
Abstract
The pathogenesis of Alzheimer’s disease (AD) is complex and involves an imbalance between production and clearance of amyloid-ß peptides (Aß), resulting in accumulation of Aß in senile plaques. Hypercholesterolemia is a major risk factor for developing AD, with cholesterol shown to accumulate in senile plaques and increase production of Aß. ABCG4 is a member of the ATP-binding cassette transporters predominantly expressed in the CNS and has been suggested to play a role in cholesterol and Aß efflux from the brain. In this study, we bred Abcg4 knockout (KO) with the APPSwe,Ind (J9) mouse model of AD to test the hypothesis that loss of Abcg4 would exacerbate the AD phenotype. Unexpectedly, no differences were observed in novel object recognition (NOR) and novel object placement (NOP) behavioral tests, or on histologic examinations of brain tissues for senile plaque numbers. Furthermore, clearance of radiolabeled Aß from the brains did not differ between Abcg4 KO and control mice. Metabolic testing by indirect calorimetry, glucose tolerance test (GTT), and insulin tolerance test (ITT) were also mostly similar between groups with only a few mild metabolic differences noted. Overall, these data suggest that the loss of ABCG4 did not exacerbate the AD phenotype.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in people over the age of 65 years. It is a progressive neurodegenerative disorder that can manifest as deficits in memory, executive function, visuospatial cognition, language function, and personality changes (Albert et al. 2011; McKhann et al. 2011). While a clinical diagnosis of probable AD dementia can be made if other causes of dementia are ruled out, a definitive diagnosis of AD neuropathological changes requires a histological examination of brain tissue. Key neuropathological features of AD include neurofibrillary tangles (NFTs), primarily composed of abnormally phosphorylated tau protein, extracellular deposition of amyloid-ß peptides (Aß) in senile plaques, and dystrophic neurites (Hyman et al. 2012; Montine et al. 2012).
The amyloid hypothesis of AD postulates that an imbalance between Aß production and clearance resulting in the accumulation of Aß is a driver of AD pathogenesis (Hardy and Selkoe 2002). While rare genetic early-onset forms of AD are associated with the over-production of abnormal proteins, including Aß and tau (Scheuner et al. 1996; Naj et al. 2017), the more common late-onset Alzheimer’s disease is associated with reduced clearance of Aß from the brain (Mawuenyega et al. 2010; Tarasoff-Conway et al. 2015; Zuroff et al. 2017). However, therapies utilizing monoclonal antibodies targeting Aß to increase efflux from the brain have been largely unsuccessful (Salloway et al. 2014; Doody et al. 2014), indicating that AD pathogenesis is not so simple.
AD is a complex disease, and its etiology is likely multifactorial. There is a growing body of work which links neuroinflammation, oxidative damage, and dysfunctional glucose and lipid metabolism to AD (Kapogiannis and Mattson 2011; Croteau et al. 2018; Butterfield and Halliwell 2019a, b; Behl et al. 2020; Leng and Edison 2020; Ionescu-Tucker and Cotman 2021; Meng et al. 2022). Accordingly, diabetes mellitus and hypercholesterolemia are major risk factors for developing AD (Shepardson et al. 2011a; Alzheimer’s Association 2013; Arnold et al. 2018; Meng et al. 2022). Patients with AD have brain insulin resistance (Arnold et al. 2018), and treatment with intranasal insulin improves cognitive function (Reger et al. 2008). Meanwhile, cholesterol has been shown to accumulate in senile plaques (Mori et al. 2001) and increase production of Aß (Shepardson et al. 2011a), but evidence on the effects of statins on cognitive function is mixed (Kurata et al. 2011; Sano et al. 2011; Shepardson et al. 2011b; Tong et al. 2012). Nevertheless, apolipoprotein E4 remains the most potent genetic risk factor for the development of AD (Naj et al. 2017). Interestingly, of the identified transporters that efflux Aß out of the brain, many, like LRP1, LRP2, ABCA1, ABCB1 (also known as P-glycoprotein-1), and ABCG4 (Shibata et al. 2000; Cirrito et al. 2005; Bell et al. 2007; Do et al. 2012; Dodacki et al. 2017), also have roles in cholesterol metabolism.
