
Abstract
Recently, the first genome-wide association (GWA) study in frontotemporal lobar degeneration (FTLD) identified common genetic variability at the TMEM106B gene on chromosome 7p21.3 as a potential important risk-modifying factor for FTLD with pathologic inclusions of TAR DNA-binding protein (FTLD-TDP), the most common pathological subtype in FTLD. To gather additional evidence for the implication of TMEM106B in FTLD risk, multiple replication studies in geographically distinct populations were set up. In this review, we revise all recent replication and follow-up studies of the FTLD-TDP GWA study and summarize the growing body of evidence that establish TMEM106B as a bona fide risk factor for FTLD. With the TMEM106B gene, a new player has been identified in the pathogenic cascade of FTLD which could hold important implications for the future development of disease-modifying therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In an attempt to resolve the missing heritability in neurodegenerative diseases, genetic research has shifted focus from linkage and positional cloning studies in multigenerational families for the identification of rare causal genes, towards genome-wide association (GWA) studies for the identification of more common genetic risk factors. In ever-growing sample sizes, these studies have become increasingly successful, e.g., in amyotrophic lateral sclerosis (ALS, (van Es et al. 2009)), Parkinson's disease (Simon-Sanchez et al. 2009), and Alzheimer's disease (Lambert et al. 2009; Harold et al. 2009). However, in FTLD research, likely due to the higher familial load in this dementia subtype (up to 30–40% of patients have a positive family history), the first GWA study was only recently undertaken. In FTLD, the known causal genes explain 10% to 20% of all patients. In our Flanders–Belgian FTLD cohort (n = 315) for example, we have observed 35 patients with a mutation in GRN (n = 13), MAPT (n = 4), VCP (n = 2), CHMP2B (n = 1), FUS (n = 1), or PSEN1 (n = 1) representing 11% of the total population and 29% of familial patients. Notwithstanding, a significant fraction of patients, including the familial patients, remains unresolved. In the beginning of 2010, the results of the first GWA study in FTLD were published (Van Deerlin et al. 2010). Although there had been skepticism about the success of such a GWA study in FTLD, because of its inherent clinical and pathologic heterogeneity, this study ensured its success by including only patients with FTLD-TDP pathology. Despite having to pay a penalty for more limited sample size, this approach dramatically reduced the genetic heterogeneity of their study population and thereby their power to identify disease association. In an international collaboration including 45 sites from 11 countries, DNA from 515 patients with confirmed FTLD-TDP pathology, either by histopathology or presence of a GRN mutation, was collected. Comparison of the FTLD-TDP patients to 2,509 control individuals identified three single-nucleotide polymorphisms (SNPs) with genome-wide significance (rs1020004, rs6966915, and rs1990622). The three top SNPs mapped to a single linkage disequilibrium (LD)-block of 68 kb at 7p21.3 encompassing the gene coding for TMEM106B, an uncharacterized transmembrane protein, and two other genes at the rim of the associated region, scinderin (SCIN) and ADP-ribosylation factor-like 4A (ARL4A) (Van Deerlin et al. 2010). The association was strongest in carriers of pathogenic GRN mutations, suggesting a disease modifying effect of TMEM106B SNPs in GRN mutation carriers. Moreover, expression studies in brain demonstrated that TMEM106B is expressed in frontal cortex (heavily affected in FTLD) and is significantly increased in FTLD-TDP patients compared to unaffected individuals, providing initial biological evidence for TMEM106B as a novel risk factor for FTLD-TDP.
