
Abstract
Age-related macular degeneration (AMD) is one of the most common causes of irreversible vision loss in the elderly. Its pathogenesis is likely multifactorial, involving a complex interaction of metabolic and environmental factors, and remains poorly understood. Previous studies have shown that mitochondrial dysfunction and oxidative stress play a crucial role in the development of AMD. Oxidative damage to the retinal pigment epithelium (RPE) has been identified as one of the major mediators in the pathogenesis of age-related macular degeneration (AMD). Therefore, this article combines transcriptome sequencing (RNA-seq) and single-cell sequencing (scRNA-seq) data to explore the role of mitochondria-related genes (MRGs) in AMD. Firstly, differential expression analysis was performed on the raw RNA-seq data. The intersection of differentially expressed genes (DEGs) and MRGs was performed. This paper proposes a deep subspace nonnegative matrix factorization (DS-NMF) algorithm to perform a multi-layer nonlinear transformation on the intersection of gene expression profiles corresponding to AMD samples. The age of AMD patients is used as prior information at the network’s top level to change the data distribution. The classification is based on reconstructed data with altered distribution. The types obtained significantly differ in scores of multiple immune-related pathways and immune cell infiltration abundance. Secondly, an optimal AMD diagnosis model was constructed using multiple machine learning algorithms for external and qRT-PCR verification. Finally, ten potential therapeutic drugs for AMD were identified based on cMAP analysis. The AMD subtypes identified in this article and the diagnostic model constructed can provide a reference for treating AMD and discovering new drug targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Age-related macular degeneration (AMD) is a chorioretinal disease closely related to age. Pathologically, the main manifestations are aging changes in the structure of the macular area and a decrease in the phagocytosis and digestion function of the retinal pigment epithelial cells on the outer disc membrane of the visual cells. Further features that increase the number and diameter of extracellular retinal deposits are called drusen (Lim et al. 2012). As the population ages, AMD is the leading cause of blindness in people over 50 years old worldwide (Newman et al. 2012). Genetic factors play an essential role in the pathogenesis of AMD, and multiple genetic variants have been associated with the risk of AMD. In a recent study, Qiao et al. identified multiple genetic susceptibility loci (including Lama5, Mtg2, Col9A3) through genome-wide association studies (GWAS) and whole-exome sequencing in older Asian people (Fan et al. 2023).
The RPE is particularly susceptible to oxidative damage because it is extremely metabolically active, highly oxidative, and exposed to photosensitizers such as the age pigment lipofuscin. This sensitivity leads to various age-related changes, ultimately leading to reduced RPE function and increased susceptibility to cell death. Oxidative stress is a recognized risk factor for AMD, in which changes in areas of focal loss of the RPE lead to photoreceptor degeneration and central vision loss (Jarrett and Boulton 2012). Increased mitochondrial damage and reactive oxygen species (ROS) production are associated with AMD, suggesting that damaged mitochondria and other oxidatively modified components are not efficiently removed by aging RPE cells (Karunadharma et al. 2010).
As a vital organ within cells, mitochondria are responsible for the energy cells, which are required and participate in biological processes such as cell metabolism and redox reactions (Kaarniranta and Salminen 2009). Therefore, mitochondria-related genes (MRGs) may play a vital role in the occurrence and development of AMD. Firstly, oxidative stress and mitochondrial dysfunction may accelerate the development of AMD (Beatty et al. 2000). Patients with AMD are often accompanied by increased oxidative stress, which may lead to oxidative damage to mitochondrial DNA, leading to mitochondrial dysfunction. Impaired mitochondrial function may further aggravate oxidative stress, forming a vicious cycle. Secondly, inflammation and immune response also impact the pathological process of AMD (Ambati et al. 2003). Abnormal mitochondrial function and oxidative stress can trigger immune system responses, leading to chronic inflammation in AMD. Finally, mitochondrial dysfunction may lead to insufficient intracellular energy supply, triggering apoptosis or necrosis (Khandhadia and Lotery 2010). These cell death processes can lead to cell loss in the macular area of AMD, further exacerbating chorioretinal damage.
Previous studies have utilized various machine learning methods to identify AMD diagnostic-related genes and construct AMD diagnostic models. Wang et al. constructed AMD diagnostic models based on DNA methylation and gene expression data using random forest models (Wang et al. 2021). Han et al. identified key modules and modular genes most relevant to AMD through weighted gene co-expression network analysis. They employed random forest, support vector machine, Xgboost, and GLM models to select predictive genes and build an AMD clinical prediction model (Han and He 2023). Additionally, Han et al. integrated weighted gene co-expression network analysis and differential expression to pinpoint genes intricately associated with Tfh cells. Using the MCODE function in Cytoscape software, they screened these genes and identified key diagnostic genes using the LASSO algorithm (Yang, et al. 2023).