ABCG4 is a member of the ATP-binding cassette transporter family that regulates cholesterol homeostasis; ABCG4 is predominantly expressed in the CNS (Yoshikawa et al. 2002; Cserepes et al. 2004; Bojanic et al. 2010). It has also been suggested to have a function in glucose-stimulated insulin secretion (GSIS) (Hou et al. 2016). In the brain, ABCG4 is expressed in neurons, astrocytes, microglia, and capillary endothelial cells at the blood-brain barrier (BBB) (Tarr and Edwards 2008; Uehara et al. 2008; Bojanic et al. 2010; Dodacki et al. 2017). Abcg4 may play a role in cholesterol efflux from the brain (Wang et al. 2004, 2008; Vaughan and Oram 2006), and in vitro studies suggest it may also play a role in the export of Aß from the brain at the BBB (Do et al. 2012; Dodacki et al. 2017). Furthermore, in in vitro studies, ABCG4 was found to inhibit γ-secretase activity, thus reducing Aß production (Sano et al. 2016). Abcg4−/− mice were reported to have a deficit in contextual memory (Bojanic et al. 2010) though no other confirmatory reports have been published. It has therefore been posited that Abcg4 may play a protective role against the development of AD.
We sought to test the hypothesis that Abcg4 may be involved in AD pathogenesis using Abcg4 knockout (KO) mice. We chose the APPSwe,Ind (also referred to as J9) mouse model of AD because it has been reported to have a slower onset AD than other models, and loss of Abcg4 on this background would be expected to accelerate onset, should the hypothesis be supported. The J9 model is a transgenic mouse that expresses human amyloid precursor protein (APP) with Swedish (K670N/M671L) and Indiana (V717F) mutations, which increases Aß formation and favors Aß42, the form more likely to be found in senile plaques (Hsia et al. 1999; Mucke et al. 2000). We crossed J9 mice with Abcg4 KO mice and assessed metabolic and behavioral effects.
Methods
Animal Care
All animal protocols were approved by the University of Cincinnati IACUC, Cincinnati, OH. Mice were group-housed in individually ventilated PIV cages maintained on 14 h:10 h light and dark cycles and fed a standard chow diet (Envigo 7912; Harlan Teklad, Madison, WI) with access to water ad libitum unless otherwise specified by individual experimental protocols.
Tg(PDGFB-APPSwInd)J9Lms embryos submitted by Gladstone Institute of Neurological Disease (San Francisco, CA) to Jackson Labs (Bar Harbor, ME) were revived from cryopreservation. Three founder mice were received, and one bred successfully establishing the TgJ9 + mouse line. The Abcg4 KO mice line was generated as previously described (Dodacki et al. 2017). Abcg4 KO mice were bred with TgJ9 + mice to produce Abcg4+/−, J9 + mice. These mice were bred with Abcg4+/−, J9- mice to produce the experimental cohort and ensure no more than a single copy of the J9 transgene was present in any animal.
Genotyping Details
Genomic DNA was isolated from a tail snip. The genotype of Abcg4 allele was determined by PCR using primers (5′-CTGCCCTCCCTTATCAATC-3′) and (5′-TATCACAAGCCAGCCTTCTCGG-3′) to detect a 423 bp fragment for the WT allele, and primers (5′-CTGCCCTCCCTTATCAATC-3′) and (5′- TTGCTCACCATGGTGGCGACCGGTGG-3′) primers were used to detect a 400 bp fragment for the mutant allele. The presence of the J9 transgene was determined by amplifying a 360 bp fragment using primers (PDAPP-F; GGTGAGTTTGTAAGTGATGCC and PDAPP-R; TCTTCTTCTTCCACCTCAGC3). PCR products were run on a 1.5% agarose gel under standard conditions using a 100 bp DNA ladder for size identification. DNA was stained using SYBR Safe DNA gel stain (S33102, Thermo Fisher Scientific, Waltham, MA) and scanned on a gel station (Universal Hood II, Bio-Rad).
Behavioral Testing
Behavioral testing was performed by the University of Cincinnati Mouse Metabolic Phenotyping Center Animal Behavior Core. Mice were assessed at 6–7 months of age and 16–19 months of age.
Open field test: Animals were placed into a novel open field environment (e.g., ~ 36 × 36 in a plastic box or circular field) for up to 30 min. Time spent in the middle and time in the periphery are recorded. Time spent in the periphery near the walls is an indicator of anxiety (Choleris et al. 2001).
Novel object recognition (NOR): Mice were placed into the open field apparatus, as described above, containing 2 different objects. Animals were given 15 min to explore the objects and returned to their home cages. While the mice were away from the arena, one object was replaced with a different object. The animals were returned to the apparatus 24 h later with one of the former objects and a new object. The difference in the amount of time exploring the new vs. familiar object reflects the memory of the previous experience and the animal’s non-spatial learning (Antunes and Biala 2012).
