
Abstract
Ataxin-2 (ATXN2) is implicated mainly in mRNA processing. Some ATXN2 associates with receptor tyrosine kinases (RTK), inhibiting their endocytic internalization through interaction of proline-rich domains (PRD) in ATXN2 with SH3 motifs in Src. Gain of function of ATXN2 leads to neuronal atrophy in the diseases spinocerebellar ataxia type 2 (SCA2) and amyotrophic lateral sclerosis (ALS). Conversely, ATXN2 knockout (KO) mice show hypertrophy and insulin resistance. To elucidate the influence of ATXN2 on trophic regulation, we surveyed interactions of ATXN2 with SH3 motifs from numerous proteins and observed a novel interaction with Grb2. Direct binding in glutathione S-transferase (GST) pull-down assays and coimmunoprecipitation of the endogenous proteins indicated a physiologically relevant association. In SCA2 patient fibroblasts, Grb2 more than Src protein levels were diminished, with an upregulation of both transcripts suggesting enhanced protein turnover. In KO mouse embryonal fibroblasts (MEF), the protein levels of Grb2 and Src were decreased. ATXN2 absence by itself was insufficient to significantly change Grb2-dependent signaling for endogenous Ras levels, Ras-GTP levels, and kinetics as well as MEK1 phosphorylation, suggesting that other factors compensate for proliferation control. In KO tissue with postmitotic neurons, a significant decrease of Src protein levels is prominent rather than Grb2. ATXN2 mutations modulate the levels of several components of the RTK endocytosis complex and may thus contribute to alter cell proliferation as well as translation and growth.
Similar content being viewed by others


Introduction
Ataxin-2 (ATXN2) became known as the disease protein of spinocerebellar ataxia type 2 (SCA2). A trinucleotide repeat in ATXN2 exon 1, which codes for a polyglutamine (polyQ) domain, was found to expand from usually 22 U to 32 or more CAG units and thus cause this multisystem atrophy of the central nervous system through a gain-of-function mechanism (Auburger 2011). Intermediate and large expansions of 27–37 trinucleotide units, usually CAG repeats with CAA interruptions, were recently shown to contribute to the risk of the motor neuron disease amyotrophic lateral sclerosis (ALS) and of the Parkinson plus syndrome Progressive Supranuclear Palsy (PSP) (Elden et al. 2010; Chen et al. 2011; Daoud et al. 2011; Gispert et al. 2011; Lee et al. 2011a, b; Ross et al. 2011; Soraru et al. 2011; Van Damme et al. 2011). In animal models, overexpression of ATXN2 potentiates toxicity from neurodegenerative disease proteins ATXN1, ATXN3, TDP-43, and MAPT, which are responsible for different variants of spinocerebellar ataxia, ALS, frontotemporal dementia, and PSP (Shulman and Feany 2003; Al-Ramahi et al. 2007; Lessing and Bonini 2008; Elden et al. 2010). Moreover, the deficiency of ATXN2 mitigates this toxicity (Elden et al. 2010). Neuroblastoma tumors are driven into apoptosis by upregulation of ATXN2 (Wiedemeyer et al. 2003). Thus, atrophy of numerous neuronal populations is enhanced by ATXN2 gain of function. The widespread effects of ATXN2 appear to affect health in the general population, since a genetic variant in ATXN2 intron 1 is associated with longevity among human centenarians (Sebastiani et al. 2010) as well as levels of chronic blood pressure in independent studies (Levy et al. 2009; Newton-Cheh et al. 2009). Conversely, the deficiency of ATXN2 was found to cause mice to grow to excessive size and weight, through modulation of insulin signaling (Kiehl et al. 2006; Lastres-Becker et al. 2008a). These findings raised the notion that ATXN2 is a relevant regulator of general cell growth.
The amino acid sequence of ATXN2 contains a conspicuous PAM2 motif known to mediate interaction with the cytoplasmic poly(A)-binding protein (PABPC1) as well as a Lsm and a Lsm-associated domain characterized for their role in RNA processing (Kozlov et al. 2001; Albrecht and Lengauer 2004; Ralser et al. 2005a; Satterfield and Pallanck 2006). The subcellular distribution of ATXN2 and its orthologues in other species is mostly cytoplasmic at the rough endoplasmic reticulum and in association with polysomes or RNA-containing stress granules (Nonhoff et al. 2007; van de Loo et al. 2009; Swisher and Parker 2010). This indicates that ATXN2 modulates RNA translation or RNA turnover, in good keeping with present concepts that RNA pathology underlies the motor neuron disease ALS (Lagier-Tourenne et al. 2010). However, some ATXN2 was detected in the nuclear and the plasma membrane fractions (Nonis et al. 2008; Hallen et al. 2011), suggesting that it can modulate cell growth at various steps. Several proline-rich domains (PRD) within the ATXN2 sequence were observed. They were found to interact with SH3 motifs within endophilin A1, CIN85, and Src, three components of the endocytosis machinery, which forms at the plasma membrane in response to growth factor signals stimulating receptor tyrosine kinases (RTK) such as the epidermal growth factor (EGF) receptor (Nonis et al. 2008) or as the insulin receptor. Interfaces between SH3 motifs and PRD are a common way of protein interaction in endocytosis and also in related cellular processes of eukaryotic organisms such as signal transduction or cytoskeleton organization (Fig. 1a). The list of proteins with SH3 motifs implicated in these processes is large; amphiphysin, actin-binding protein 1, Grb2, intersectin, P85-PI3 kinase, and phospholipase C are just some examples. Thus, the altered posttranscriptional regulation of insulin receptors observed in tissues with ATXN2 knockout (KO) (Lastres-Becker et al. 2008a) might be explained by altered internalization and degradation of the insulin receptor through the ATXN2 interaction with SH3 proteins. The endocytic internalization of the EGF receptor was indeed found to be modulated by ATXN2; it is delayed by ATXN2 overexpression and enhanced by its deficiency (Nonis et al. 2008).