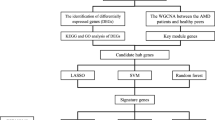
Figure 1 shows the technical roadmap of this article. This paper aims to deeply explore the role of MRGs in AMD through bioinformatics analysis and experimental verification. We obtained MRGs from previous literature (Pei et al. 2023), and their expression levels were extracted from transcriptome data for differential expression analysis. AMD subtypes were then identified based on the deep subspace nonnegative matrix factorization algorithm (DS-NMF). This method can use the age of AMD patients as prior information, thereby changing the original data distribution so that patients of different age groups are distributed far away from each other. Furthermore, multiple machine learning algorithms were used to identify hub genes and construct a diagnostic model for AMD. Finally, the different patterns of multiple immune cells in trajectory analysis and cell communication were explored through AMD’s single-cell sequencing (scRNA-seq) data. This research is expected to provide a new theoretical basis for treating AMD and provide more critical insights into MRGs for future biomedical research.

The technical roadmap of the paper
Method
Acquisition of Data Sets
This article downloaded two macular degeneration RNA-seq data sets (GSE29801 (Newman et al. 2012) and GSE135092 (Jones et al. 2023; Orozco et al. 2020)) from the Gene Expression Omnibus (GEO) database. The GSE29801 data set is used as an internal data set containing 151 normal samples and 142 AMD samples. The GSE135092 data set is used as an external data set, which includes 50 normal samples and 50 AMD samples. scRNA-seq data sets of two AMD samples were collected from the GSE210543 data set.
Differential Expression Analysis and GO Enrichment Analysis
Differentially expressed genes (DEGs) between the control and diseased groups were identified based on the limma package (Ritchie et al. 2015). The parameters are set as follows: the absolute value of logFC is more than 0.25, and the p-value is less than 0.05. Gene Ontology (GO) enrichment analysis was performed on DEGs based on the R package “clusterProfiler” (Yu et al. 2012).
Lim et al. 2012) Ritchie et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research, 43(7), e47. https://doi.org/https://doi.org/10.1093/nar/gkv007
Newman et al. 2012) Yu et al. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics: a journal of integrative biology, 16(5), 284–287. https://doi.org/https://doi.org/10.1089/omi.2011.0118
DS-NMF Algorithm
The DS-NMF algorithm consists of two parts: deep subspace reconstruction and NMF algorithm. Given data points \({\left\{{x}_{i}\right\}}_{i=1,\cdots ,N}\) extracted from multiple linear subspaces \({\left\{{R}_{i}\right\}}_{i=1,\cdots ,K}\), specific points within a particular subspace can be represented as linear combinations of other points within the same subspace. This property is known as self-expression. For a data matrix \(X\), the self-expression property can be formulated as \(X=\text{C}X\), where \(C\) is the coefficient matrix capturing the linear relationships. Under certain permutation conditions, \(C\) should exhibit a block diagonal structure, with each block corresponding to samples from the same subspace (Fan et al. 2008).
This method differs notably from autoencoders. Self-expression networks focus on discovering subspace structures and clustering information in data through the self-expression property. They optimize the coefficient matrix WWW by leveraging linear combinations and sparse regularization to uncover relationships among data points. In contrast, autoencoders emphasize nonlinear dimensionality reduction and reconstruction of data. They achieve this through encoder and decoder networks for nonlinear mapping and reconstruction. In summary, while both self-expression networks and autoencoders are used for data representation and reconstruction, self-expression networks are more concerned with subspace structures and clustering in data, while autoencoders prioritize nonlinear dimensionality reduction and reconstruction performance.
In the deep subspace reconstruction part, the original input is reconstructed using the self-expression properties of the data. Define the gene expression matrix \(X\in {R}^{N*p}\). \(N\) represents the number of genes. \(p\) represents the sample size. Let \(X=\left[{x}_{1},{x}_{2},\cdots ,{x}_{\text{Nz}}\right]\), where \({x}_{1}\) is the first sample of the first label, and \(z\) represents the total number of categories. \({x}_{\text{Nz}}\) is the last sample of the \({z}_{\text{th}}\) label.
First, the original data is put into a multi-layer feedforward neural network, and the nonlinearly transformed matrix \({H}_{i}^{M}\) is output in the output layer. \(M\) indicates the total number of layers on the network. Iterative calculations are performed on the top \(M+1\) layer to achieve subspace reconstruction. Below is the definition of network parameters.
Among them, \(m=\text{1,2},,...,\) M indicates the number of network layers. \({W}^{(m)}\) and \({b}^{(m)}\) represent the weight and bias of the \(m-th\) layer network respectively. \({H}^{(M)}=\left[{h}_{1}^{(M)},{h}_{2}^{(M)},\cdots ,{h}_{{N}_{z}}^{\left(M\right)}\right]\), where \({h}_{{N}_{z}}^{\left(M\right)}\) represents the \(N\) sample of the \({\text{z}}_{th}\) class after undergoing a nonlinear transformation by the multi-layer neural network. Finally, the objective function of the subspace reconstruction algorithm of the \({z}_{\text{th}}\) class samples is:
where \({c}_{l}\) represents the vector of self-expression coefficients of layer l and \({||\cdot ||}_{\text{F}}\) represents the Frobenius norm. \({h}_{l}^{(M)}\) is the output of the \(l-th\) feature of the top-level network after nonlinear transformation. \({H}^{(M)}\) is the output of the original data. The expression of \({h}_{l}^{(m)}\) is as follows:
where G(·) indicates the sigmoid activation function. The specific definitions are as follows.
