2% Glucose and 100 µg/ml Ampicillin was inoculated with 20 µl of antibody library. The culture was incubated at 37 °C overnight. The next day, 50 µl of pre-culture was inoculated into 5 ml 2xYT media supplemented with 2% Glucose and 100 µg/ml Ampicillin and cultured at 37 °C until OD600nm of 0.4 was obtained. Then, 1010 helper phage, M13KO7 (New England Biolab, MA, USA), was added to the culture and incubated at 37 °C for 30 min without and 60 min with shaking. Next, to remove glucose, bacterial culture was centrifuged, the supernatant was discarded, and the bacterial pellet was resuspended in fresh 2xYT media supplemented with 100 µg/ml Ampicillin and 50 µg/ml Kanamycin without Glucose. The culture was incubated at 37 °C overnight to release recombinant phages. The next day, the culture was centrifuged down at 8,000 xg for 10 min at 4 °C. Next, the supernatant was transferred into a sterile tube and incubated at 4 °C for 30 min. Next, PEG-NaCl was added to the supernatant and incubated on ice for 3 h. Afterward, the recombinant phages were harvested by centrifugation at 21,000 xg for 30 min at 4 °C. Phages were washed twice using DPBS to remove cell debris. The recombinant phages were used in polyclonal phage panning to enrich the library against CD20.
Panning
After recombinant phage propagation, the scFv library was enriched against CD20 via on-column panning. In this experiment, GST-CD20 (Abnova, USA) was used as a bait protein to capture anti-CD20 scFv. The library underwent three negative and positive selection cycles to isolate highly specific scFv with the maximum affinity.
Negative selection
Eight wells of ELISA plated were coated with 200 ng/ml of lab-made GST protein [27]. After blocking using 4% skimmed milk for 1 h at 37 °C, around 1011 phages were added to the plate and incubated for 1 h at 37 °C. In this step, the phages with high- to-medium affinity to GST tag were eliminated. The supernatant, harboring the phages, was used in positive selection.
Positive selection
First, 200 ng GST-CD20 was mixed with 1011 propagated phage and incubated at 37 °C for 30 min while shaking. In this step, all the phages against CD20 were bound to CD20-GST protein. Meanwhile, ER2738 E. coli was cultured in 20 ml of 2xYT until the OD600nm of 0.4 was obtained. Then, the bacteria were stored at 4 °C until needed. To capture and isolate the phages of interest, the mentioned scFv-GST-CD20 complex was passed through a pre-packed GSTrap FF Column (Cytiva Life Sciences, MA, USA). This column is an affinity column to purify GST-tagged protein. Using this column, the phages bound to GST-CD20 were trapped in the column, and all other phages were washed out using 20 ml of PBS. The entrapped phages were eluted using 3 ml of elution buffer (50 mM Tris–Cl, 150 mM NaCl, 0.1 mM EDTA, 10 mM reduced glutathione, pH8.0). The eluate was added to 5 ml of ER2738 E. coli at OD600nm of 0.4 in 2xYT media without shaking for 30 min at 37 °C. Next, the infected ER2738 was cultured in 50 ml of LB supplemented 100 µg/ml Ampicillin at 37 °C overnight to propagate the library.
Polyclonal phage ELISA
To check the enrichment of anti-CD20 scFv before and after each panning, polyclonal phage ELISA was performed. First, either Bovine Serum Albumin (BSA) or 200 ng/well rCD20 was coated in an ELISA plate (SPL Life Sciences, South Korea). Next, the coated plated was blocked using 4% skimmed milk (MilliporeSigma, USA) for 1 h at 37 °C. Around, 1010 recombinant phages were added to each well and incubated at 37 °C for 1 h. Following three washing steps by PBS-T, the secondary antibody, anti-M13 HRP conjugated (LSbio, USA). 1:5000 dilution, was added and incubated for another hour at 37 °C. The excess antibodies were removed by washing six times. Then, 100 µl of TMB was added, and the reaction was terminated by adding 100 µl of stopping solution (2 N HCl). The plate was read at 405/650 nm.
Monoclonal phage ELISA
To find the scFv with the highest affinity and specificity, 100 colonies were selected for monoclonal phage ELISA. After the third round of panning, 200 colonies were randomly selected for colony PCR using M13 primers (Table 1). Next, 100 positive colonies were selected and cultured in 0.5 ml LB media supplemented with 100 µg/ml Ampicillin and cultured at 37 °C overnight. The next day, 3 µl of preculture was inoculated to 200 µl 2xYT media supplemented with 100 µg/ml Ampicillin and cultured at 37 °C for 2 h. Then, 109 of helper phage was added and incubated for 30 min at 37 °C without shaking. Next, 50 µg/ml Kanamycin was added to each well and incubated at 37 °C overnight. The next day, the plate was centrifuged, and the supernatant was transferred into a new 96-well plate for ELISA.