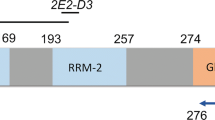
ATXN2 interactions with SH3 containing proteins of the RTK-driven signaling pathways. a Schematic presentation of RTK-driven pathways. Activated receptor tyrosine kinases stimulate several downstream signaling cascades via the activation of Src. The cell fate is determined depending on the pathway activated. The signals can maintain survival via PI3K/Akt, the cell can undergo proliferation via the Grb2/Ras pathway, it can decide to migrate or be driven into apoptosis via JNK signaling, or its differentiation is modulated via STAT5 signals. b Schematic representation of ATXN2 highlighting the proline-rich domains (PXXP) in its amino acid sequence. The arrows indicate the polyproline domains implicated in the interaction with endophilin A and the bars indicate the potential regions for interaction with other SH3 motifs. c Arrays with spotted SH3 motifs identified numerous candidate interactors of ATXN2 (see Table 1). Membranes with SH3 motifs from different proteins were incubated with HEK-293 cell extracts with (left side) or without (right side) transfected Myc-ATXN2. After washing, the membranes were incubated with anti-Myc antibody. The red square indicates the positions of the Grb2 SH3 Domains #1 and #2
Trophic factors, which are contained in the serum, activate RTK to control crucial decisions of cell fate. After RTK dimerization and autophosphorylation, the kinase Src is the central hub that initiates signaling in several parallel cascades (Fig. 1a). The endocytic internalization of activated RTK was reported to decide which of these pathways is activated (Goh et al. 2010). Survival, mRNA translation, and cell growth depend on Src signals via the PI3K/Akt pathway and also on nutrient availability through the mTOR pathway. For proliferation, Src signals need to be transduced by Grb2/SOS via the Ras/Erk pathway and trigger nuclear transcriptional responses, such as the activation of c-Jun or ATF2 (Baichwal et al. 1991; Yu et al. 1993). The crucial RTK endocytosis events are mediated by association steps between SH3 motif-containing proteins such as Grb2/CIN85/endophilin A and PRD in interactors such as Cbl/Sprouty/Alix (Smit and Borst 1997; Dikic and Giordano 2003; Schmidt and Dikic 2005). Dozens of interacting proteins allow this complex to perform a highly regulated control, where mutations in only few components lead to monogenic phenotypes, while variants of most components result in additive polygenic pathology.
In this system, several criteria suggest ATXN2 to act similarly as Sprouty, since both have PRD, associate with Cbl, and inhibit endocytosis (Fong et al. 2003; Nonis et al. 2008); are implicated in H-Ras- and K-Ras-mediated cell transformation (Shaw et al. 2007; Sutterluty et al. 2007; Holgren et al. 2010; Bertoni et al. 2011); play a role as tumor suppressors (Wiedemeyer et al. 2003; Taniguchi et al. 2009); and induce neuronal cell death, while their deficiency favors neuronal survival (Gross et al. 2007; Elden et al. 2010). ATXN2 also has similarities with Alix, since both are constitutively associated with the RTK and both inhibit endocytosis (Schmidt et al. 2004).
Interestingly, ATXN2 contains additional PRD, possibly to interact with other SH3-motif proteins (Fig. 1b). Examining the occurrence of further candidate interactors would help to clarify the physiological role of ATXN2 in endocytosis and/or in other processes where ATXN2 could be relevant. To elucidate the precise role of ATXN2 in the trophic regulation of cell fate, we now studied ATXN2 fragments containing these PRD and tested their interaction with an array of SH3 motifs from diverse proteins. In addition, we investigated the question of whether altered interactions of ATXN2 and SH3-motif proteins affect their levels to provide mechanistic insight in the effects of ATXN2 on cell trophism. SCA2 patient cells were used to test whether the ATXN2 gain of function has relevant effects on these interactors to explain cell atrophy. Finally, we sought to determine whether the absence of ATXN2 alone is able to alter cell proliferation signaling and to explore whether ATXN2 plays a major role in carcinogenesis. Our data do support a role of ATXN2 for Src and Grb2 levels but clearly contradict claims from recent immunofluorescent studies in a single SCA2 patient fibroblast line that Ras signaling is modulated strongly, which were published during the course of our study (Bertoni et al. 2011).
Results
In Vitro, ATXN2 Binds Diverse SH3 Domains
To screen for ATXN2 association with additional SH3 motifs, commercially available array membranes with 304 spots carrying SH3 motifs from diverse proteins were exposed to HEK-293 protein extracts containing Myc-ATXN2 with the polyQ domain of physiological size (Q22). Several SH3 motifs bound with varying intensities to the ATXN2 fusion protein (Fig. 1c). A set of proteins was selected for validation (Table 1), taking into account the ATXN2 binding strength, the occurrence in the brain, and the antibody availability.
ATXN2 is Associated with Grb2 in Vivo
To test the physiological association of ATXN2 with our candidates in mouse brain cytosolic fractions, endogenous ATXN2 was immunoprecipitated and the proteins of interest detected. Only the association between endogenous ATXN2 and endogenous growth factor receptor-bound protein 2 (Grb2) could be verified (Fig. 2a). In order to identify the Grb2 domains needed for the interaction with ATXN2, GST pull-down assays were carried out. A series of deletion variants expressing the different domains of Grb2 was generated as GST fusion proteins (Fig. 2b) and used. ATXN2 with the wildtype (WT) polyQ domain (Q22) was pulled down only by Grb2 full length and by the carboxy-terminal SH3 motif, in accordance with the result obtained in the array (Fig. 2c). Similar results were obtained upon pull-down of ATXN2 with the pathogenic polyQ expansion (Q74). These in vitro data suggest that the Grb2 association is preserved when ATXN2 contains a pathological expansion (Fig. 2d).