The following equation can be obtained after calculating the partial derivatives of Eq. 2 and sorting it out (for the specific derivation process, see Supplementary material 1.1).
The self-expression coefficient matrix for all samples is defined as follows.
Finally, the reconstructed data can be expressed as the following formula.
The variable \(\widetilde{X}\) represents the data after deep subspace reconstruction. Leveraging neural networks to uncover nonlinear features in the data enables the incorporation of existing clinical information to reflect grouping information that reflects similar data structures. Incorporating prior information into the data as input to the NMF algorithm can enhance the algorithm’s performance to a certain extent. The NMF algorithm is a low-rank decomposition algorithm. This paper clusters the reconstructed data based on the NMF algorithm. The objective function of the NMF algorithm is given below. In Supplementary Material 1.2, we provide the parameters related to the neural network component in the DS-NMF algorithm. In Supplementary Material 1.3, the optimization and solving process of the algorithm is detailed.
Among them, \(W\in {R}^{S*K}\) and \(H\in {R}^{K*M}\) are the basis and coefficient matrices, respectively. \(W\) and \(H\) need to be guaranteed to be nonnegative. The sum needs to be guaranteed to be nonnegative. \(K\) is the number of clusters. Spectral clustering is performed on \(W\) to obtain the final clustering result.
Analysis of Gene Set Variation Between Subtypes and Immunoassays
This article implements gene set variation analysis of different subtypes based on the R package “GSVA” (Hänzelmann et al. 2013). Based on the “c2.cp.kegg.v7.5.1.symbols.gmt” reference gene set, multiple Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with significantly different scores between subtypes were identified. The infiltration abundance of 22 immune cells in different subtypes was evaluated using the “CIBERSORT” algorithm (Chen et al. 2018). In addition, differences in the expression of immune checkpoints and HLA genes among different subtypes were evaluated. The above analyses all used the Wilcoxon method to compare differences.
Diagnostic Model Construction Methods
This paper implements the construction of diagnostic models through random forest (RF), support vector machine recursive feature elimination (SVM-RFE), K nearest neighbor (KNN), and adaptive boosting (Adaboost) algorithms. Python’s scikit-learn package (Pedregosa, et al. 2011) implements RF, KNN, and Adaboost. SVM-RFE is implemented by the R package “e1071.” After the internal data set was randomly divided into the training set and the test set at a ratio of 7:3, a tenfold cross-validation method was used on the training set to obtain the gene set with the highest accuracy in the test set by selecting the top features and perform AUC verification. In the RF algorithm, criteria are set to entropy, and n_estimators are set to 500. n_neighbors is set to 3 in the KNN algorithm. n_estimators is set to 500 in the Adaboost algorithm. The random seed of the SVM-RFE algorithm is 13,579. The remaining parameters of all algorithms involved are default parameters.
Nomogram Model Construction
This paper builds a nomogram model based on the R package “rms” and uses diagnostic genes. The construction effect of the nomogram model was evaluated through calibration curves. Decision curve analysis (DCA) is implemented based on the decision_curve function in the R package “rmda.” Clinical impact curves were also plotted to predict high-risk probability stratification for a population of 1000.
Analysis Methods of scRNA-seq Data
This article is based on the CreateSeuratObject function of the R package “Seurat” (Hao et al. 2024) to convert the original count matrix into a format readable by the Seurat package. In the quality control process, cells that meet the requirements of nFeature_RNA greater than 200 and less than 7000, nCount_RNA less than 5000, mitochondrial gene proportion less than 3%, and red blood cell proportion less than 0.2% are retained based on the subset function. The LogNormalize method based on the NormalizeData function implements standardization in the standardization process. The first 20 principal components are selected for clustering based on the RunPCA function in the dimensionality reduction and clustering process. After setting the resolution to 1, 21 cell clusters are obtained based on the FindClusters function. The removal of double cells is realized based on the R package “DoubletFinder” (Stoeckius et al. 2018). Subsequently, cell type annotation was implemented based on the R package “singleR” (Aran et al. 2019). Cell trajectory analysis is implemented based on the R package “monocle.” Cell communication analysis is implemented based on the “CellChat” (Jin et al. 2021) package.
Connectivity Map (cMAP) Analysis
The cMAP database (https://clue.io/) can explore associations between diseases, genes, and small-molecule compounds based on gene expression. The dysregulated genes from the differential analysis were entered into the cMAP database to identify potential small-molecule drugs for AMD treatment. Potential small-molecule compounds are entered into the Pubchem database (https://pubchem.ncbi.nlm.nih.gov/) to obtain the compound’s molecular structure.