ELISA was performed according to Sect. 2.1.7. Briefly, 200 µg/ml of rCD20 was coated. The next day, the plate was blocked by 4% skimmed milk. After washing the plate trice, 100 µl of monoclonal phage was added to each well. Next, the plate was washed six times to remove nonspecific or low-affinity binders. Afterward, 100 µl of secondary antibody, anti M13 antibody HRP conjugated, was added. After washing, 100 µl of TMB was added, and the reaction was stopped after 10 min using 2 N HCl. The plate was read at 405/650 nm.
Cloning, expression, and purification anti-CD20 scFv
The sequence of selected scFv was sub-cloned into pET30a (Novagen, USA) for overexpression. pET expression vectors take advantage of the powerful T7 promoter. The gene of scFv was PCR amplified and cloned between BamHI and XhoI (New England Biolab, USA) restriction sites. The product of the ligation reaction was chemically transformed into BL21 Star™ (DE3).
The scFv was expressed in 100 ml LB media supplemented with 50 µg/ml Kanamycin. After adding IPTG at OD600nm of 1, expression continued overnight at 25 °C. The next day, the bacteria were harvested by centrifugation at 10,000 xg for 10 min at 4 °C. First, the pellet was washed twice with basal purification buffer (500 mM NaCl, 20 mM NaH2PO4, pH 8) supplemented with 0.05% Tween 20. Next, the pellet was resuspended in 6 M urea and sonicated on ice for 8 min (5 s on, 3 s off, 70% amplitude). Next, the cell lysate was loaded onto Ni–NTA column. Afterward, the column was washed twice with 30 ml of 50 mM imidazole without urea to remove impurities. Then, the protein was eluted using 400 mM imidazole. The purified protein was analyzed by 12% SDS-PAGE. Finally, the purified protein was buffer exchanged, and the concentration was measured by the Bradford reagent (Bio-Rad, USA).
Characterization of anti-CD20 scFv
The selected scFv, G7, was scrutinized by ELISA, western blot, and flow cytometry to evaluate specificity and affinity. By Beatty et al. method, the affinity of scFv was calculated. Using flow cytometry, the binding of G7 scFv to native CD20 on Raji cells (ATCC, VA, USA) was analyzed and compared to commercialized mAb, Rituximab. The generated scFv can also be used in western blot to detect CD20 receptor.
Affinity estimation
The affinity of G7 scFv was measured by ELISA as described by Beatty et al. In this experiment, two concentrations of CD20, 10 and 20 ng/ml, were coated in ELISA plate overnight at 4 °C. The next day, the plate was washed using PBS-T followed by blocking using 4% skimmed milk for 2 h at RT. Next, four different concentrations of G7 scFv 142.86, 71.42, 35.71, and 17.85 nM, was added and incubated at 37 °C. After washing six times, 100 µl anti-c-myc HRP-conjugated antibody (R&DBiosystems, MN, USA) (1:10,000 dilution) was added and incubated at 37 °C for 1 h. After washing six times, 100 µl TMB was added, and the reaction was stopped after 15 min using 2 N HCl. The plate was read at 405/650 nm. The affinity was calculated as the concentration that 50% of maximum binding was obtained. To analyze the data, the maximum binding (142.86 nM) was assumed as 100% binding. The rest of the binding percentage was calculated based on the maximum binding. The data were analyzed using GraphPad Prism version 7.
Western blot
This experiment examined if G7 scFv can also be used as a primary antibody to detect CD20 in western blot. First, 10 µg of rCD20, K562 (ATCC, VA, USA), and Raji cell lysates were applied to 12% SDS-PAGE. Next, the resolved proteins were transferred to PVDF membrane and blocked using 5% skim milk for 2 h at RT. Then, 5 µg/ml of scFv was added to the membrane and incubated at 4 °C overnight. The next day, after washing thrice using 0.05% PBS-T, the S-tag antibody HRP conjugated (Abcam, USA) was added and incubated at RT for 1 h and washed thrice. Finally, the ECL (Invitrogen, USA) substrate was added to the membrane and imaged by a CCD camera.
Flow cytometry
In this experiment, the ability of selected scFv to bind to the native CD20 on Raji cells was measured. The Rituximab, a commercial anti-CD20 monoclonal antibody, was used as a positive control. First, cells were detached using 0.25% Trypsin–EDTA and counted using Hemocytometer. Then, for each antibody staining, 1 × 106 cells were washed twice with 1% PBS-BSA. All the antibody staining were performed for 1 h at RT in darkness. Cells were washed twice after each antibody staining. Next, cells were stained with 100 nM either Rituximab (Kindly provided by Tehran University Hospital) or scFv. Afterward, the secondary antibody was added to each tube, anti-Human (Abcam, USA) and anti-his tag (Abcam, USA) FITC conjugated for Rituximab and scFv stained samples, respectively. Next, one sample of each cell line was stained with Isotype as a negative control. Finally, the cells were analyzed by FACS-Calibur flow cytometer and CellQuest software (Becton Dickinson, San Jose, CA, USA).