Co-IP analysis of the ATXN2–Grb2 complex. a Endogenous ATXN2 is associated with endogenous Grb2 in mouse brain. Brain homogenates were fractionated by differential velocity centrifugation. The cytosolic fraction was utilized to carry out coimmunoprecipitation assays with anti-ATXN2 antibody and the corresponding Ig isotype as control. Immunoblotting was performed using antibodies against ATXN2 and Grb2. (b–d) Grb2 interacts with ATXN2 through its C-terminal SH3 motif. b Diagram of human Grb2 and its different domains. c Myc-ATXN2 overexpressed in HEK-293 cells was incubated with GSH affinity beads charged with GST-Grb2 or fusions with its respective subfragments. Loading density of GST or GST-fused proteins on the affinity beads was controlled with anti-GST antibody. d A repetition of this experiment with overexpressed mutant ATXN2 (Q74) yielded identical results
ATXN2 PolyQ Expansion Alters Steady-state Levels of Src and Grb2
To further explore whether the ATXN2 polyQ expansions implicated in neural atrophy diseases could modify interaction strength, stability, and turnover of the protein complex, the protein levels of SH3-containing components of the RTK endocytosis machinery were quantified in primary skin fibroblasts of four SCA2 patients in comparison to four healthy matched controls. In view of the inherent growth differences of untransformed clonal fibroblast lines, the largest available number of WT vs. mutant cell lines was compared in the subsequent human and murine studies. A significant reduction of Grb2 protein to 0.62-fold levels was observed (Fig. 3a). In testing Src as the crucial established ATXN2 interactor within the RTK endocytosis machinery (Nonis et al. 2008), only a trend towards reduction was apparent for Src protein levels. Since it remained unclear whether this subtle change exerts a functional effect, the activation of Src was studied. Src is regulated at the phosphorylation sites Tyr416 and Tyr527 with opposing effects on its activation (Salter and Kalia 2004). In the inactive state, the SH2 domain of Src interacts with phosphorylated Tyr527, while Tyr416 in the activation loop is dephosphorylated. After dephosphorylation of Tyr527 by protein tyrosine phosphatases, the conformation of Src changes, leading to subsequent autophosphorylation of Tyr416 and a conformational change of the activation loop, which causes the kinase to be fully activated. Parallel increases in the dephosphorylated state of both principal Src sites (Fig. 3b) reflected the altered levels. In order to understand whether the decreased protein levels are caused by enhanced protein degradation or reduced protein synthesis, the mRNA levels were studied and a trend towards upregulation was observed for GRB2 transcript (Fig. 3a), accompanied by a significant upregulation to 1.29-fold levels for SRC transcript (Fig. 3b), suggesting that the deficit in steady-state protein levels prevails in spite of a compensatory upregulation of the respective mRNAs.

ATXN2 polyQ expansion alters Grb2 and Src steady-state levels. Investigation of Grb2 (a) and Src (b) endogenous protein levels in SCA2 patient skin fibroblasts showed a significant reduction for Grb2 and a trend towards reduction for Src. The dephosphorylated state of activating and inhibiting Src sites was increased in parallel. The mRNA levels showed a trend towards upregulation for GRB2 and significant upregulation for SRC (four patient fibroblast lines vs. four control fibroblast lines, n = 2 for Western blotting, n = 3 for qRT-PCR). As loading control, β-actin was used in immunoblots, TBP in qRT-PCR. Representative Western blots are shown for illustration, and unpaired Student’s t test was used for statistical analysis (*p < 0.05; **p < 0.01; ***p < 0.001)
ATXN2 Deficiency Alters the Steady-state Levels of Grb2 and Src
To test whether the levels of SH3-containing interactors change in the absence of ATXN2, steady-state Grb2 protein levels were documented. A significant reduction to 0.68-fold levels was observed for Grb2 protein in seven WT vs. eight KO mouse embryonal fibroblast (MEF) lines but not in the mixed tissue cerebellum at adult age from nine WT vs. nine KO mice (Fig. 4a). For Src, a similar effect with significant reduction to 0.67-fold in MEF and a stronger effect with significant reduction to 0.75-fold also in adult cerebellum were observed (Fig. 4b). The analysis of the dephosphorylated state of both sites in KO MEF again showed a parallel trend towards upregulation. In adult KO cerebellum, this parallel upregulation was significant. These data indicate that a particularly strong reduction of Src endogenous levels characterizes this ATXN2-KO tissue with postmitotic neurons and that it is accompanied by significantly less steady-state phosphorylation at both sites, evidently balanced between the inactive and active states.

ATXN2 loss of function decreases protein levels of Grb2 and Src. a In mouse embryonal fibroblasts (MEFs) with ATXN2 knockout (KO or −/−) vs. littermate wildtype (WT), Grb2 protein levels were reduced (seven WT vs. eight KO, n = 2). In the mixed tissue cerebellum from adult ATXN2−/− mice, Grb2 protein levels did not differ (nine WT vs. nine KO). As loading control, β-actin was used in immunoblots. b In KO-MEFs, Src protein levels were reduced (seven WT vs. eight KO, n = 2). In cerebellum from adult ATXN2−/− mice, a significant decrease of total Src protein levels was observed (nine WT vs. nine KO). As loading control, β-actin was used in immunoblots. Representative Western blots are shown for illustration, and unpaired Student’s t test was used for statistical analysis (*p < 0.05; **p < 0.01; ***p < 0.001)
ATXN2 Deficiency Does Not Block Grb2-dependent Ras Signaling
The association of Grb2 with RTK and SOS initiates signaling in the Ras pathway (Fig. 1a), which depends on the large mammalian Ras subfamily of small GTPases. In view of a recent publication from semiquantitative immunocytochemical data in a single SCA2 fibroblast line claiming that pathogenic expansion of ATXN2 increases HRas expression and that expanded ATXN2 effects on DNA damage are antagonized by HRas silencing (Bertoni et al. 2011), we used quantitative immunoblots and several fibroblast lines from four SCA2 patients and seven ATXN2−/− mice, assessing the expression levels of all three principal Ras family members. In contrast to the report mentioned, for HRas, KRas, and NRas, no significant expression changes were detectable at the protein level in the murine or human fibroblasts with ATXN2 mutations (Fig. 5a and b). The discordance with the previous report might be attributed to differences in cell line differentiation in ATXN2 expansion lengths or to the assay tools employed.