When establishing new risk genes identified through GWA studies of complex diseases, replications in independent study populations are paramount. Hereto, we set out to replicate the association of FTLD with the TMEM106B locus in a Flanders–Belgian patient-control cohort (van der Zee et al. 2011). Whereas the original study purposely selected patients with FTLD-TDP pathology, we investigated a cohort of patients with primarily clinical diagnoses of FTLD, in the absence of postmortem histopathology. One might argue that this inclusion criterion would result in a too heterogeneous patient cohort. We addressed this concern by reducing as much as possible the fraction of FTLD-tau in our sample. Patients of whom histopathology was available and was indicative of non-TDP pathology, such as FTLD-tau or FTLD-FUS, were excluded. Furthermore, patients with non-TDP associated mutations, such as MAPT, CHMP2B, or PSEN1 mutations, were excluded. Also, limiting inclusion to patients with a clinical diagnosis of the three major FTLD subtypes FTD, PNFA, and SD, but not of CBS or PSP further reduced the presence of FTLD-tau in our patient cohort. Finally, replication in a Flanders–Belgian population has the added advantage that patients and control individuals originate from a small geographical region and have shown limited geographic mobility over time, in turn reducing the genetic heterogeneity as exemplified by recurrence of Flanders–Belgian founder mutations (Cruts et al. 2006; Nuytemans et al. 2008). In 288 patients who were not previously included in the GWA study and 595 unaffected patients, we replicated association of FTLD with the three 7p21.3 top SNPs (van der Zee et al. 2011). Allelic association reached significant p values of p = 0.008, p = 0.013, and p = 0.041 for rs1990622, rs6966915, and rs1020004, respectively (Table 1). For SNP rs1990622 showing the strongest evidence of association, the odds ratio (OR) for the minor allele was 0.75 [95% confidence interval (CI) 0.61–0.93]. Comparing genotype frequencies and associated effect sizes, a minimal OR was calculated for rs1020004 (ORCC = 0.51, [95% CI 0.29–0.91], p = 0.023) (Table 2). For rs1990622, one copy of the risk allele was sufficient to achieve a significantly decreased risk effect (ORCT = 0.72, [95% CI 0.53–0.98], p = 0.036; ORCC = 0.59, [95% CI 0.38–0.91], p = 0.019) (Table 2). In other words, carriers of one or two copies of the TMEM106B protective alleles, have a 30% to 50% reduced risk of developing FTLD. To screen for potential functional variants and to investigate whether TMEM106B could not only be a risk factor but also a causative gene for FTLD, we performed exon sequencing analysis of TMEM106B in the population of Flanders–Belgian FTLD patients. Of the 61 detected sequence variants, merely 2 predicted amino acid substitutions, S134N and T185S (Fig. 1). S134N is located in exon 5 and was detected with equal frequency of 2% in patients as in control individuals. T185S (rs3173615), located in exon 6, was observed with a frequency of 35% in patients versus 42% in control individuals. Of the remainder of the SNPs, none were predicted to be near splice sites, transcription factor binding sites, or microRNA binding sites, indicating that TMEM106B does not seem to bear highly penetrant mutations leading to FTLD in our patient sample. SNP saturation of the TMEM106B genomic region indicated that the association is likely restricted to a 36 kb genomic region containing only the TMEM106B gene (van der Zee et al. 2011). However, fine mapping did not identify additional independent associated SNPs, other than rs1990622 and rs1020004. Importantly, rs1990622 and rs1020004, for which the correlation (r 2) was 0.79 in the Flanders–Belgian population, were in high LD with 30 more SNPs, including rs6966915 (third GWA top SNP, r 2 = 0.97 with rs1990622) and rs3173615 (r 2 = 0.98 with rs1990622) coding for the missense mutation T185S that we previously identified in the sequencing analysis. The functional relevance of these SNPs to FTLD remains unknown at the present. However, the fact that they are all located in or very close to TMEM106B, and that no other genes are present in or near the associated region, makes TMEM106B the most likely candidate underlying FTLD susceptibility. In silico prediction of the functional properties of the 32 SNPs anticipated the highest impact for rs3173615, T185S, though the predicted effect on protein function was modest. The missense mutation is positioned at codon 185, C-terminal of the transmembrane domain (codon 97–117). Except for being a single-pass transmembrane protein, little is currently known about the function of TMEM106B, making it difficult to estimate the potential impact of amino acid substitutions on its biological function. Analysis of protein conservation, however, indicated that the T-residue at position 185 is highly conserved among vertebrates.