Sensitivity
The sensitivity of antibody means the minimum concentration of paratope that can be detected. Here we used ELISA to assess the sensitivity of G7 scFv. First, 400 ng of G7 scFv was blocked with different concentrations of CD20, from 0 ng/ml to 65 ng/ml in microtubes at RT for 60 min. Meanwhile, a 96-well ELISA plate coated with CD20 was blocked using 2% skimmed milk. Next, blocked G7 scFv was applied to ELISA as the primary antibody. After six steps of washing, the secondary antibody, S-tag antibody HRP conjugated (Abcam, USA), was added and incubated at RT for 60 min. After washing and TMB, the reaction was stopped using 2 N HCl, and the plate was read at 405/650 nm.
Bioinformatic analysis
The 3-dementional protein structure of CD20 was modeled by I-TASSER server (http://zhang.bioinformatics.ku.edu/I-TASSER). The structure of G7 scFv was predicted by Swiss Model and Rosie Graylab software (https://rosie.graylab.jhu.edu/). The complementarity determining regions (CDRs) of selected scFv was specified via ImmunoGenetics (IMGT)/V-Quest database [28]. The 3D model of G7 scFv was refined using GalaxyRefine structure refinement server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) to balance any possible error that might have occurred [29]. The docking of G7 scFv to CD20 was performed by ClusPro (https://cluspro.bu.edu/login.php), in which the predicted scFv and CD20 were employed in docking [30]. The result was examined and visualized using PyMOL Molecular Graphics System, Version 2.3.2 Schrödinger, LLC [31].
Results
Production and characterization of CD20 large loop
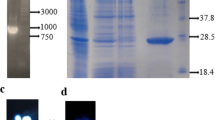
The tertiary structure of CD20 presents a membrane bound receptor with two extracellular loops strikingly different in size. The majority of the therapeutic mAbs target the large loop [32,33,34] (Fig. 2A). Thus, we expressed and purified CD20 large loop for panning step to isolate the scFvs that only interact with this region. A membrane protein can be either isolated from the membrane or produced as a recombinant protein using eukaryotic or prokaryotic expression system. As CD20 spans the membrane four times, it is challenging to solubilize and stabilize the protein. In 2019, Agez et al. showed that CD20 can be purified from SUDHL4 and RAMOS cell lines using the calixarene-based detergent approach [35]. The complication is that this approach require laborious optimization steps to prevent protein aggregation [36, 37]. Recently, a group of scientists applied RIEDL tag system, a pentapeptide composed of Arg79, Ile80, Asp81, Glu82, and Leu83, for CD20 purification from the cell membrane [38], but the production yield was very meager. The other option was to produce the protein using mammalian cell line [39]. The CD20 large loop has only one disulfide bond with no other post-translational modification or N-glycosylation consensus sequence. Hence, we used prokaryotic protein expression which is straightforward and cost effective. So as to reach the maximum expression, the larger loop, encompasses 47 residues (Fig. 2A), was codon-optimized for E. coli and subcloned into pET28a. The large loop of CD20 is 5.6 kDa, but including all pET28a tags, it became around 10.5 kDa with an isoelectric point (pI) of 8.72. The SDS-PAGE analysis showed a band around 12 kDa, which was > 95% pure (Fig. 2B). The total yield of the protein was 750 µg/L of the original culture.

The large loop of CD20 (rCD20) was produced in the lab to be used in library screenings. A a schematic view of CD20 receptor including four transmembrane domains and two extracellular loops, small and large. The large loop is the main target of most FDA-approved monoclonal antibodies against CD20. This loop is only composed of 47 residues, 141–188. B The SDS-PAGE analysis presented a purified rCD20 with maximum purity. As shown here, no impurity was detected after staining with Coomassie Brilliant Blue R-250 with the sensitivity of staining 20-30 ng protein/band. C To authenticate the purified protein, ELISA was performed using a commercial monoclonal antibody, Obinutuzumab. The results showed that Obinutuzumab can detect the sequence at different dilutions. Therefore, as a commercial antibody validated the purified protein, we used this protein for library screening
Following the purification of rCD20, an ELISA test was performed using Obinutuzumab to validate the sequence [40]. According to ELISA result, Obinutuzumab interacted with rCD20 in different dilutions, from 1:50 to 1:2000 (Fig. 2C). Thus, the result verified that CD20 large loop was selected and produced as expected and encompassed all the epitopes.