ATXN2 mutations do not influence endogenous RAS expression levels in cultured fibroblasts. a ATXN2 deficiency did not change HRAS, KRAS, or NRAS protein levels in MEFs (seven WT vs. eight KO). As loading control, β-actin was used in immunoblots. b ATXN2 polyQ expansion in SCA2 patient skin fibroblasts also did not influence HRAS, KRAS, or NRAS protein levels (four patient fibroblast lines vs. four control fibroblast lines, n = 2 for Western blotting). As loading control, β-actin was used in immunoblots. Representative Western blots are shown for illustration, and unpaired Student’s t test was used for statistical analysis (*p < 0.05; **p < 0.01; ***p < 0.001)
For proliferation signaling, not only the RAS levels but also the activation state is relevant. Ras-GTPases cycle between their cytoplasmic inactive GDP form and their plasma membrane-bound active GTP form (Hancock 2003). To further examine the role of ATXN2 in tissue rather than cell lines and on the downstream Ras signaling pathway, pull-down of total Ras and activated GTP-Ras was performed in KO compared to WT cerebellum (Fig. 6a). Since no relevant changes were detectable at steady-state in tissue, we decided to assess dynamic alterations of Ras activation, studying ATXN2−/− MEFs in a time course after EGF stimulation. Ras activation was now documented through the capability of Raf-1 to bind to GTP-Ras. Ras activation was observed after 2 min of EGF stimulation, reached a peak at 5 min, and started to decrease by 10 min (Fig. 6b). No significant differences between KO and WT cells could be detected at any of the time points. A major downstream effector target of Ras-GTP is the MAP-kinase Raf-1 that activates the MEK/Erk gene regulation cascade (Schlessinger 2000; Hancock 2003). Since MEK1 is expressed at much higher levels in the brain than MEK2, we investigated the phosphorylation of MEK1 in ATXN2−/− mice (Fig. 6c). After serum deprivation of MEF over 24 h and EGF stimulation, changes in total and phosphorylated MEK1 were documented by enzyme -linked immunosorbent assay (ELISA). Consistent differences were not detected. Altogether these data suggest that absence of ATXN2 alone is not sufficient to significantly alter the Ras/Erk pathway control of cell proliferation. These molecular data are in agreement with cell biology studies, where significant differences in the doubling times were neither observed between ATXN2−/− and control MEF nor between SCA2 and control skin fibroblasts (data not shown).

Ras pathway signaling appears unaffected by ATXN2. a Ras pull-down experiments in cerebellum of ATXN2−/− mice showed no changes of total Ras amounts. Cerebellum of 6-month-old ATXN2−/− and WT mice were dissected and directly frozen in liquid nitrogen prior to the pull-down experiments (10 KO vs. 10 WT). Tubulin was used as loading control. Statistical analysis was performed with unpaired Student’s t test (*p < 0.05; **p < 0.01; ***p < 0.001). b Time course of Ras activation in MEFs showed no difference in absence of ATXN2. KO and WT MEF were serum deprived for 24 h and afterwards stimulated with 100 ng/ml of EGF for the indicated time (four WT vs. four KO). The activation of Ras was analyzed employing a Ras GTPase activation ELISA kit and measuring chemiluminescence. c MEK1 activation in MEF was not modulated by ATXN2 deficiency. KO and WT MEF were serum deprived for 24 h and stimulated with 100 ng/ml EGF for the indicated time (two WT vs. two KO). The activation of MEK1 was analyzed by ELISA. Total amounts of MEK1 were not different in KO compared to WT mice
Discussion
ATXN2 was previously shown to be constitutively associated with the RTK protein complex and to inhibit its endocytic internalization via direct interaction with the SH3 motif-containing proteins endophilin A1, CIN85, and Src (Ralser et al. 2005b; Nonis et al. 2008). However, it is unclear whether these findings are relevant for proliferating cells and carcinogenesis, or for the translation control in postmitotic neurons and for neurodegenerative disorders. A recent publication (Bertoni et al. 2011) claiming ATXN2 expansions to increase HRas expression and to be antagonized by HRas silencing resulting in the generation of DNA damage, therefore, seemed to shed credible light on the relationship between ATXN2 and cell stress.
Our present data show for the first time that ATXN2 indeed interacts with the SH3 motif containing protein Grb2 in the endocytosis machinery and that ATXN2 loss-of-function and gain-of-function mutations in fibroblasts modulate Grb2 protein levels. Grb2 is a broadly expressed adaptor protein of 217 amino acids (25 kDa) that provides a critical link between cell surface RTK and downstream proliferation pathways (Downward 1994). While the gain of function of Grb2 promotes cell proliferation and transformation, decreased Grb2 function impairs developmental processes and Grb2 null mice are lethal at early embryonic stage (Cheng et al. 1998; Tari and Lopez-Berestein 2001; Giubellino et al. 2008). Grb2 is composed of two SH3 and one SH2 motifs and can associate with activated tyrosine-phosphorylated receptors via its SH2 domain (Fig. 2b); among these receptors are EGF, platelet-derived growth factor (PDGF), and insulin receptors (Buday and Downward 1993; Schlessinger 2000). Subsequently, Grb2 binds to Cbl and SOS via its SH3 motifs to activate the Ras/Erk, phosphatidylinositol (PI)-3 kinase (PI3K) and ubiquitination–degradation pathways that control proliferation (van der Geer et al. 1994; Gotoh 2008).
However, our analyses could not detect changes in the Ras activation and downstream MEK1 phosphorylation, both in tissue steady-state analyses and in cellular time course experiments, contradicting current knowledge (Bertoni et al. 2011). Our observations that ATXN2 expansions as well as ATXN2 deficiency result in mildly reduced levels of Grb2 and Src protein while Ras signaling is not affected strongly by ATXN2 deficiency provide a molecular basis for the previous observations (Wiedemeyer et al. 2003) that ATXN2 mutations antagonize rather than promote carcinogenesis.
In SCA2 patient skin fibroblasts, both Grb2 and Src protein levels appeared reduced in spite of a compensatory upregulation of the transcripts. These data are compatible with two alternative concepts. ATXN2 expansions could lead to altered stability of the endocytosis complex with increased turnover and degradation of some components, such as Grb2 and Src, insufficiently compensated by enhanced synthesis. This scenario might suggest a role of ATXN2 as scaffold for this protein complex. Alternatively, ATXN2 expansions might impair the mRNA translation rate, leading to reduced protein synthesis in spite of compensatory high transcript levels. At present, the interaction between ATXN2 and PABPC1 makes a role in mRNA translation credible, but it is not clear whether the impact of mutations would be inhibitory or stimulatory, and it is unclear which specific mRNAs are regulated.