Cell Cultures and Treatment
Human retinal pigment epithelial cells (ARPE-19) were purchased as frozen vials from Shanghai EK-Bioscience Biotechnology Co., Ltd. (Shanghai, China), and all cell experiments were performed between the third and fifth generations. The cells were cultured in DMEM/F-12 (supplemented with 10% FBS, 1% streptomycin/penicillin) at 37 °C in a humidified atmosphere containing 95% air and 5% CO2. Cells at 80–90% confluence were selected for subculture and subsequent experimentation. For H2O2-induced oxidative damage studies, the cells were treated with a serum-free medium containing various concentrations of H2O2 (400 μM) for 24 h.
Real-Time Quantitative Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from RPE cells using Trizol reagent (Invitrogen, USA). Total RNA was reverse transcribed into cDNA using HiScript II Q Select RT SuperMix for qPCR (Vazyme, China). qPCR was performed using SYBR green reagent (Vazyme, China) on Roche 96 (Roche, USA). The gene expression level was quantified using the 2−ΔΔCt method. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as an internal control gene. Analysis of each sample was performed in triplicate. Primer sequences are listed in Supplement Table 1. Statistical analysis was performed in GraphPad Prism software. Statistical difference between groups was assessed by Student’s t-test. p < 0.05 was considered statistically significant.
Result
Acquisition and Analysis of DEMRGs
First, this article conducts differential expression analysis on samples from the normal and diseased groups in the internal data set. A total of 528 DEGs were obtained. Figure 2A shows the volcano plot obtained from differential expression analysis. The differentially expressed genes are in the gene diff.xls file in the Supplementary Material. Figure 3C is a bar graph obtained by GO enrichment analysis of DEGs. We will discuss in part an analysis of the role of these pathways in the development of AMD. Subsequently, 31 intersection genes were obtained from the intersection of MRGs and DEGs collected from previous literature (Chang et al. 2023) (Fig. 2B–E).

Results of differential expression analysis. A The volcano plot obtained by differential expression analysis. B A bar graph of GO enrichment analysis of DEGs. C The Venn diagram of the intersection of DEGs and mitochondria-related genes. D A box plot of intersection gene expression in normal and diseased groups. E A circle diagram of the chromosomal location of intersection genes

Clustering results based on DS-NMF algorithm. A, B PCA analysis scatter plots obtained before and after reconstruction using the deep subspace reconstruction algorithm. C A line chart of changes in cophenetic, dispersion, evar, residuals, rss, silhouette, and sparseness of the DS-NMF algorithm as the number of clusters increases. D The consensus matrix obtained by the DS-NMF algorithm when the number of clusters is 2. E A PCA analysis scatter plot obtained by clustering with the DS-NMF algorithm
Identification of Subtypes of Age-Related Macular Degeneration
To use age as prior information to change the distribution of the original data, this article first reconstructs AMD samples based on the DS-NMF algorithm. The input data are the expression profiles of 31 intersection genes and two group labels of AMD divided by the median age. This method performs multi-layer nonlinear mapping of gene expression profiles through multi-layer feedforward neural networks. It is then reconstructed on top of the network. During reconstruction, AMD samples were divided into two groups according to the median age. Figure 3A and B show the distribution changes of the original data before and after reconstruction. Labels 1 and 2 represent samples younger and older than the median age of all AMD samples, respectively. The NMF algorithm was performed on the reconstructed data. The final number of clusters was set to 2 according to cophenetic (Fig. 3C). Figure 3D shows the consensus matrix with a cluster number of 2. Figure 3E shows the final clustering results for AMD samples. The two subtypes were effectively distinguished. In addition, visualization results of different subtypes of samples based on t-SNE dimensionality reduction are presented in Supplementary materials Figure S3.
To explore the differences between the two isoforms in terms of enrichment pathways and immune landscapes, differential expression analysis of the two isoforms was first performed (Fig. 4A). Subsequent GSVA analysis identified multiple pathways with significantly different scores between the two subtypes (Fig. 4B). We will analyze the biological significance of these pathways in the “Discussion” section. Furthermore, the CIBERSORT algorithm was used to evaluate the difference in infiltration abundance of 22 immune cells between the two subtypes. The infiltrating abundance of most immune cells in the two subtypes was significantly different (Fig. 4C). Finally, immune checkpoints and HLA genes were collected from previous literature and found that the expression of most genes was significantly different in the two groups (Fig. 4D, E) (Liu et al. 2024). The results of enrichment and immunoassays confirmed the reliability of AMD subtype identification. Furthermore, in Supplementary Material Sect. 1.4, we present the AMD clustering results obtained using other clustering algorithms and highlight the differences in immune infiltration abundance across subtypes. Additionally, we included the GSVA results of the two baseline clustering algorithms in Supplementary Material 1.4, further confirming the biological significance of the subtypes identified by the DS-NMF algorithm.