A fully functional anti-CD20 scFv was selected from Naïve human immune repertoire by phage display
Antibody phage display is a well-established procedure to isolate high affinity and specific antibodies [41] targeting the protein of interest. This library named Yama I and used to isolate scFvs targeting CD20 large loop. Although animal immunization is mandatory to raise antibodies with nanomolar affinity, some studies report isolation of high-affinity scFvs from a naïve antibody library. In 2020, Yuan et al. reported the isolation of three specific scFvs against Covid-19 spike protein from a human naïve antibody library [22]. To evaluate the specificity of isolated scFv, they used three closely related proteins, the spike protein of SARS-COV-1, MERS-COV, and SARS-COV-2 (Covid-19) in ELISA. The result showed that the isolated scFv was highly specific to the Covid-19 spike protein.
In this study, phage display was performed using the gifted library to isolate anti-CD20 scFvs. To obtain a CD20 specific library, three round of bio-panning was performed using GST-CD20 as a bait protein. In this experiment, on-column bio-panning was performed by which GST-CD20 was passed through a GST HiTrap column. One of the advantages of on-column panning, also called chromato-panning, over the traditional approach using ELISA plate is that all the surface area of the bait molecule is accessible for binding for interacting with recombinant phages [42]. Hence, the diversity of the final round of panning in on-column panning is higher than in traditional approaches [43]. Next, the phage library was passed through the column to bind to CD20 and eluted. As shown in Fig. 3A, the polyclonal phage ELISA showed that a higher affinity library was obtained after each round of panning. After the first round, the optical densities at 450 nm (OD450nm) of library binding to Bovine Serum Albumin (BSA) and CD20 were almost equal. But after the third round, the OD450nm of CD20 binders was three times higher than BSA. This result presented that the third round of panning libraries was enriched in high-affinity CD20 binders. The polyclonal phage ELISA showed that the ratio of binder over non-binders was elevated after each round of panning. As shown in Fig. 3B, an equal number of phages, 1011, was applied to each panning, and the output was significantly lower, which means none or low-affinity binders were eliminated, and a more refined repertoire was acquired. Scrutinizing 96 colonies using monoclonal phage ELISA revealed that all selected colonies were CD20 binders. In this step, the number of phages of each colony was not quantified to select the scFv with the highest OD and decent expression and solubility. We assumed that the samples with the higher number of phages may carry scFvs with decent protein expression. Hence, quantifying the number of phages at this point impose the possibility of selecting the scFvs with highest affinity but poor protein expression. This result showed that the number of binders becomes dominant after each round of panning, but the library’s diversity becomes thin. The OD of selected scFvs varied from 0.2 to 1.5 (Fig. 3C). Then the colonies were ranked based on the highest OD, and the top 5 were selected for further study.

The phage display was performed to enrich the library of CD20 binders. A the polyclonal phage ELISA after each round of phage display revealed a higher OD than a negative control, BSA. This clearly showed that the non-specific scFvs were eliminated from the library after each round, and the repertoire was enriched with high-affinity scFv against CD20. B The number of input and output phages also attested to library enrichment. 1011 recombinant phages were used for each phage display round, but the output was significantly lower than the initial input. C after three rounds of phage display, 96 colonies were randomly selected for monoclonal phage ELISA. The results showed that almost all the scFv were specific for CD20. These results successfully showed that the repertoire is utterly enriched in anti-CD20 scFv
Characterization of G7 scFv
Next, to find the top binder among the selected scFv G7, G8, F7, D8, and B7, were selected for another round of monoclonal phage ELISA using equal number phages. As illustrated in Fig. 4A, G7 presented the highest OD450nm compared to other top candidates. Hence, this scFv was selected for further characterizations. The selected anti-CD20 scFv, G7, was subcloned into pET30a expression vector and transformed into BL21 Star™ (DE3) for overexpression. Some studies showed that scFv expression in E. coli leads to inclusion body and protein aggregation [44]. Aguiar et al. generated an scFv against FGF2. They tested a prokaryotic expression system for protein production, pET26b in BL21 Star™ (DE3), but almost all the expressed proteins were stuck in the inclusion body. Hence, they changed to mammalian expression system, pCDNA3.1 in HEK293 [45]. Our results showed that only 50% of G7 scFv was accumulated inside bacteria, and the rest was in soluble fraction. Furthermore, no protein aggregation was observed after purification. It showed that G7 scFv is highly soluble and compatible with the prokaryotic expression system. As the majority of scFvs are stabilized by at least one intramolecular disulfide bond, selecting an appropriate expression system is a crucial step for production, otherwise free cysteines in protein may haphazardly establish intermolecular disulfide bonds and cause aggregation. There are some options to express a disulfide-bonded protein in a prokaryotic expression system, such as Origami [46], Shuffle [47], and Periplasmic space [48, 49]. Some papers repost that scFv disulfide bond is not necessary for the functional protein. Javadian et al. showed that scFv could be expressed in different strains of E. coli, including BL21 Star™ (DE3), Shuffle, and Origami, in a fully functional and soluble configuration [50]. Moreover, as periplasmic is a tiny space between the cytoplasm and cell wall, the expression yield is lower than cytoplasmic expression [51]. Since our result showed a fully functional and soluble scFv in BL21 Star™ (DE3), we pursued this system to obtain the maximum yield. The yield of G7 was 1.2 ± 0.3 mg/L of the culture. Using the purification protocol mentioned above, > 95% purity was obtained according to SDS-PAGE gel stained with Coomassie Brilliant Blue R250 (Fig. 4B).