It is very interesting that the decreased protein levels of Grb2 and Src were not only caused by ATXN2 deficiency but also by the ATXN2 polyQ expansion. While polyQ neurodegenerative diseases such as Huntington’s disease and spinobulbar muscle atrophy are known to be driven by a toxic gain of function of the expanded protein, some disease features are compatible with a partial loss of function of the disease protein (Lieberman et al. 2002; Cary and La Spada 2008; Zuccato et al. 2010), indicating that multiple pathways of pathogenesis coexist. Thus, deficient RTK signaling may be expected to occur in polyQ-ATXN2 modulated neural atrophies such as SCA2/ALS/PSP. Src as a central hub for glutamate receptor regulation modulates neural communication at a crucial step (Salter and Kalia 2004), while Grb2 via Erk is thought to influence the volume and number of cells, neurites, and synapses (Vaudry et al. 2002; Kelleher et al. 2004). In this context, it is interesting to note that Grb2 through its SH3 motifs interacts also with proline-rich motifs within huntingtin (Liu et al. 1997), the disease protein responsible for the polyQ-mediated neurodegeneration Huntington’s disease. The polyQ expansion of huntingtin leads to blockade of NGF receptor-mediated neurite outgrowth in rat pheochromocytoma cells (Song et al. 2002). Moreover, Grb2 was observed to interact with amyloid precursor protein (APP) and presenilins 1 and 2, the disease proteins responsible for Alzheimer’s dementia (Nizzari et al. 2007). These observations strengthen the concept that RTK endocytosis and signaling are relevant in neurodegenerative diseases.
Conclusions
Our data confirm the previously reported role of ATXN2 as an interactor of SH3-motif proteins that modulate RTK endocytosis (Nonis et al. 2008) and suggest a specific effect of ATXN2 deficiency to reduce the levels of Grb2 and Src proteins in mouse mutants. A novel analysis of patient skin fibroblasts with ATXN2 polyQ expansion also detected reduced Grb2 and Src protein levels, compatible with the idea of distorted RTK signaling as a modulator of neural atrophy in human disease. However, the effects were found to be insufficient for a distortion of Ras signaling by themselves, in disagreement with current literature, while they might act as modifiers within the polygenic control of cell proliferation. A more relevant role of ATXN2 may, therefore, lie in the many neural signaling pathways controlled by Src.
Materials and Methods
Screen for ATXN2-interacting Proteins with SH3 Domain
Commercially available SH3 domain array membranes (Panomics Inc., Fremont, CA, USA) were utilized to screen for ATXN2 putative interacting proteins. The experiments were performed according to the manufacturer’s instructions with one modification. The big size of ATXN2 made ATXN2 unsuitable to be expressed in bacteria because the protein yield is very low and the degradation products make up the majority of the purified protein. Therefore, ATXN2 was expressed in the mammalian system. Human Myc-ATXN2 was transiently transfected with Lipofectamine 2000 (Invitrogen) in HEK-293 cells and after 24 h, protein extracts were dissolved in binding buffer [150 mM NaCl, 10 mM HEPES pH 7, 10 mM KCl, 5 mM MgCl2, 0.5 mM phenylmethanesulfonylfluoride (PMSF), 1 % Igepal CA-630, protease inhibitor cocktail (Roche)]. Myc-ATXN2 binding to the SH3 domains was detected with anti-Myc antibody and the corresponding secondary horseradish peroxidase conjugate antibody.
Preparation of GST Fusion Proteins
Plasmid coding for GST-Grb2 and its subdomain fusion proteins, GST-SH3 N, GST-SH3 C, GST-SH2, and GST-Raf1-RDB, were introduced by heat pulse at 42 °C for 20 s into E. coli BL21-Gold (DE3) pLysS (Stratagene, cat # 230134). For the starting solutions, individual colonies were selected and placed into 2 ml of 2XYTA medium in the presence of 50 μg/ml ampicillin and 50 μg/ml chloramphenicol and incubated overnight at 37 °C rotating at 250 rpm. The starter solution was added to 150 ml of fresh 2XYTA medium in the presence of ampicillin and chloramphenicol and grown at 37 °C by rotating at 250 rpm. The OD at 600 nm was measured every 30 min. When the OD reached 0.8–1, induction with isopropyl β-D-1-thiogalactopyranoside (IPTG) 1 mM final concentration was carried out for 3–4 h at 37 °C by rotating 250 rpm. Then, the cells were collected and centrifuged for 15 min at 16,000 ×g at 4 °C. The pellet was resuspended in 4 ml of cold phosphate buffered saline (PBS) with protease inhibitor cocktail (Roche) including 1 mM PMSF and incubated on ice for 20 min. Afterwards, the cells were disrupted by sonication and the subsequent addition of Triton X-100 to a final concentration of 0.5 %. The solution was incubated for 20 min on ice and then centrifuged for 15 min at 16,000 ×g at 4 °C.
In Vitro Protein Binding Experiments with GST Fusion Proteins
Glutathione sepharose 4B (Amersham Biosciences) beads were washed once with PBS and then charged with extracts containing GST fusion protein full length or subdomains. The beads with the extracts were incubated with end-over-end rotation for 1 h at 4 °C and then washed once with binding buffer (25 mM HEPES pH 8, 150 mM NaCl, 2 mM MgCl2, 1 mM dithiothreitol (DTT), 5 % glycerol, 0.1 % Igepal CA-630). Cell lysates or tissue homogenates previously dialyzed against binding buffer were added and incubated at 4 °C for 2 h or overnight with end-over-end rotation. After extensive washing with binding buffer, bound proteins were eluted by boiling in sodium dodecyl sulfate (SDS) sample buffer and analyzed by Western blotting.
Mammalian Expression Vectors
Human ATXN2 cDNA was inserted as a NotI-KpnI fragment (positions 172–4,348) (Pulst et al. 1996) into the N-terminal tag vectors pCMV-HA and pCMV-Myc (BD Biosciences) with the aid of a small oligonucleotide linker to provide appropriate restriction sites and to adjust the reading frame. In addition to the WT ATXN2 form with a (CAG)22 repeat, a pathogenic ATXN2 version with (CAG)74, was fused to these vectors. 5′ and 3′ deletion variants of ATXN2 were cloned in pCMV-Myc. The remaining fragments of ATXN2 were amplified by polymerase chain reaction (PCR) and inserted into the N-terminal tag vector pCMV-Myc with the addition of a small oligonucleotide linker to provide appropriate restriction sites and to adjust the reading frame.
Cell Cultures and Transient Transfection
HEK-293 cells were grown in Dulbecco’s modified eagle medium (DMEM) supplemented with 10 % fetal calf serum (Sigma-Aldrich) under standard conditions. HEK-293 cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Transient expression over 48 h was used for the preparation of protein extracts.