Novel object placement (NOP): Mice were placed into the open field apparatus, as described above, containing 2 different objects. Animals were given 15 min to explore the objects and returned to their home cages. While the mice were away from the arena, one object was moved to a different location within the apparatus. The animals were returned to the apparatus 24 h later with the same objects, one object in the same location and one object in a different location. The difference in the amount of time exploring the moved vs. unmoved object reflects the memory of the previous experience and the animals’ spatial learning (Antunes and Biala 2012).
Indirect Calorimetry
Mice were individually housed in chambers maintained at 23 °C with 12 h:12 h light:dark cycle for simultaneous measurement of oxygen consumption (vO2, ml/h), carbon dioxide production (vCO2, ml/h), energy expenditure, respiratory exchange ratio (RER), and locomotor activity via indirect calorimetry (TSE Systems, Chesterfield, MO, USA). Energy expenditure was calculated using the simplified Weir equation (H = 1.44 (3.94 VO2 + 1.11 VCO2)), and energy expenditure or heat (H) was expressed as kcal/h. Previously, the instrument was calibrated with gas cylinders containing nitrogen, 1% carbon dioxide, and atmospheric air mixture (oxygen 20.7%, carbon dioxide 0.03%). Data were acquired every 20 min using the LabMaster software (TSE Systems).
Food Intake
Food intake was assessed with a BioDAQ Food intake monitoring system (Research Diets, Inc., New Brunswick, NJ) between the ages 8 and 12 months and 16 and 18 months. Mice were individually housed in BioDAQ cages, which monitored the weight of food in the hopper. Water was provided ad libitum. Cumulative food intake over 3 days was calculated for each mouse in each trial. For each age group, mice were tested twice with a 1-week gap between trials to assess if potential differences in cognitive ability affected the feeding behavior of the mice due to the novel environment of the BioDAQ cage.
Glucose (GTT) and Insulin (ITT) Tolerance Tests
Mice were fasted for 4–6 h and then administered via intraperitoneal injection 2 g/kg glucose for GTT or 0.6 IU/kg human insulin, Humulin R (HI-213; Lilly, Indianapolis, IN) for ITT. Blood glucose was measured with Accu-Chek Nano electronic glucometer (Roche Applied Science, Indianapolis, IN, USA) at 0, 30, 60, 90, and 120 min after intraperitoneal administration of glucose or insulin. If blood glucose dropped below 40 mg/dL, the test was terminated, and the animal was administered glucose.
Measurement of Aß Clearance
A guide stainless steel cannula (22-gauge, Plastics One, Roanoke, VA) was implanted stereotaxically into the right caudate putamen of anesthetized mice. The cannula tip coordinates for placement were 0.9 mm anterior from bregma, 1.9 mm lateral from midline, and 2.9 mm below the surface. Animals were allowed to recover for 4–5 h to allow for some blood-brain barrier recovery, but before substantial inflammatory response develops. Tracer fluid (1.0 μl) containing 50nCi of [14C]-inulin (as reference marker) and 50nCi of [3H]Aβ1–42 was injected with a Motorized Integrated Stereotaxic Injector (iSi) system (Stoelting Co.) into the interstitial fluid (ISF) over 5 min. After injection, the needle was left in place for 5 min. CSF was collected 60 min after injection, and brain tissue from the caudate putamen was collected immediately after.
Thio-S Staining for Plaques
After completing the experiments, the mice were euthanized by CO2 anesthesia followed by thoracotomy to expose the heart. Mice were perfused with chilled PBS followed by 4%PFA via the intracardiac route. The perfused brains were collected and fixed in 4% PFA for another 24 h. Then transferred to 30% sucrose and stored at 4 °C for at least 48 h. Sagittal sections (~ 30 µm) through the hippocampus were obtained and stained with 0.5% ThioS in 50% EtOH for 10 min at room temperature. Sections were rinsed with 50% EtOH, followed by PBS, and mounted on glass slides with aqueous mounting media, then were protected from light until visualized by fluorescence microscopy and the number of plaques counted. Separate sections were additionally stained with AntiNeuN antibody conjugated to Alexa Fluor555 (Millipore Sigma, Cat#MAB377A5) at 1:100 dilution to visualize neuronal structure.