Difference analysis, enrichment analysis, and immune analysis between subtypes. A The top 20 DEGs between the two subtypes. B The differential expression heat map of pathway scores obtained by GSVA analysis of the two subtypes. C A box plot of the difference in infiltration abundance of immune cells between different types based on CIBERSORT analysis. D, E Box plots of differential expression of immune checkpoints and HLA genes in different subtypes, respectively
Results of Correlation Analysis Between Diagnostic Genes and Immune Cells
In order to explore the diagnostic value of mRGs in the macula, this article constructed a diagnostic model based on the expression profiles of 31 intersection genes and using multiple machine learning algorithms. The diagnostic model can effectively classify AMD and control group. Specifically, this article selects different numbers of top features to build diagnostic models based on RF, Adaboost, KNN, and SVM-RFE algorithms. The RF, Adaboost, KNN, and SVM-RFE algorithms achieved the maximum accuracy in the internal test set when selecting the first 9, 26, 14, and 13 features, respectively (Fig. 5A, C, E, and G). Figure 5B, D, F, and H are the AUCs of the four algorithms on the internal test set, respectively. Among them, the SVM-RFE algorithm reached the largest AUC (0.806). Therefore, we further tested the diagnostic model constructed on the external test set, and its AUC was 0.816 (Fig. 5I). In addition, we give the maximum AUC of the deep neural network and the Rogeist regression algorithm in the supplementary material Figure S2.

A diagnostic model based on multiple machine learning algorithms. A, C, E, G The accuracy change curves of the diagnostic model built based on RF, Adaboost, KNN, and SVM-RFE algorithms, respectively, after selecting different top features. B, D, F, H The ROC curves of the diagnostic models constructed by the four methods respectively. I The ROC curve used to verify the diagnostic model built by SVM on the external test set
Furthermore, the columnar line chart model is a graphical risk prediction tool that integrates multiple predictive factors into a single predictive model, providing intuitive and clinically accessible prediction outcomes. In our study, the columnar line chart model was used for AMD risk assessment and personalized prediction. We constructed the columnar line chart model based on the first 13 diagnostic genes (CYP27B1, FUS, FZD5, GLS2, GLYATL1, HSPA1A, NDUFA4L2, PDPN, SLC2A1, SNN, SPR, GSTZ1 and TRAF6) (Fig. 6A). Figure 6B gives the calibration curve of the nomogram model. The nomogram model based on 13 diagnostic genes agreed with the ideal model. DCA analysis showed that although both the nomogram model and individual diagnostic genes produced net benefits, the net usage of the nomogram model was significantly greater than that of individual diagnostic genes. This suggests that nomogram models may have more clinical value than individual diagnostic genes (Fig. 6C). Clinical impact curve analysis showed that the nomogram model had high diagnostic ability (Fig. 6D). In addition, to explore the correlation between diagnostic genes and immune cell infiltration abundance, this article screened the correlation results with p < 0.0001 based on the Spearman correlation coefficient of the two. Figure 7A–U show each gene and its two most strongly correlated immune cells. The remaining correlation analysis results are shown in Supplementary material Figures S4-S6.

Construction of nomogram model and DCA analysis results. A A nomogram model built based on diagnostic genes. B The calibration curve of the nomogram model. C DCA analysis. D The clinical decision curve

Scatter plot of correlation between diagnostic genes and immune cell infiltration abundance. A, B Correlation scatter plots between CYP27B1 and the two cells with the strongest correlation. C, D Correlation scatter plots between FUS and the two cells with the strongest correlation. E, F Correlation scatter plots between FZD5 and the two cells with the strongest correlation. G, H Correlation scatter plots between GLS2 and the two cells with the strongest correlation. I, J Correlation scatter plots between GLYATL1 and the two cells with the strongest correlation. K A scatter plot of the correlation between HSPA1A and macrophage M1. L A scatter plot of the correlation between NDUFA4L2 and T cells follicular helper. M, N The correlation scatter plot between PDPN and the two cells with the strongest correlation. O, P The correlation scatter plot between SLC2A1 and the two cells with the strongest correlation. Q, R A correlation scatter plot between SNN and the two cells with the strongest correlation. S, T A correlation scatter plot between SPR and the two cells with the strongest correlation. U A scatter plot of the correlation between TRAF6 and T cells follicular helper
Analysis Results of scRNA-seq Data
Immune infiltration analysis found that the infiltration abundance of various immune cells was significantly different between AMD and normal groups. This article further explores the interaction between various immune cells based on scRNA-seq data. Specifically, this article performs quality control, standardization, scaling, dimensionality reduction, clustering, and cell type identification on scRNA-seq of two AMD samples. Figure 8A and B show violin plots of critical indicators before and after quality control. Figure 8C shows the score heat map for identifying cell types based on the singleR algorithm. Figure 8D offers two-dimensional plan views of different types of cells after nonlinear dimensionality reduction using uniform manifold approximation and projection (UMAP). A total of nine cell types (chondrocytes, CMP, endothelial cells, tissue stem cells, neurons, T cells, monocytes, NK cells, and fibroblasts) were identified in this article. Figure 9A and B show the expression landscape of some hub genes in immune cells (T cells, NK cells, and monocytes). Among them, FUS is highly expressed in NK cells and T cells. HSPA1A and SNN are highly expressed in monocytes.