Embryonic skin fibroblasts from mice (MEFs) with constitutive deletion of exon 1 of the ATXN2 gene resulting in absence of ATXN2 mRNA and protein were isolated around embryonal day 17 (Lastres-Becker et al. 2008a). Skin pieces were minced in 3-cm Petri dishes and underwent enzymatic digestion at 37 °C for 30 min in Falcon tubes with 1 ml HyQTase (HyClone). The digested tissue was passed through a 160 μm Nitex filter into 3-cm Petri dishes, and the enzymes neutralized with DMEM + 15 % bovine growth serum (Sigma-Aldrich). The resulting suspension was centrifuged at 450× g for 5 min, and the cells were plated at a density of 1 × 106 cells/cm2 in culture flasks. The fibroblasts were placed in an incubator at 37 °C supplemented with 5 % CO2, and the medium was changed daily for the first 3 days. SCA2 patient fibroblasts were grown in DMEM supplemented with 15 % fetal calf serum (Sigma-Aldrich), 1 % l-glutamine (Gibco) and 1 % penicillin/streptomycin (Gibco).
Preparation of Protein Extracts and Immunoblotting
Mouse tissue or cells were lysed in 5 μl RIPA buffer per milligram wet weight (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM ethylene diamine tetraacetic acid (EDTA), 1 mM ethylene glycol tetraacetic acid (EGTA), 1 % Igepal CA-630, 0.5 % sodium deoxycholate, 0.1 % SDS, protease inhibitor cocktail (Roche), and 1 mM PMSF). For coimmunoprecipitation experiments, cellular extracts were prepared with a lysis buffer containing 50 mM HEPES–NaOH, pH 7.5, 150 mM NaCl, 1 mM EDTA and 1 mM EGTA, 5 % glycerol, 0.1 % Igepal CA-630, protease inhibitor cocktail (Roche), 1 mM PMSF, and 1 mM NaF. Protein concentration was determined using a BCA Protein Quantification Kit (Interchim) according to the manufacturer’s protocol. Twenty micrograms of protein per sample was separated on 7.5 % to 10 % polyacrylamide gels followed by transfer to polyvinylidene fluoride (PVDF) membranes. Blots were blocked with 5 % nonfat milk in PBS/0.05 % Tween 20 for 1 h at room temperature and further incubated with primary antibodies for 1 h at room temperature (RT) or overnight at 4 °C. After washing the membranes, immune complexes were detected with peroxidase-conjugated anti-IgG secondary antibodies and SuperSignal West Pico chemiluminescence substrate (Pierce), always with a range of exposure times and selection of near-linear quantification. A densitometric analysis of the best immunoblots was carried out using the program ImageJ (NIH). For quantification, the protein expression changes were calculated by establishing the integrated density of each individual band. The value measured for the protein of interest was divided by its respective β-actin value. Bar graphs show the mean ± SEM of normalized protein expression levels from all available control or WT samples vs. the average protein expression level from all available patient or KO samples. The following antibodies were used: anti-ATXN2 (BD Biosciences), anti-Src (Cell Signaling), anti-non-phospho Src (Tyr416) (Cell Signaling), anti-non-phospho Src (Tyr527) (Cell Signaling), anti-Grb2 (BD Biosciences), anti-HRas (Abcam), anti-KRas (Santa Cruz), anti-NRas (Abcam), and anti-β-actin (Sigma-Aldrich).
Subcellular Fractionation and Coimmunoprecipitation with Mouse Brain Tissue
Freshly dissected C57/BL6 mouse brains were disrupted with a Dounce homogenizer in 5 vol./wet weight fractionation buffer. Separation of cellular organelles by differential velocity centrifugation followed exactly the scheme given by Feng et al. (1997), yielding fractions that predominantly contained nuclei, mitochondria/heavy membranes, light membranes/polysomes, and cytosol. The cytoplasmic fraction was concentrated using a centrifugal Amicon Ultra 30,000MWCO filter device (Millipore) by centrifugation at 3,400 ×g for 45 min at 4 °C. The light membrane fraction was diluted in binding buffer and then centrifuged at 16,000 ×g for 15 min at 4 °C and the supernatant stored for further immunoprecipitation assays. Cytosolic and light membrane fractions were incubated with appropriate amounts of antibodies overnight (approximately 12 μg/100–150 μl of tissue lysate). For the immunoprecipitation of ATXN2, a commercial antibody (BD Biosciences) was used. Antibody–protein complexes were precipitated with protein G-agarose (for monoclonal antibodies) at 4 °C for at least 2 h. The agarose beads were sedimented by centrifugation and extensively washed with ice-cold binding buffer. The bead sediment was boiled in SDS sample buffer, and the supernatant was analyzed by immunoblotting.
Coimmunoprecipitation with Transfected HEK-293 Cells
After 48 h of transfection, cells were washed once with cold PBS and lysed in 500 μl of binding buffer per 10 cm diameter dishes. The cells were incubated in binding buffer for 10 min on ice, collected in 2 ml reaction tubes, and centrifuged at 16,000 ×g for 10 min at 4 °C. The supernatants were incubated with appropriate amounts of antibodies for at least 2 h at 4 °C. Antibody–protein complexes were precipitated with protein A/G or G-agarose at 4 °C overnight. The agarose beads were sedimented by centrifugation and extensively washed with ice-cold binding buffer. The bead sediments were boiled in SDS sample buffer, and the supernatants were analyzed by Western blotting. Anti-Myc tag (BD Biosciences) and anti-ATXN2 (BD Biosciences) antibodies were used for the immunoprecipitations (approximately 12 μg/300 μl of cell extract).
Enzyme-linked Immunosorbent Assay
Ras activation assays and phosphorylation of MEK1 were investigated in ATXN2−/− MEFs after EGF stimulation. For that purpose, a Ras GTPase activation ELISA kit (Millipore) was employed and a MEK1/phospho-MEK1 ELISA kit (Cell Signaling), respectively. Eighty percent of confluent MEFs were serum deprived for 24 h and then stimulated for different periods of time with 100 ng/ml of EGF. The stimulation was stopped by transferring the Petri dishes with MEFs on ice. The cell extraction and further steps were carried out according to the manufacturer’s instructions.