The top CD20 binders were selected and characterized. A the top anti-CD20 scFvs were selected using monoclonal phage ELISA. The result showed that G7 scFv was statistically superior to other scFvs. B G7 scFv was cloned into pET30a and expressed in BL21 Star™ (DE3). As shown in SDS-PAGE, G7 scFv was more than 95% pure. C next, the affinity of G7 scFv was estimated according to the method developed by Beatty et al. The affinity was calculated around 63.41 ± 3.9 nM. D the binding of G7 scFv was also studied in western blot using rCD20 (lane 1), K562 (lane 2) and Raji (lane 3) cell lysate. The result proved that G7 scFv could bind to denatured CD20 in western blot. As the protein used in western blot was denatured, it unveiled that G7 scFv detects a linear epitope. E finally, the sensitivity of G7 scFv in detecting CD20 was calculated to be 2 ng/ml
Next, we used an enzyme-linked immunoadsorbent assay (EIA) to determine the affinity of selected scFv to CD20. The method was initially developed by Beatty et al. [52, 53] has been corroborated by many different studies [54,55,56]. Here we calculated Ka, the concentration at which the antibody of interest occupies 50% of epitopes. The higher the Ka, the higher the affinity. As suggested by Beatty el al., CD20 was coated in two different concentrations to achieve maximum precision (Fig. 4C). The calculated affinity based on the approach developed by Beatty et al. was 63.41 ± 3.9 nM. While nanomolar affinity antibodies are always preferred, some papers revealed the antibodies with moderate affinity showed a superior tumor penetration and distribution compared to high-affinity ones [57, 58].
Next, we used the purified G7 scFv in western blot to detect CD20, using rCD20 (Fig. 4D, line 1), K562 (Fig. 4D, line 2) and Raji (Fig. 4D, line 3) cell lysates. The result showed that the selected scFv could bind to denature CD20. This result proved that G7 detects a linear epitope on the larger loop of CD20. No band was detected in K562 cell lysate as this cell line was CD20─. Finally, we studied the sensitivity of G7 scFv in detecting CD20. In this approach, 400 ng of purified scFv was incubated with different concentrations of rCD20. Then, the blocked scFvs were used in ELISA to bind coated rCD20. The maximum signal, 100%, was obtained when G7 scFv was blocked with 0 ng of rCD20, and the lowest signal was obtained for 65 ng rCD20. As 10% of signal dropped when G7 scFv was incubated with 2 ng/ml of rCD20, the sensitivity was calculated 2 ng/ml of rCD20. This result demonstrates that G7 scFv can detect as low as 2 ng/ml of CD20.
The binding efficiency of G7 scFv to CD20.+ cell line
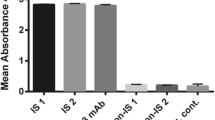
Here, we used ELISA to study the specificity of G7 scFv in binding to non-related and closely related proteins. As shown in Fig. 5A, the selected scFv showed maximum binding to rCD20 and commercial CD20, while insignificant interaction was recorded with CD19 (Abcam, USA) as a closely related receptor on B cells. As both rCD20 and the commercial one contained his tag, we also included commercial 10X his tag peptide to show no interaction between G7 scFv and his tag. The result shows the baseline OD for 10X his tag stained with G7 scFv. Furthermore, as the commercial CD20 used in the phage display was fused to GST tag, we also included the commercial GST tag in this study to ensure no interaction with GST moiety. Furthermore, the result also showed that G7 scFv had no interaction with non-related proteins including, skimmed milk, BSA, and casein.