Results
Presence of Abcg4 Did Not Make a Difference in Long-Term Memory
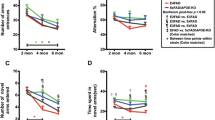
In order to assess spatial and non-spatial memory, we performed novel object placement (NOP) and novel object recognition (NOR) tests. Animals were tested at 2 time points in an attempt to quantify if there was a progressive decline in cognitive function over time. The mice were first tested at 6–7 months of age and then at 16–19 months of age. At both time points, the performance of Abcg4−/−, J9 + mice (G4KO J9 +) did not differ from the performance of the Abcg4+/+, J9 + mice (G4WT J9 +). In both the NOP and NOR tasks, neither group of mice showed a preference for the novel objects or the moved objects (Fig. 1). Wild-type mice are expected to have a preference for novel objects, suggesting a defect with memory in the J9 + mice, even at the early timepoint, since the animals did not prefer the more familiar object or placement.

Behavioral testing. Novel object recognition (NOR) (A–D) and novel object placement (NOP) (E–H) tests were performed on Abcg4+/+ J9 + and Abcg4−/− J9 + mice at ages 6–7 months and 16–19 months to assess spatial and non-spatial long-term memory. Total time with each object is shown. Symbols represent individual animals. Lines represent mean ± 1SD
Metabolic Effects
Despite not finding any differences in behavior, due to the association of AD with metabolic dysfunction, we were also interested in the metabolic effects of Abcg4 KO and the J9 model. Body weights were measured monthly, and body composition every 2–4 months. Throughout the course of the experiment, both male and female Abcg4−/−, J9 + mice had similar body weights and body composition as their respective Abcg4+/+, J9 + counterparts (Fig. 2). The presence or absence of Abcg4 in the J9 strain (Abcg4+/+, J9 + vs. Abcg4−/−, J9 +) did not demonstrate any difference in body weight or composition. Similarly, as expected, there was no significant difference in mean food intake, energy expenditure, activity, or respiratory exchange rate (RER) between groups at either time point (Figs. 3, 4, 5).

Body weight and composition. Abcg4+/+J9 + and Abcg4−/−J9 mice were weighed monthly, and body composition was measured by EchoMRI every 2–4 months. Symbols represent individual animals. The error bars represent mean ± 1SD. No statistically significant differences were observed between genotypes

Food consumption. Food consumption of Abcg4+/+ J9 + and Abcg4−/−J9 mice was assessed with a BioDAQ Food intake monitoring system between ages 8–12 months (A–B) and 16–18 months (C–D). Cumulative food intake over 3 days is displayed for each trial with a 1-week gap between trials. Symbols represent individual animals. The error bars represent mean ± 1SD. No statistically significant differences were observed between genotypes or between trials

Metabolic profile 8–10 months. Metabolic profile of male (A–C) and female (D–F) Abcg4+/+ J9 + (G4WT J9 + , n = 6 male, 7 female) and Abcg4−/− J9 + (G4KO J9 + , n = 11 male, 9 female) mice were assessed by indirect calorimetry between 8 and 10 months of age. Each line represents the mean of each respective genotype measured every 20 min. The shaded area indicates the dark cycle. No consistent patterns to distinguish between the different genotypes were noted

Metabolic profile 16–19 months. Metabolic profile of male (A–C) and female (D–E) Abcg4+/+ J9 + (G4WT J9 + , n = 7 male, 4–5 female) and Abcg4−/− J9 + (G4KO J9 + , n = 7 male, 7 female); mice were assessed by indirect calorimetry between 16 and 19 months of age. Each line represents the mean of each respective genotype measured every 20 min. The shaded area indicates the dark cycle
When comparing energy expenditure to body weight, Abcg4−/−, J9 + and Abcg+/+, J9 + female mice displayed the expected pattern of increasing EE as body weight increases (Fig. 6C and D). Interestingly, Abcg4−/−, J9 + male mice also maintained this pattern when younger; however, when tested again at 16–19 months of age, this relationship inverted and demonstrated decreased EE with increased body weight (Fig. 6B). While the average body weights and EE were similar between groups, Abcg4+/+, J9 + male mice did not demonstrate this inverse relationship (Fig. 6A and B).