Ras Pull-down Assay
Cerebellum of ATXN2-KO and WT mice obtained from 6-month-old animals was used to perform the Ras pull-down assay. Homogenized tissue (in 50 mM Tris–HCl, pH 7.4, 100 mM NaCl, 2 mM MgCl2, 10 % glycerol, and 1 % Nonidet P-40) was centrifuged at 14,000 g, 4 °C, and the supernatant was used for further experiments. The protein concentration was determined using the DC protein assay (Bio-Rad Laboratories). For the detection of total Ras, equal amounts of proteins (10 μg/slot) were analyzed with SDS polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. The immunoreactive bands were detected using the primary antibody anti-Ras IgG (Upstate, diluted 1:10,000 in tris-buffered saline), anti-mouse horseradish peroxidase-conjugated secondary antibody (Sigma-Aldrich) and an enhanced chemiluminescence substrate detection kit (Amersham Pharmacia Biotech). Blots were analyzed by densitometric measurement and quantified using the program ImageJ (NIH). Two hundred micrograms of protein from lysates was used for Ras pull-down experiments, which have been performed as described previously in detail (de Rooij and Bos 1997; Lastres-Becker et al. 2008b). The ratio between optic densities of GTP-Ras and total Ras immunoblots served as an index for the relative levels of active Ras within the probes. The band density was quantified using the program ImageJ (NIH).
RNA Preparation and First-strand cDNA Synthesis
RNA was extracted from cerebellum of 6-month-old ATXN2−/− mice using the TRIzol reagent (Invitrogen) method. The extraction of RNA from cultured cells was done using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol.
One microgram total RNA was digested with DNase I [1 U/μl] Amplication Grade and 10× DNaseI buffer (Invitrogen) in reaction volumes of 10 μl. cDNA synthesis was performed using a First-Strand cDNA Synthesis Kit (SuperScript III, Invitrogen).
Quantitative Real-time RT-PCR
Twenty-five nanograms of cDNA was used to perform the quantitative real-time RT-PCR (qRT-PCR) in a final reaction volume of 20 μl. The expression analysis was carried out using a StepOnePlus Real-Time PCR system (Applied Biosystems) and 96-well microplates (Applied Biosystems). All reactions were carried out using the TaqMan Universal PCR Master Mix (Applied Biosystems). Primers and probes for TaqMan PCR were obtained from Applied Biosystems and the following predesigned TaqMan Gene Expression Assays were used: GRB2 Hs00157817_m1 and SRC Hs00178494_m1. The PCR conditions were 50 °C for 2 min and 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s and 60 °C for 40 s. All assays were run in duplicate. An analysis of relative gene expression data was performed using the ∆∆CT method with TATA box binding protein (TBP) (Hs99999910_m1) as endogenous control (Livak and Schmittgen 2001).
Statistics
The statistical analyses were carried out with the GraphPad software package (version 4.03, GraphPad Software Inc., San Diego, CA, USA) to perform unpaired Student’s t tests when normal distribution and equal variances were fulfilled and to display data as bar graphs, illustrating mean values and standard error of the mean (SEM). Significant differences were highlighted with asterisks (*p < 0.05; **p < 0.01; ***p < 0.001).
References
Albrecht M, Lengauer T (2004) Survey on the PABC recognition motif PAM2. Biochem Biophys Res Commun 316(1):129–138
Al-Ramahi I, Perez AM et al (2007) dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet 3(12):e234
Auburger GW (2011) Spinocerebellar ataxia type 2. Handb Clin Neurol 103:423–436
Baichwal VR, Park A et al (1991) v-Src and EJ Ras alleviate repression of c-Jun by a cell-specific inhibitor. Nature 352(6331):165–168
Bertoni A, Giuliano P et al (2011) Early and late events induced by polyQ-expanded proteins: identification of a common pathogenic property of polyQ-expanded proteins. J Biol Chem 286(6):4727–4741
Buday L, Downward J (1993) Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 73(3):611–620
Cary GA, La Spada AR (2008) Androgen receptor function in motor neuron survival and degeneration. Phys Med Rehabil Clin N Am 19(3):479–494
Chen Y, Huang R et al (2011) Ataxin-2 intermediate-length polyglutamine: a possible risk factor for Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging 32(10):1925.e1–1925.e5
Cheng AM, Saxton TM et al (1998) Mammalian Grb2 regulates multiple steps in embryonic development and malignant transformation. Cell 95(6):793–803
Daoud H, Belzil V et al (2011) Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch Neurol 68(6):739–742
de Rooij J, Bos JL (1997) Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene 14(5):623–625
Dikic I, Giordano S (2003) Negative receptor signalling. Curr Opin Cell Biol 15(2):128–135
Downward J (1994) The GRB2/Sem-5 adaptor protein. FEBS Lett 338(2):113–117
Elden AC, Kim HJ et al (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466(7310):1069–1075
Feng Y, Gutekunst CA et al (1997) Fragile × mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci 17(5):1539–1547
Fong CW, Leong HF et al (2003) Tyrosine phosphorylation of Sprouty2 enhances its interaction with c-Cbl and is crucial for its function. J Biol Chem 278(35):33456–33464
Gispert S, Kurz A et al (2011) The modulation of amyotrophic lateral sclerosis risk by ataxin-2 intermediate polyglutamine expansions is a specific effect. Neurobiol Dis
Giubellino A, Burke TR Jr et al (2008) Grb2 signaling in cell motility and cancer. Expert Opin Ther Targets 12(8):1021–1033
Goh LK, Huang F et al (2010) Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor. J Cell Biol 189(5):871–883
Gotoh N (2008) Regulation of growth factor signaling by FRS2 family docking/scaffold adaptor proteins. Cancer Sci 99(7):1319–1325
Gross I, Armant O et al (2007) Sprouty2 inhibits BDNF-induced signaling and modulates neuronal differentiation and survival. Cell Death Differ 14(10):1802–1812
Hallen L, Klein H et al (2011) The KRAB-containing zinc-finger transcriptional regulator ZBRK1 activates SCA2 gene transcription through direct interaction with its gene product, ataxin-2. Hum Mol Genet 20(1):104–114
Hancock JF (2003) Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 4(5):373–384