The specificity of G7 scFv was studied by ELISA and flow cytometry. A the ELISA result proved that G7 specifically interacted with the large loop of CD20, while no interaction was recorded for GST and 10 × his tags. B–G to show the specificity of anti-CD20 scFv, Raji (CD20+) and K562 (CD20—) cell lines were stained with Rituximab and G7 scFv. The flow cytometry histograms proved that G7 scFv was specifically bound to CD20+ cell line while the baseline interaction was observed with CD20— cell line. H the bar chart illustrating the mean fluorescence intensity (MFI) of each sample. As shown here, the MFI of Raji cells stained with G7 scFv and Rituximab were statistically higher that isotype (p-value 0.0005)
Flow cytometry was performed further to characterize the binding of G7 scFv to CD20. The advantage of flow cytometry over ELISA is that CD20 expressed on CD20+ cells is in native conformation. In this experiment, Rituximab was used as a positive control, which interact with CD20 large loop. Two cell lines were selected, Raji (CD20+) and K562 (CD20—) cell lines. As shown in Fig. 5B and E, the gating was applied based on isotype to eliminate any background and non-specific binding of the secondary antibody. Next, G7 scFv showed specific binding to Raji cell and shifted the histogram between 101 and 102, and 71.9% of the cells were positive Fig. 5F, while only 5.95% of K562 cells were positive Fig. 5C. This clearly shows that the binding of G7 scFv is highly specific. Binding to 5.95% of K562 cells was not due to the non-specific binding of G7 scFv to other receptors because using Rituximab also revealed a similar result, 17.2% positive K562 cells (Fig. 4D). Although K562 cell line is considered a CD20—, it might express a very shallow level of this receptor. Staining Raji cells with Rituximab showed a significant shift toward 103, almost two folds more than G7 scFv, and 93.4% cells became positive Fig. 5G. Furthermore, the analysis of the mean fluorescence intensity of cells stained with antibody showed that G7 scFv and Rituximab caused a significant (pvalue 0.0005) shift of the histogram to the right side, while two aforementioned antibodies did not boost the MFI in K562 compared to isotype (Fig. 5H).
In-silico analysis of G7 scFv unveiled tertiary structure and interaction with CD20
After in-vitro characterization of G7 scFv in depth, we studied the tertiary structure of G7 and its interaction with CD20 molecule. As scFv is composed of one VH and on VL domain, it has totally six complementary determining regions (CDRs) and eight frameworks (Fig. 6A). The 3D structure of G7 scFv presented a root-mean-square deviation (RMSD) of 0.284 ångström (Å), which revealed insignificant structure modification compared to the native model (Fig. 6B). This structure clearly revealed antiparallel β-sheet connected through some loops. Furthermore, the interaction of G7 scFv with CD20 was modeled using Z-Dock. As illustrated in Fig. 6C, G7 can engaged to large loop of CD20.

The tertiary structure of G7 scFv and its docking to CD20 receptor. A sequence of heavy and light chain of G7 scFv. B the 3D structure of G7 scFv was modeled so as to visualize the structure of the protein. As shown here, the structure of G7 scFv is close to the structure of all scFvs, including VH and VL. The 3D model clearly shows that each variable domain is composed of several anti-parallel β-strands upheld by some protruded loops. C the interaction of G7 scFv and CD20 was modeled by ClusPro server. The model illustrated that scFv (red) interact with CD20 (blue). H-CDR, variable heavy chain CDR L-CDR, variable light chain CDR
Conclusion
Herein, we report isolation and characterization of high-affinity scFv targeting large loop of CD20 through phage display using human naïve antibody repertoire. Human antibody lacks any foreign amino acid sequences that may lead to immunogenicity. The interaction of G7 scFv to CD20 was characterized by ELISA, western blot, and flow cytometry. Next, the affinity of G7 scFv was calculated around 63.41 ± 3.9 nM and sensitivity of 2 ng/ml. These results collectively illustrated a high-affinity interaction between G7 scFv and CD20. As the G7 scFv was isolated from the human antibody library, no further humanization step is required. scFv are miniaturized antibody fragments that can be applied to a wide range of clinical applications such as BiTE (bispecific T-cell engager), BiKE (bispecific NK-cell engager), chimeric antigen receptor (CAR) T and NK cells, nanomedicine, and many more. As mentioned above, G7 scFv can directly be used in clinics without further modification. Nonetheless, some experiments are yet to be done regarding pharmacokinetics and pharmacodynamics, tolerability, and therapeutic index of G7 scFv. Furthermore, the serum half-life of this molecule must be determined to optimize this molecule for tumor immuno-imaging.
References
Bashford-Rogers R, et al. Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature. 2019;574(7776):122–6.
Greenfield AL, Hauser SL. B-cell therapy for multiple sclerosis: entering an era. Ann Neurol. 2018;83(1):13–26.
Payandeh Z, et al. The applications of anti-CD20 antibodies to treat various B cells disorders. Biomed Pharmacother. 2019;109:2415–26.
Casan J, et al. Anti-CD20 monoclonal antibodies: reviewing a revolution. Hum Vaccin Immunother. 2018;14(12):2820–41.