scRNA-seq data preprocessing and cell type annotation results. A, B Violin plots of nFeature_RNA, nCount_RNA, mitochondrial gene proportion (percent.mt), and red blood cell gene proportion (percent.HB) before and after quality control respectively. C A scoring heat map for cell type scoring of different cell clusters based on the singleR algorithm. D A two-dimensional plan view of cell types annotated after nonlinear dimensionality reduction of scRNA-seq based on the UMAP algorithm.

The expression landscape of some hub genes in immune cells (T cells, NK cells, and monocytes) is shown. Among them, FUS is highly expressed in NK cells and T cells. HSPA1A and SNN are highly expressed in monocytes
Furthermore, this article conducts a pseudo-chronological analysis of different types of cells after annotation (Fig. 10 A, B). Monocytes, T cells, and chondrocytes are in the early stages of differentiation. NK cells differentiate last. The remaining cells are located at multiple stages of differentiation. Cell communication analysis revealed that chondrocytes had stronger signaling than other cell types (Fig. 10 C, D). We found multiple significant pathways using each immune cell group (T cells, monocytes, and NK cells) as source and target, respectively. Then, we conducted communication analysis with other cell groups (Fig. 10 E, F). We will explore the role of these pathways in the development and progression of AMD in detail in the “Discussion” section.

Results of cell trajectory analysis and communication analysis between immune cells. A, B The results of grouping cells according to pseudo-chronological order and cell type respectively. C, D Network diagrams of the number and intensity of signaling pathways in the communication process between immune cells respectively. The size of the nodes in the graph reflects the number of cells of this type. The thickness of the line reflects the amount/strength of communication between cells. E, F Bubble diagrams of the pathways in which each immune cell acts as a source and target and communicates with other cells respectively
Potential Drug Identification
To identify potential small-molecule drugs that could treat AMD patients, we imported the top 150 upregulated DEGs and the top 150 downregulated DEGs into the cMAP database. The results showed that the top ten highest-scoring compounds included triamterene, LE-135, eflornithine, ellipticine, L-690330, IOX2, SAL-1, NF-449, miglitol, and SR-57227A, which are potential therapeutic candidates for AMD patients but have not yet been validated by existing literature (Fig. 11 A-J).

Screening of potential small-molecule compounds for AMD by cMAP analysis. A–J Eflornithine, ellipticine, IOX2, L-690330, LE-135, miglitol, NF-449, SAL-1, SR-57227A, and triamterene
Validation Results of the Hub Genes by qRT-PCR
To further verify the results of bioinformatics analysis, the mRNA levels of the 13 hub genes were determined with qRT-PCR. As illustrated in Fig.12 , the SNN, PDPN, GLYATL1, CYP27B1, GLS2, NDUFA4L2, FUS, and SLC2A1 were significantly downregulated in H2O2-treated ARPE‐19 cells compared to normal cells (all p < 0.05), while the HSPA1A and TRAF6 were significantly upregulated in H2O2-induced ARPE‐19 cells (all p < 0.05), as predicted by the bioinformatics analysis. Although the expression of FZD5, GSTZ1, and SPR showed no statistical difference between H2O2-treated cells and normal cells, their expression trend was consistent with bioinformatics analysis.

Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) for the expression of the hub genes in ARPE-19 cells of oxidative damage and the controls. Expression of hub genes was normalized against GAPDH expression (p < 0.05). ***p < 0.001, **p < 0.01, *p < 0.05
Discussion
From the bioinformatics perspective, this article explores the role of MRGs and the signaling pathways involved in the progression of AMD through RNA-seq, scRNA-seq, and other data. Firstly, this paper intersects the DEGs of the normal group and the AMD group. After conducting GO enrichment analysis on 31 intersection genes, it was found that some top pathways have been confirmed to play a critical role in AMD. Eszter Emri conducted a combined transcriptome, proteome, and secretome analysis from three genetically distinct human donors and found that AMD samples were involved in the unique pathway of the extracellular matrix (Emri, et al. 2020). Zhao et al. detected 44 and 53 significantly different metabolites in positive and negative ion modes in the AMD and control groups, respectively (Zhao et al. 2023). Retinal ganglion cell (RGC) death is the leading cause of AMD. The study by Zhong et al. found that K + channels, including ether-à-go-go (Eag), may contribute to dendritic repolarization during excitatory postsynaptic potentials and the attenuation of action potential backpropagation and protect RGCs (Zhong et al. 2013).
Secondly, this paper proposes a DS-NMF algorithm. This method reconstructs the input gene expression profile using the traditional NMF algorithm. Specifically, the original gene expression profile is passed through a multi-layer feedforward neural network during reconstruction. The sample age is then used as prior information at the network’s top level to change the data distribution. Finally, the reconstructed data is used as the input of the NMF algorithm. Based on the DS-NMF algorithm, this article identified two subtypes with apparent differences in enrichment pathways and immune cell infiltration. Most of the pathways with significant differences between the two subtypes have been confirmed to play a critical role in the progression of AMD. Research by Santos et al. revealed that complement and coagulation components and adhesion factors are differential biomarkers for vitreoretinal eye diseases, including AMD (Santos et al. 2023). Subramanian et al. found that OCT biomarkers were associated with visual impairment and vitreomacular adhesion in patients with diabetic macular edema (Subramanian et al. 2023). Chen et al. found that fenofibrate inhibited subretinal fibrosis by inhibiting TGF-β-Smad2/3 signaling and Wnt signaling in neovascular AMD (Chen et al. 2020).