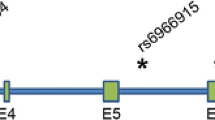
Gene structure of the TMEM106B gene. The relative position of the three replicated SNPs from the FTLD-TDP GWA study is indicated. Mutation analysis by exonic sequencing in 288 Flanders–Belgian FTLD patients identified two coding variants, one rare missense variant S134N in exon 5 and one common missense variant T185S (rs3173615) in exon 6. T185S was found in high linkage disequilibrium with rs1990622 in the Flanders–Belgian population (r 2 = 98)
In addition to the Flanders–Belgian replication study, other replication studies of TMEM106B are being reported in geographically distinct populations. Whereas our case–control study only included ten unrelated patients with a GRN mutation, Finch and colleagues (2011) focused on the risk-modifying effect of TMEM106B on GRN-related FTLD. In contrast to our findings, in 482 clinical FTLD patients without GRN mutations compared to 822 control individuals, no association with the TMEM106B SNPs was observed. However, in a relatively small sample of 78 GRN-positive patients, the TMEM106B variants appeared to be highly significantly associated, with homozygotes of the minor alleles decreased to 3% of patients versus 19% of controls (p = 0.009) (Finch et al. 2011). Finch et al. (2011) attributed this to a protective effect of the TMEM106B SNPs specific to GRN mutation carriers. We observed a similar trend in the subset of 97 familial Flemish patients (counting only ten GRN mutation carriers). Here, homozygotes of the rs1990622 protective C allele were decreased to 8% (n = 7) in patients (compared to 13% in the total patient population) versus 18% in controls, with a reduced disease risk of 68% (ORCC = 0.32, [95% CI 0.14–0.73], p = 0.007). As both in the original GWA study by Van Deerlin et al. and the replication study by Finch et al., the effect was clearly most pronounced in GRN mutation carriers; it is likely that the TMEM106B protective effect is strong in GRN-related FTLD. However, our data seem to indicate that although the effect is clearly present in the GRN-positive patients, it is not limited to GRN-related FTLD. The higher genetic load in familial patients is probably at the basis of the observed stronger genetic association. Although we have to be mindful of over-stratifying the cohorts in, e.g., subsets of familial patients or of GRN-positive patients, as this may result in small, statistically less reliable sample sizes in which the allele frequencies may be skewed.
In a first step to find biological evidence for the association of TMEM106B and FTLD-TDP, Van Deerlin et al. (2010) were able to demonstrate an up to 2.5 increase of TMEM106B expression in brain frontal cortex of FTLD-TDP patients compared to controls. This led them to suggest that TMEM106B confers FTLD-TDP risk by increased expression. Obtaining high-quality RNA from a significant number of brain samples is challenging. In our study, we were able to analyze brain frontal cortex samples from 11 FTLD-TDP patients and 12 control individuals. In this sample set, we observed no increased TMEM106B expression in FTLD-TDP patients compared to controls. Although our number of brain samples was limited, it was in the same range of the original study (n = 25) (van der Zee et al. 2011). Furthermore, in brain frontal cortex cDNA of 40 unaffected controls, Cruchaga et al. (2011) also failed to find evidence of correlation between TMEM106B expression and genotype. Expression analyses of larger number of patient and control brains will be vital to resolve whether or not TMEM106B pathogenesis is mediated by variable gene expression.
Ever since the identification of GRN as a major disease gene in FTLD, researchers have speculated about the biological basis of the observed variability in onset age associated with GRN mutations. GRN mutations lead to FTLD through haploinsufficiency; still, onset age is highly variable within and between families and ranges from mid-forties to mid-seventies (Cruts and Van Broeckhoven 2008). Furthermore, mutation carriers who died without symptoms in their 80s have been reported. This strongly suggests that other genes and/or environmental factors affect onset age in GRN-related FTLD. Moreover, identification of such genetic modifiers may expose valuable molecular targets for disease-modifying therapies. It is in this light that Cruchaga and colleagues (2011) hypothesized that TMEM106B variants might explain (part of) this phenotypic variability. They analyzed 27 symptomatic and 23 presymptomatic GRN mutation carriers from four previously reported GRN-loss of function (LOF) families (HDDD1 - Ala237fs, FD1 - Ala237fs, HDDD2 - Ala9Asp, and the Karolinska family - Gly35fs) and observed a strong association between rs1990622 and disease onset, with a mean decrease in onset age of 13 years for patients homozygote for the risk allele (T) compared to patients with one or two copies of the minor protective allele (C) (Cruchaga et al. 2011). Moreover, they additionally demonstrated a correlation between rs1990622 and GRN plasma levels in 73 healthy individuals (Cruchaga et al. 2011). In a much more extended control series, Finch and coworkers (2011) also considered if associated TMEM106B variants modified disease penetrance in GRN-positive FTLD by modulating GRN protein levels. In 1,013 plasma samples from healthy controls, they