Energy expenditure vs. body weight. Linear regression of the relationship between cumulative energy expenditure (EE) measured by indirect calorimetry and body weight was calculated for male (A–B) and female (C–D) Abcg4+/+ J9 + and Abcg4−/− J9 mice. For male mice, a p-value for the null hypothesis that the slopes are the same is displayed. P-value < 0.05 was considered significant. The was no significant difference in the slopes of female mice
To assess the glucose metabolism, GTT and ITT were performed at 8–12 months and 16–18 months. When compared to male Abcg4+/+, J9 + mice, male Abcg4−/−, J9 + mice demonstrated slightly elevated blood glucose levels 30 min after glucose injection (Fig. 7A), but not enough to cause statistically significant changes in overall GTT AUC at 8–12 months (Fig. 7C) or at 16–19 months (Fig. 8A). Female Abcg4−/−, J9 + mice and Abcg4+/+, J9 + mice did not show significant differences in glucose tolerance at either time point (Figs. 7 and 8). However, female Abcg4−/−, J9 + mice did demonstrate reduced insulin sensitivity at the 60-min time-point after insulin injection (Fig. 8E). The biological relevance of this finding is unclear. Male Abcg4−/−, J9 + mice and Abcg4+/+, J9 + mice did not show any difference in insulin sensitivity at either time point (Figs. 7 and 8).

Glucose tolerance and insulin resistance 8–12 months. Glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed between 8 and 12 months of age on Abcg4+/+J9 + and Abcg4−/−J9 + male and female mice. The error bars denote ± 1SD. Panels C (GTT AUC) and F (ITT AUC) represent the area under the curve (AUC) respective analyses. Symbols represent individual animals. The error bars represent mean ± 1SD

Glucose tolerance and insulin resistance 16–18 months. Glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed between 16 and 18 months of age on Abcg4+/+ J9 + and Abcg4−/− J9 + male and female mice. The error bars denote ± 1 SD. Panels C (GTT AUC) and F (ITT AUC) represent the area under the curve (AUC) respective analyses. Symbols represent individual animals. The error bars represent mean ± 1SD
Senile Plaque Formation
After behavioral and metabolic testing was complete, the mice were euthanized, and a subset of the brains was cryopreserved for ThioflavinS staining to visualize amyloid plaques. Plaques were counted in the whole brain and hippocampus to quantify any differences. Plaque numbers varied greatly, but no significant differences in plaque number were noted in the whole brain or hippocampi of J9 + mice regardless of the presence or absence of Abcg4 (Fig. 9).

Amyloid plaque formation. The brains of Abcg4+/+J9 + , Abcg4−/−J9 + , and Abcg4−/−J9- mice were stained with ThioflavinS (blue) to visualize Aß plaques and anti-NeuN Ab conjugated with Alexa555 (pink) to visualize neurons (A). Arrows indicated Aß plaques. The number of plaques was counted in whole brain (B) and hippocampus (C) of Abcg4+/+ J9 + and Abcg4−/− J9 + mice. Symbols represent individual animals. Lines denote the mean ± 1 SD. (N = 5 per group). Abbreviations: CC corpus callosum, CA cornu ammonis, den dentate gyrus
Aß Clearance
In light of the lack of difference in AD neuropathologic change with Abcg4 KO, we directly assessed Aß clearance. Radiolabeled Aß was injected into the brains of Abcg4 KO and control mice and then measured 60 min later. There was no difference in the amount of radiolabeled Aß recovered in the brain and CSF of Abcg4 KO vs Abcg4 WT mice (Fig. 10), indicating that Aß clearance rate is unchanged with knockout of Abcg4. Radiolabeled inulin, which is cleared only by passive clearance, was injected simultaneously as a control. There was no difference in the amount of radiolabeled inulin recovered in the brain and the CSF of Abcg4 KO vs. Abcg4 WT mice either.