This article builds a robust diagnostic model based on various machine learning algorithms to assist the clinical diagnosis of AMD. Among them, the AUC of the diagnostic model built using the SVM-RFE algorithm reached 0.806 and 0.816 in the internal and external test sets, respectively. We screened a total of 13 diagnostic genes (CYP27B1, FUS, FZD5, GLS2, GLYATL1, HSPA1A, NDUFA4L2, PDPN, SLC2A1, SNN, SPR, GSTZ1 and TRAF6). Some genes have been confirmed to be closely related to AMD. McKay and others found that two SNPs in the CYP27B1 gene are associated with early AMD (McKay et al. 2017). The findings of Choudhury et al. suggest that the interaction between HSPA1A and FHL-1 may impact AMD. This may mean that the expression and function of HSPA1A may be related to the onset and progression of AMD (Choudhury et al. 2021). In a multicenter cohort association study of SLC2A1 single nucleotide polymorphisms and AMD, Baas et al. found population-dependent genetic risk heterogeneity in AMD (Baas et al. 2012). Choroidal neovascularization (CNV) is a form of wet AMD. Ding et al. found that inhibiting TRAF6 can alleviate choroidal neovascularization in vivo (Ding et al. 2018).
Since various immune cells are significantly related to diagnostic genes, this article uses scRNA-seq data to analyze the differences in pseudo-chronology and communication of different immune cells. This article discovered the significant signaling pathways conducted through cell communication analysis when three immune cells communicate with other cells. The retinal pigment epithelium (RPE) performs many functions critical to retinal health and visual function and is implicated in the development of AMD. Studies by Jadeja et al. have shown that the loss of NAMPT in the aging RPE will promote cell senescence (Jadeja et al. 2018). Schlecht and others discovered that regulating the SPP1 pathway provides new opportunities for AMD therapeutic intervention by establishing a mouse model (Schlecht et al. 2020). Chandola et al. found that CD44 aptamer-mediated cargo delivery to retinal pigment epithelial cell lysosomes can prevent AMD (Chandola et al. 2019). Lee et al. found that COE and BP exert anti-angiogenic effects on retinal neovascularization by inhibiting the expression of AREG and other genes (Lee et al. 2016). Finally, qPCR validation was performed on all diagnostic genes. The expression trends of most genes were confirmed.
Conclusion
Aging changes in macular structure may cause different subtypes of AMD. This article proposes a DS-NMF algorithm to identify two subtypes of AMD. The two subtypes have significant differences in enriched pathways and immune infiltration. Based on the MRGs between subtypes, this paper constructed an AMD diagnosis model based on four machine learning methods. The diagnostic model constructed by the SVM-RFE algorithm can reasonably predict the occurrence of AMD. The communication patterns between immune cells and other cells in AMD samples were explored through scRNA-seq data sets. The subtypes and pathways identified in this article and the diagnostic model constructed in this article can provide new insights into the precise treatment of AMD.
Data Availability
The data used in the paper was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/).
References
Ambati J et al (2003) An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med 9(11):1390–1397
Aran D et al (2019) Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20(2):163–172
Baas DC et al (2012) Multicenter cohort association study of SLC2A1 single nucleotide polymorphisms and age-related macular degeneration. Mol vis 18:657–674
Beatty S et al (2000) The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol 45(2):115–134
Chandola C et al (2019) CD44 aptamer mediated cargo delivery to lysosomes of retinal pigment epithelial cells to prevent age-related macular degeneration. Biochem Biophys Rep 18:100642
Chang J et al (2023) Constructing a novel mitochondrial-related gene signature for evaluating the tumor immune microenvironment and predicting survival in stomach adenocarcinoma. J Transl Med 21(1):191
Chen B et al (2018) Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol 1711:243–259
Chen Q et al (2020) Fenofibrate inhibits subretinal fibrosis through suppressing TGF-β-Smad2/3 signaling and Wnt signaling in neovascular age-related macular degeneration. Front Pharmacol 11:580884
Choudhury R et al (2021) FHL-1 interacts with human RPE cells through the α5β1 integrin and confers protection against oxidative stress. Sci Rep 11(1):14175
Ding D et al (2018) Inhibition of TRAF6 alleviates choroidal neovascularization in vivo. Biochem Biophys Res Commun 503(4):2742–2748
Emri E et al (2020) A multi-omics approach identifies key regulatory pathways induced by long-term zinc supplementation in human primary retinal pigment epithelium. Nutrients 12(10). https://doi.org/10.3390/nu12103051
Fan Y et al (2008) Spatial patterns of brain atrophy in MCI patients, identified via high-dimensional pattern classification, predict subsequent cognitive decline. Neuroimage 39(4):1731–1743