Petrie RJ, Deans JP. Colocalization of the B cell receptor and CD20 followed by activation-dependent dissociation in distinct lipid rafts. J Immunol. 2002;169(6):2886–91.
Polyak MJ, et al. CD20 homo-oligomers physically associate with the B cell antigen receptor: dissociation upon receptor engagement and recruitment of phosphoproteins and calmodulin-binding proteins. J Biol Chem. 2008;283(27):18545–52.
Luo C, et al. Efficacy and safety of new anti-CD20 monoclonal antibodies versus rituximab for induction therapy of CD20+ B-cell non-Hodgkin lymphomas: a systematic review and meta-analysis. Sci Rep. 2021;11(1):1–14.
Fanale, M.A., et al. (2018) Safety and efficacy of anti-CD20 immunotoxin MT-3724 in relapsed/refractory (R/R) B-cell non-Hodgkin lymphoma (NHL) in a phase I study. Am Soc Clin Oncol.
Leonard JP, et al. AUGMENT: a phase III study of lenalidomide plus rituximab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J Clin Oncol. 2019;37(14):1188.
Davids MS, et al. Efficacy and safety of duvelisib following disease progression on ofatumumab in patients with relapsed/refractory CLL or SLL in the DUO crossover extension study. Clin Cancer Res. 2020;26(9):2096–103.
Fox E, et al. A phase 2 multicenter study of ublituximab, a novel glycoengineered anti-CD20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mult Scler J. 2021;27(3):420–9.
Marinov AD, et al. The type II anti-CD20 antibody obinutuzumab (GA101) is more effective than rituximab at depleting B cells and treating disease in a Murine Lupus model. Arthritis Rheumatol. 2021;73(5):826–36.
Golay J, et al. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood, J Am Soc Hematol. 2001;98(12):3383–9.
Golay J, et al. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood, The Journal of the American Society of Hematology. 2000;95(12):3900–8.
Clynes RA, et al. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6(4):443–6.
Reff ME, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435–45.
Tang X, et al. Rituximab (anti-CD20)-modified AZD-2014-encapsulated nanoparticles killing of B lymphoma cells. Artificial Cells Nanomed Biotechnol. 2018;46(sup2):1063–73.
Shadman M, et al. CD20 targeted CAR-T for high-risk B-cell non-Hodgkin lymphomas. Blood. 2019;134:3235.
Ye S, et al. Early clinical results of a novel anti-CD20 chimeric antigen receptor (CAR)-T cell therapy for B-cell NHL patients who are relapsed/resistant following CD19 CAR-T therapy. Blood. 2020;136:8–9.
Hosseini I, et al. Mitigating the risk of cytokine release syndrome in a Phase I trial of CD20/CD3 bispecific antibody mosunetuzumab in NHL: impact of translational system modeling. NPJ Syst Biol App. 2020;6(1):1–11.
Hutchings M, et al. Epcoritamab (GEN3013; DuoBody-CD3× CD20) to induce complete response in patients with relapsed/refractory B-cell non-Hodgkin lymphoma (B-NHL): complete dose escalation data and efficacy results from a phase I/II trial. J Clin Oncol. 2020;38:8009.
Yuan AQ, et al. Isolation of and characterization of neutralizing antibodies to Covid-19 from a large human naïve scFv phage display library. BioRxiv. 2020;25:527.
Khajeh S, et al. Phage display selection of fully human antibody fragments to inhibit growth-promoting effects of glycine-extended gastrin 17 on human colorectal cancer cells. Artif Cells, Nanomed Biotechnol. 2018;46(sup2):1082–90.
Glumac PM, et al. The identification of a novel antibody for CD133 using human antibody phage display. Prostate. 2018;78(13):981–91.
Xu M, et al. Development of a novel, fully human, anti-PCSK9 antibody with potent hypolipidemic activity by utilizing phage display-based strategy. EBioMedicine. 2021;65:103250.
Pansri P, et al. A compact phage display human scFv library for selection of antibodies to a wide variety of antigens. BMC Biotechnol. 2009;9(1):1–16.
Harper S, Speicher DW. Purification of proteins fused to glutathione S-transferase. In: Protein chromatography. Springer; 2011. p. 259–80.
Brochet X, Lefranc M-P, Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized VJ and VDJ sequence analysis. Nucleic Acids Res. 2008;36:W503–8.
Ko J, et al. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40(W1):W294–7.
Comeau SR, et al. ClusPro: a fully automated algorithm for protein–protein docking. Nucleic Acids Res. 2004;32:W96–9.
DeLano WL. Pymol: an open-source molecular graphics tool. CCP4 Newslett Protein Crystallogr. 2002;40(1):82–92.