Aβ kinetics. The brains of Abcg4 KO and WT mice were injected with radiolabeled amyloid β protein (Aβ), and the amount remained was measured to assess the Aβ clearance (A). Aβ in CSF was measured to assess passive clearance (C). Radiolabeled inulin, which is cleared only by passive clearance, was measured as a control (B and D). Symbols represent individual animals. Lines denote the mean ± 1 SD
Discussion
We set out to determine the effects of Abcg4 KO in the J9 model of AD, and unexpectedly did not find any difference in cognitive function or AD-related neuropathologic changes (Figs. 1 and 9). Knockout of Abcg4 did not change the rate of Aβ clearance (Fig. 10), despite previous studies demonstrating that ABCG4 functions in the efflux of Aβ both in in vitro and in vivo studies (Do et al. 2012; Dodacki et al. 2017). This suggests that the loss of ABCG4 may have been compensated for by an unidentified mechanism(s), possibly by upregulation of alternate transporters, including its natural binding partner ABCG1 (Cserepes et al. 2004). Regardless of these possibilities, loss of Abcg4 alone is not sufficient to accelerate AD pathology. If Abcg4 is still involved in the pathophysiology, it would require additional pathogenic processes, such as defects in cholesterol metabolism. For example, the cholesterol precursor, desmosterol, inhibits Aβ clearance (Dodacki et al. 2017) and is exported from the brain by ABCG4 (Wang et al. 2008; Dodacki et al. 2017). Therefore, the presence of defects that increase the accumulation of desmosterol, like loss of DHCR24 (Allen et al. 2019; Kanuri et al. 2021), could potentially make the role of ABCG4 more important. However, these remain speculative at this juncture.
While effects on AD-related behavior and neuropathologic changes were not noted in these studies, there were some mild metabolic changes, mainly some minor differences in glucose and insulin tolerance (Figs. 7 and 8). While it is not clear if these differences are biologically significant or simply type I error, there are several possible mechanisms by which loss of ABCG4 could have metabolic consequences, including changes in cholesterol homeostasis and defects with GSIS. Although the natural functions of ABCG4 are not completely defined, they include cholesterol and sterol transport. We should keep in mind that the expression of Abcg4 is not just limited to the CNS. It is also expressed in the retina, hematopoietic cells, spleen, and pancreatic islets (Bojanic et al. 2010; Hou et al. 2016). ABCG4 was discovered to be the target of miR-463-3P, whose expression reduced GSIS (Hou et al. 2016), which suggests that ABCG4 may have a role in GSIS. This mechanism could explain glucose intolerance and insulin resistance in mice with knockout of Abcg4. These studies were designed to investigate the effect of the presence or absence of Abcg4 in the context of the J9 strain (Abcg4+/+, J9 + vs. Abcg4−/−, J9 +), so it is not possible to draw a conclusion from this data about the individual effects of Abcg4 KO or J9 transgene alone.
In conclusion, our data show that loss of Abcg4 did not lead to acceleration of the pathology in a mouse model of Alzheimer’s disease, and that the kinetics of Aß clearance from the brain were not affected by the presence or absence of Abcg4.
Data Availability
Relevant data generated or analyzed during this study are included in this published article; raw datasets are available from the corresponding author upon reasonable request.
References
Albert MS, DeKosky ST, Dickson D et al (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:270. https://doi.org/10.1016/J.JALZ.2011.03.008
Allen LB, Genaro-Mattos TC, Porter NA et al (2019) Desmosterolosis and desmosterol homeostasis in the developing mouse brain. J Inherit Metab Dis 42:934. https://doi.org/10.1002/JIMD.12088
Alzheimer’s Association (2013) 2013 Alzheimer’s disease facts and figures. Alzheimer’s Dement 9:208–245. https://doi.org/10.1016/J.JALZ.2013.02.003
Antunes M, Biala G (2012) The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process 13:93. https://doi.org/10.1007/S10339-011-0430-Z
Arnold SE, Arvanitakis Z, Macauley-Rambach SL et al (2018) Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14:168. https://doi.org/10.1038/NRNEUROL.2017.185
Behl T, Kaur I, Sehgal A et al (2020) The interplay of ABC transporters in Aβ translocation and cholesterol metabolism: implicating their roles in Alzheimer’s disease. Mol Neurobiol 584(58):1564–1582. https://doi.org/10.1007/S12035-020-02211-X