Holgren C, Dougherty U et al (2010) Sprouty-2 controls c-Met expression and metastatic potential of colon cancer cells: sprouty/c-Met upregulation in human colonic adenocarcinomas. Oncogene 29(38):5241–5253
Kelleher RJ 3rd, Govindarajan A et al (2004) Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44(1):59–73
Kiehl TR, Nechiporuk A et al (2006) Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem Biophys Res Commun 339(1):17–24
Kozlov G, Trempe JF et al (2001) Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc Natl Acad Sci U S A 98(8):4409–4413
Lagier-Tourenne C, Polymenidou M et al (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19(R1):R46–R64
Lastres-Becker I, Brodesser S et al (2008a) Insulin receptor and lipid metabolism pathology in ataxin-2 knock-out mice. Hum Mol Genet 17(10):1465–1481
Lastres-Becker I, Fernandez-Perez A et al (2008b) Pituitary adenylate cyclase-activating polypeptide stimulates glial fibrillary acidic protein gene expression in cortical precursor cells by activating Ras and Rap1. Mol Cell Neurosci 39(3):291–301
Lee T, Li YR et al (2011a) Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology 76(24):2062–2065
Lee T, Li YR et al (2011b) Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum Mol Genet 20(9):1697–1700
Lessing D, Bonini NM (2008) Polyglutamine genes interact to modulate the severity and progression of neurodegeneration in Drosophila. PLoS Biol 6(2):e29
Levy D, Ehret GB et al (2009) Genome-wide association study of blood pressure and hypertension. Nat Genet 41(6):677–687
Lieberman AP, Harmison G et al (2002) Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet 11(17):1967–1976
Liu YF, Deth RC et al (1997) SH3 domain-dependent association of huntingtin with epidermal growth factor receptor signaling complexes. J Biol Chem 272(13):8121–8124
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25(4):402–408
Newton-Cheh C, Johnson T et al (2009) Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet 41(6):666–676
Nizzari M, Venezia V et al (2007) Amyloid precursor protein and presenilin1 interact with the adaptor GRB2 and modulate ERK 1,2 signaling. J Biol Chem 282(18):13833–13844
Nonhoff U, Ralser M et al (2007) Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell 18(4):1385–1396
Nonis D, Schmidt MH et al (2008) Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal 20(10):1725–1739
Pulst SM, Nechiporuk A et al (1996) Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14(3):269–276
Ralser M, Albrecht M et al (2005a) An integrative approach to gain insights into the cellular function of human ataxin-2. J Mol Biol 346(1):203–214
Ralser M, Nonhoff U et al (2005b) Ataxin-2 and huntingtin interact with endophilin-A complexes to function in plastin-associated pathways. Hum Mol Genet 14(19):2893–2909
Ross OA, Rutherford NJ et al (2011) Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet
Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5(4):317–328
Satterfield TF, Pallanck LJ (2006) Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet 15(16):2523–2532
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103(2):211–225
Schmidt MH, Dikic I (2005) The Cbl interactome and its functions. Nat Rev Mol Cell Biol 6(12):907–918
Schmidt MH, Hoeller D et al (2004) Alix/AIP1 antagonizes epidermal growth factor receptor downregulation by the Cbl-SETA/CIN85 complex. Mol Cell Biol 24(20):8981–8993
Sebastiani P, Solovieff N et al (2010) Genetic signatures of exceptional longevity in humans. Science
Shaw AT, Meissner A et al (2007) Sprouty-2 regulates oncogenic K-ras in lung development and tumorigenesis. Genes Dev 21(6):694–707
Shulman JM, Feany MB (2003) Genetic modifiers of tauopathy in Drosophila. Genetics 165(3):1233–1242
Smit L, Borst J (1997) The Cbl family of signal transduction molecules. Crit Rev Oncog 8(4):359–379
Song C, Perides G et al (2002) Expression of full-length polyglutamine-expanded huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem 277(8):6703–6707
Soraru G, Clementi M et al (2011) ALS risk but not phenotype is affected by ataxin-2 intermediate length polyglutamine expansion. Neurology 76(23):2030–2031
Sutterluty H, Mayer CE et al (2007) Down-regulation of Sprouty2 in non-small cell lung cancer contributes to tumor malignancy via extracellular signal-regulated kinase pathway-dependent and -independent mechanisms. Mol Cancer Res 5(5):509–520
Swisher KD, Parker R (2010) Localization to, and effects of Pbp1, Pbp4, Lsm12, Dhh1, and Pab1 on stress granules in Saccharomyces cerevisiae. PLoS One 5(4):e10006
Taniguchi K, Ishizaki T et al (2009) Sprouty4 deficiency potentiates Ras-independent angiogenic signals and tumor growth. Cancer Sci 100(9):1648–1654
Tari AM, Lopez-Berestein G (2001) GRB2: a pivotal protein in signal transduction. Semin Oncol 28(5 Suppl 16):142–147
Van Damme P, Veldink JH et al (2011) Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology 76(24):2066–2072
van de Loo S, Eich F et al (2009) Ataxin-2 associates with rough endoplasmic reticulum. Exp Neurol 215(1):110–118
van der Geer P, Hunter T et al (1994) Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol 10:251–337
Vaudry D, Stork PJ et al (2002) Signaling pathways for PC12 cell differentiation: making the right connections. Science 296(5573):1648–1649
Wiedemeyer R, Westermann F et al (2003) Ataxin-2 promotes apoptosis of human neuroblastoma cells. Oncogene 22(3):401–411
Yu CL, Prochownik EV et al (1993) Attenuation of serum inducibility of immediate early genes by oncoproteins in tyrosine kinase signaling pathways. Mol Cell Biol 13(4):2011–2019
Zuccato C, Valenza M et al (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev 90(3):905–981
Acknowledgments
We are grateful to Bianca Scholz and Dr. Mekhman Azizov for technical assistance; to Dr. José María Rojas, Madrid for the GST-Grb2 subdomain fusion protein constructs; and to Dr. Mario Vallejo, Instituto de Investigaciones Biomédicas “Alberto Sols”, Madrid for the GST-Raf1-RDB plasmid. The project was supported by the DFG (AU96/11-1, 13-1 and 14-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Drost, J., Nonis, D., Eich, F. et al. Ataxin-2 Modulates the Levels of Grb2 and Src but Not Ras Signaling. J Mol Neurosci 51, 68–81 (2013). https://doi.org/10.1007/s12031-012-9949-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-012-9949-4