Fan Q et al (2023) Contribution of common and rare variants to Asian neovascular age-related macular degeneration subtypes. Nat Commun 14(1):5574
Han D, He X (2023) Screening for biomarkers in age-related macular degeneration. Heliyon 9(7):e16981
Hänzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14:7
Hao Y et al (2024) Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat Biotechnol 42(2):293–304
Jadeja RN et al (2018) Loss of NAMPT in aging retinal pigment epithelium reduces NAD(+) availability and promotes cellular senescence. Aging (albany NY) 10(6):1306–1323
Jarrett SG, Boulton ME (2012) Consequences of oxidative stress in age-related macular degeneration. Mol Aspects Med 33(4):399–417
Jin S et al (2021) Inference and analysis of cell-cell communication using Cell Chat. Nat Commun 12(1):1088
Jones MK et al (2023) Integration of human stem cell-derived in vitro systems and mouse preclinical models identifies complex pathophysiologic mechanisms in retinal dystrophy. Front Cell Dev Biol 11:1252547
Kaarniranta K, Salminen A (2009) Age-related macular degeneration: activation of innate immunity system via pattern recognition receptors. J Mol Med (berl) 87(2):117–123
Karunadharma PP et al (2010) Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol vis Sci 51(11):5470–5479
Khandhadia S, Lotery A (2010) Oxidation and age-related macular degeneration: insights from molecular biology. Expert Rev Mol Med 12:e34
Lee YM et al (2016) Cnidium officinale extract and butylidenephthalide inhibits retinal neovascularization in vitro and in vivo. BMC Complement Altern Med 16:231
Lim LS et al (2012) Age-related macular degeneration. Lancet 379(9827):1728–1738
Liu Y et al (2024) The prognostic genes model of breast cancer drug resistance based on single-cell sequencing analysis and transcriptome analysis. Clin Exp Med 24(1):113
McKay GJ et al (2017) Associations between serum vitamin D and genetic variants in vitamin D pathways and age-related macular degeneration in the European Eye Study. Ophthalmology 124(1):90–96
Newman AM et al (2012) Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med 4(2):16
Orozco LD et al (2020) Integration of eQTL and a single-cell atlas in the human eye identifies causal genes for age-related macular degeneration. Cell Rep 30(4):1246-1259.e6
Pedregosa F et al (2011) Scikit-learn: machine learning in Python. J Mach Learn Res 12(null):2825–2830
Pei Y et al (2023) Construction and evaluation of Alzheimer’s disease diagnostic prediction model based on genes involved in mitophagy. Front Aging Neurosci 15:1146660
Ritchie ME et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43(7):e47
Santos FM et al (2023) Proteomics profiling of vitreous humor reveals complement and coagulation components, adhesion factors, and neurodegeneration markers as discriminatory biomarkers of vitreoretinal eye diseases. Front Immunol 14:1107295
Schlecht A et al (2020) Secreted phosphoprotein 1 expression in retinal mononuclear phagocytes links murine to human choroidal neovascularization. Front Cell Dev Biol 8:618598
Stoeckius M et al (2018) Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19(1):224
Subramanian B et al (2023) Association of OCT biomarkers and visual impairment in patients with diabetic macular oedema with vitreomacular adhesion. PLoS ONE 18(7):e0288879
Wang Z et al (2021) Integrated Analysis of DNA methylation and transcriptome profile to identify key features of age-related macular degeneration. Bioengineered 12(1):7061–7078
Yang Y et al (2023) Identification of the immune landscapes and follicular helper T cell-related genes for the diagnosis of age-related macular degeneration. Diagnostics (Basel) 13(17). https://doi.org/10.3390/diagnostics13172732
Yu G et al (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16(5):284–287
Zhao T et al (2023) Integrative metabolome and lipidome analyses of plasma in neovascular macular degeneration. Heliyon 9(10):e20329
Zhong YS et al (2013) Potassium ion channels in retinal ganglion cells (review). Mol Med Rep 8(2):311–319
Funding
This study was supported by the Jiangsu Graduate Practice and Innovation program (NO. 134422631103).
Author information
Authors and Affiliations
Contributions
Shenglai Zhang: Conceptualization, Methodology, Software, Visualization, Writing – Original Draft Preparation, Writing-review & editing.
Ying Yang: Conceptualization, Methodology, Software, Visualization,Writing-review and editing.
Jia Chen: Conceptualization, Methodology, Software, Writing-review and editing.
Shu Su: Data curation, Software, Writing-review & editing.
Yu Cai: Visualization, Writing-review and editing.
Xiaowei Yang: Data curation, Writing-review, Funding and editing.
All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, S., Yang, Y., Chen, J. et al. Integrating Multi-omics to Identify Age-Related Macular Degeneration Subtypes and Biomarkers. J Mol Neurosci 74, 74 (2024). https://doi.org/10.1007/s12031-024-02249-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12031-024-02249-9