Bell RD, Sagare AP, Friedman AE et al (2007) Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27:909. https://doi.org/10.1038/SJ.JCBFM.9600419
Bojanic DD, Tarr PT, Gale GD et al (2010) Differential expression and function of ABCG1 and ABCG4 during development and aging. J Lipid Res 51:169. https://doi.org/10.1194/JLR.M900250-JLR200
Butterfield DA, Halliwell B (2019a) Oxidative stress, dysfunctional glucose metabolism, and Alzheimer disease. Nat Rev Neurosci 20:148. https://doi.org/10.1038/S41583-019-0132-6
Butterfield DA, Halliwell B (2019b) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20:148–160
Choleris E, Thomas AW, Kavaliers M, Prato FS (2001) A detailed ethological analysis of the mouse open field test: effects of diazepam, chlordiazepoxide and an extremely low frequency pulsed magnetic field. Neurosci Biobehav Rev 25:235–260. https://doi.org/10.1016/S0149-7634(01)00011-2
Cirrito JR, Deane R, Fagan AM et al (2005) P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J Clin Invest 115:3285. https://doi.org/10.1172/JCI25247
Croteau E, Castellano CA, Fortier M et al (2018) A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp Gerontol 107:18–26. https://doi.org/10.1016/J.EXGER.2017.07.004
Cserepes J, Szentpétery Z, Seres L et al (2004) Functional expression and characterization of the human ABCG1 and ABCG4 proteins: indications for heterodimerization. Biochem Biophys Res Commun 320:860–867. https://doi.org/10.1016/J.BBRC.2004.06.037
Do TM, Noel-Hudson MS, Ribes S et al (2012) ABCG2- and ABCG4-mediated efflux of amyloid-β peptide 1–40 at the mouse blood-brain barrier. J Alzheimers Dis 30:155–166. https://doi.org/10.3233/JAD-2012-112189
Dodacki A, Wortman M, Saubaméa B et al (2017) Expression and function of Abcg4 in the mouse blood-brain barrier: role in restricting the brain entry of amyloid-β peptide. Sci Rep. https://doi.org/10.1038/S41598-017-13750-0
Doody RS, Thomas RG, Farlow M et al (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370:311–321. https://doi.org/10.1056/NEJMOA1312889/SUPPL_FILE/NEJMOA1312889_DISCLOSURES.PDF
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(80):353–356. https://doi.org/10.1126/SCIENCE.1072994
Hou X, Wu W, Yin B et al (2016) MicroRNA-463-3p/ABCG4: a new axis in glucose-stimulated insulin secretion. Obesity 24:2368–2376. https://doi.org/10.1002/OBY.21655
Hsia AY, Masliah E, Mcconlogue L et al (1999) Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A 96:3228. https://doi.org/10.1073/PNAS.96.6.3228
Hyman BT, Phelps CH, Beach TG et al (2012) National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8:1. https://doi.org/10.1016/J.JALZ.2011.10.007
Ionescu-Tucker A, Cotman CW (2021) Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol Aging 107:86–95. https://doi.org/10.1016/J.NEUROBIOLAGING.2021.07.014
Kanuri B, Fong V, Ponny SR et al (2021) Generation and validation of a conditional knockout mouse model for desmosterolosis. J Lipid Res. https://doi.org/10.1016/J.JLR.2021.100028
Kapogiannis D, Mattson MP (2011) Perturbed energy metabolism and neuronal circuit dysfunction in cognitive impairment. Lancet Neurol 10:187. https://doi.org/10.1016/S1474-4422(10)70277-5
Kurata T, Miyazaki K, Kozuki M et al (2011) Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res 1371:161–170. https://doi.org/10.1016/J.BRAINRES.2010.11.067
Leng F, Edison P (2020) Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 173(17):157–172. https://doi.org/10.1038/s41582-020-00435-y
Mawuenyega KG, Sigurdson W, Ovod V et al (2010) Decreased clearance of CNS amyloid-β in Alzheimer’s disease. Science 330:1774. https://doi.org/10.1126/SCIENCE.1197623
McKhann GM, Knopman DS, Chertkow H et al (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:263. https://doi.org/10.1016/J.JALZ.2011.03.005
Meng L, Wang Z, Ji HF, Shen L (2022) Causal association evaluation of diabetes with Alzheimer’s disease and genetic analysis of antidiabetic drugs against Alzheimer’s disease. Cell Biosci. https://doi.org/10.1186/S13578-022-00768-9
Montine TJ, Phelps CH, Beach TG et al (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123:1. https://doi.org/10.1007/S00401-011-0910-3
Mori T, Paris D, Town T et al (2001) Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APPsw mice. J Neuropathol Exp Neurol 60:778–785. https://doi.org/10.1093/JNEN/60.8.778