Klein C, et al. Response to: monoclonal antibodies targeting CD20. MAbs. 2013;5:337–8.
Klein C, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5:22–33.
Rougé L, et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science. 2020;367(6483):1224–30.
Agez M, et al. Biochemical and biophysical characterization of purified native CD20 alone and in complex with rituximab and obinutuzumab. Sci Rep. 2019;9(1):1–13.
Pandey A, et al. Current strategies for protein production and purification enabling membrane protein structural biology. Biochem Cell Biol. 2016;94(6):507–27.
Lacapère J-J, Robert J-C, Thomas-soumarmon A. Efficient solubilization and purification of the gastric H+, K+-ATPase for functional and structural studies. Biochem J. 2000;345(2):239–45.
Asano T, Kaneko MK, Kato Y. RIEDL tag: a novel pentapeptide tagging system for transmembrane protein purification. Biochem Biophys Rep. 2020;23:100780.
Vogler I, et al. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol Ther. 2010;18(7):1330–8.
Niederfellner G, et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood J Am Soc Hematol. 2011;118(2):358–67.
Clackson T, et al. Making antibody fragments using phage display libraries. Nature. 1991;352(6336):624–8.
Noppe W, et al. Chromato-panning: an efficient new mode of identifying suitable ligands from phage display libraries. BMC Biotechnol. 2009;9(1):1–9.
Bratkovič T. Progress in phage display: evolution of the technique and its applications. Cell Mol Life Sci. 2010;67(5):749–67.
Kang TH, Seong BL. Solubility, stability, and avidity of recombinant antibody fragments expressed in microorganisms. Front Microbiol. 2020. https://doi.org/10.3389/fmicb.2020.01927.
de Aguiar RB, et al. Generation and functional characterization of a single-chain variable fragment (scFv) of the anti-FGF2 3F12E7 monoclonal antibody. Sci Rep. 2021;11(1):1–11.
Xiong S, et al. Solubility of disulfide-bonded proteins in the cytoplasm of Escherichia coli and its “oxidizing” mutant. World J Gastroenterol: WJG. 2005;11(7):1077.
Nikkhoi SK, Rahbarizadeh F, Ahmadvand D. Oligo-clonal nanobodies as an innovative targeting agent for cancer therapy: new biology and novel targeting systems. Protein Expr Purif. 2017;129:115–21.
Berkmen M. Production of disulfide-bonded proteins in Escherichia coli. Protein Expr Purif. 2012;82(1):240–51.
Manta B, Boyd D, Berkmen M. Disulfide bond formation in the periplasm of Escherichia coli. EcoSal Plus. 2019. https://doi.org/10.1128/ecosalplus.ESP-0012-2018.
Javadian FS, et al. Solubility assessment of single-chain antibody fragment against epithelial cell adhesion molecule extracellular domain in four Escherichia coli strains. J Genet Eng Biotechnol. 2021;19(1):1–8.
Sandomenico A, Sivaccumar JP, Ruvo M. Evolution of Escherichia coli expression system in producing antibody recombinant fragments. Int J Mol Sci. 2020;21(17):6324.
Beatty JD, Beatty BG, Vlahos WG. Measurement of monoclonal antibody affinity by non-competitive enzyme immunoassay. J Immunol Methods. 1987;100(1–2):173–9.
Beatty JD, et al. Method of analysis of non-competitive enzyme immunoassays for antibody quantification. J Immunol Methods. 1987;100(1–2):161–72.
Kazemi-Lomedasht F, et al. Production and characterization of novel camel single domain antibody targeting mouse vascular endothelial growth factor. Monoclonal Antibodies in Immunodiagnosis and Immunotherapy. 2016;35(3):167–71.
Raghava G, Agrewala JN. Method for determining the affinity of monoclonal antibody using non-competitive ELISA: a computer program. J Immunoassay Immunochem. 1994;15(2):115–28.
Fadlalla MH, et al. Development of ELISA and lateral flow immunoassays for ochratoxins (OTA and OTB) detection based on monoclonal antibody. Front Cell Infect Microbiol. 2020;10:80.
de Ridder GG, Ray R, Pizzo SV. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Res. 2012;22(3):225–35.
Adams GP, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Can Res. 2001;61(12):4750–5.
Acknowledgements
The authors thank the Iran National Science Foundation (INSF) for supporting this research project (Grant Number: 96010981). This article partly supported by Faculty of Medical Sciences, Tarbiat Modares University, Tehran, I.R. Iran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shams, N., Khoshtinat Nikkhoi, S., Gu, Z. et al. Isolation and characterization of human anti-CD20 single-chain variable fragment (scFv) from a Naive human scFv library. Med Oncol 39, 177 (2022). https://doi.org/10.1007/s12032-022-01757-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-022-01757-1