Mucke L, Masliah E, Yu GQ et al (2000) High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20:4050. https://doi.org/10.1523/JNEUROSCI.20-11-04050.2000
Naj AC, Schellenberg GD, (ADGC) for the ADGC, (2017) Genomic variants, genes, and pathways of Alzheimer’s disease: an overview. Am J Med Genet B Neuropsychiatr Genet 174:5. https://doi.org/10.1002/AJMG.B.32499
Reger MA, Watson GS, Green PS et al (2008) Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 70:440–448. https://doi.org/10.1212/01.WNL.0000265401.62434.36
Salloway S, Sperling R, Fox NC et al (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370:322. https://doi.org/10.1056/NEJMOA1304839
Sano M, Bell KL, Galasko D et al (2011) A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77:556–563. https://doi.org/10.1212/WNL.0B013E318228BF11
Sano O, Tsujita M, Shimizu Y et al (2016) ABCG1 and ABCG4 suppress γ-secretase activity and amyloid β production. PLoS One. https://doi.org/10.1371/JOURNAL.PONE.0155400
Scheuner D, Eckman C, Jensen M et al (1996) Secreted amyloid β–protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 28(2):864–870. https://doi.org/10.1038/nm0896-864
Shepardson NE, Shankar GM, Selkoe DJ (2011a) Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch Neurol 68:1239–1244. https://doi.org/10.1001/ARCHNEUROL.2011.203
Shepardson NE, Shankar GM, Selkoe DJ (2011b) Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch Neurol 68:1385–1392. https://doi.org/10.1001/ARCHNEUROL.2011.242
Shibata M, Yamada S, Ram Kumar S et al (2000) Clearance of Alzheimer’s amyloid-β1-40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J Clin Invest 106:1489. https://doi.org/10.1172/JCI10498
Tarasoff-Conway JM, Carare RO, Osorio RS et al (2015) Clearance systems in the brain—implications for Alzheimer disease. Nat Rev Neurol 11:457. https://doi.org/10.1038/NRNEUROL.2015.119
Tarr PT, Edwards PA (2008) ABCG1 and ABCG4 are coexpressed in neurons and astrocytes of the CNS and regulate cholesterol homeostasis through SREBP-2. J Lipid Res 49:169–182. https://doi.org/10.1194/JLR.M700364-JLR200
Tong X-K, Lecrux C, Hamel E (2012). Neurobiology of Disease Age-Dependent Rescue by Simvastatin of Alzheimer’s Disease Cerebrovascular and Memory Deficits. https://doi.org/10.1523/JNEUROSCI.0169-12.2012
Uehara Y, Yamada T, Baba Y et al (2008) ATP-binding cassette transporter G4 is highly expressed in microglia in Alzheimer’s brain. Brain Res 1217:239–246. https://doi.org/10.1016/J.BRAINRES.2008.04.048
Vaughan AM, Oram JF (2006) ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J Lipid Res 47:2433–2443. https://doi.org/10.1194/JLR.M600218-JLR200
Wang N, Lan D, Chen W et al (2004) ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A 101:9774–9779. https://doi.org/10.1073/PNAS.0403506101/ASSET/5458F681-8920-4DA2-9690-67B073CB9E65/ASSETS/GRAPHIC/ZPQ0270452910005.JPEG
Wang N, Yvan-Charvet L, Lütjohann D et al (2008) ATP-binding cassette transporters G1 and G4 mediate cholesterol and desmosterol efflux to HDL and regulate sterol accumulation in the brain. FASEB J 22:1073–1082. https://doi.org/10.1096/FJ.07-9944COM
Yoshikawa M, Yabuuchi H, Kuroiwa A et al (2002) Molecular and cytogenetic characterization of the mouse ATP-binding cassette transporter Abcg4. Gene 293:67–75. https://doi.org/10.1016/S0378-1119(02)00719-9
Zuroff L, Daley D, Black KL, Koronyo-Hamaoui M (2017) Clearance of cerebral Aβ in Alzheimer’s disease: reassessing the role of microglia and monocytes. Cell Mol Life Sci 74:2167. https://doi.org/10.1007/S00018-017-2463-7
Acknowledgements
We thank Dr. Diego Perez-Tilve and his laboratory members for assistance with the TSE system and indirect calorimetry.
Funding
This work was supported by the National Institutes of Health (#R03AG063322).
Author information
Authors and Affiliations
Contributions
Study conception, design, and supervision were done by SBP; data collection and analysis were performed by VF, BK, OT, and ML. The writing, original draft preparation, and incorporation of edits for this manuscript were done by VF. All authors had the opportunity to read, edit, and approve the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Fong, V., Kanuri, B., Traubert, O. et al. Behavioral and Metabolic Effects of ABCG4 KO in the APPswe,Ind (J9) Mouse Model of Alzheimer’s Disease. J Mol Neurosci 74, 49 (2024). https://doi.org/10.1007/s12031-024-02214-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12031-024-02214-6