showed that the TMEM106B SNPS are associated with a modest but significant increase in GRN protein levels. The findings on onset age by Cruchaga et al. were not in line with what we observed in the extended Flanders–Belgian FTLD founder pedigree segregating the GRN IVS1+5G>C splice donor site mutation (Cruts et al. 2006; van der Zee et al. 2011). The founder pedigree consists of 226 clinically well-documented individuals, who are related in at least seven generations, including 41 patients and at least 37 asymptomatic at risk mutation carriers. Onset age among patients ranges from 45 to 76 years, and at least 7 mutation carriers were still free of symptoms while older than the mean onset age in the pedigree, including an obligate mutation carrier who died at 81 years of age (Cruts et al. 2006; Brouwers et al. 2007). Onset age analysis in this powerful multigeneration pedigree demonstrated no correlation of the TMEM160B variants rs1040002 and rs1990622 with onset age, failing to explain the >30 years onset age range in the GRN IVS1 + 5 G > C carriers (van der Zee et al. 2011). Therefore, in the Flanders–Belgian GRN founder family, we see no evidence for a disease-modifying effect of TMEM106B. Possibly multiple GRN-LOF modifiers exist and another modifying factor is contributing to the phenotype in our founder family compared to the other GRN-LOF families. Although the control studies by Finch and Cruchaga seem to indicate that TMEM106B variants influence GRN protein levels and thereby disease penetrance in GRN-related FTLD, a direct correlation between GRN plasma levels and onset age has not been demonstrated to date. Also, the observed increase in GRN protein levels in healthy homozygous carriers of the protective TMEM106B alleles seems very limited to be able to rescue the extreme drop in GRN levels that is observed in GRN-LOF patients (Ghidoni et al. 2008; Finch et al. 2009; Sleegers et al. 2009; Sleegers et al. 2010). Further more elaborate studies in affected versus unaffected GRN-LOF mutation carriers will be required to gather conclusive evidence for whether or not TMEM106B is able to modify disease penetrance and onset age in GRN mutation carriers by modulating GRN plasma levels.
In a follow-up study by the principal authors of the FTLD-TDP GWA study, Vass et al. (2010) investigated whether the FTLD-TDP associated TMEM106B risk genotypes could also contribute to the risk and disease presentation of ALS. Although they could not demonstrate association between the TMEM106B genotypes and ALS, the protective allele of rs1990622 did appear to be associated with preserved cognition in ALS patients. Correlation with cognitive performance was strong as measured by the phonemic verbal fluency test, but could however not be confirmed by other cognitive tests. Their findings, although preliminary, are of interest in light of the TDP-43 proteinopathy spectrum that binds FTLD and ALS. It appears that TMEM106B, as a risk factor for FTLD, is also able to influence cognitive impairment in ALS patients. Along this line, Rollinson et al. (2011) set out to replicate not only the genome-wide associated TMEM106B locus at 7p21.3 but also investigated the other suggestive loci identified by the FTLD-TDP GWAS on chromosomes 1, 8, 9, 10, and 11 that reached sub-threshold significance. In a clinical cohort of 470 British patients, replication at chromosomes 1, 7, 8, 9, 10, and 11 was negative. However, when restricting the analysis to the 84 patients with an FTLD-ALS phenotype, convincing evidence for association was found at multiple SNPs on the 9p locus, a locus well known to be linked to multiple FTLD-ALS families (ALSFTD2, (Boxer et al. 2011; Gijselinck et al. 2010; Le Ber et al. 2009; Luty et al. 2008; Valdmanis et al. 2008; Morita et al. 2006; Vance et al. 2006)) as well as being associated to ALS risk (van Es et al. 2009). This study again suggests that FTLD and ALS share a common genetic basis that also includes shared genetic risk factors.
In conclusion, although the multiple replication and follow-up studies differed somewhat in research focus and therefore resulting observations, overall, the findings from the different research groups in geographically distinct populations seem to point out TMEM106B as bona fide risk factor for FTLD. Further research is needed to learn whether T185S, variable gene expression or another mechanism contributes to the disease biology driving the association of TMEM106B with FTLD. Hereto, targeted resequencing of the TMEM106B genomic region may uncover other potential functional variants that explain the association. Elucidating the biological pathways through which TMEM106B is able to affect disease risk of FTLD, whether or not by influencing GRN protein levels, will shed new light on the pathogenic cascade of FTLD-TDP and FTLD in general. These novel insights will bring us one step closer in understanding this devastating dementia disorder and in developing targeted therapeutic strategies.
References
Boxer AL, Mackenzie IR, Boeve BF et al (2011) Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry 82:196–203
Brouwers N, Nuytemans K, van der Zee J et al (2007) Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 64:1436–1446
Cruchaga C, Graff C, Chiang HH et al. (2011) Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch. Neurol
Cruts M, Van Broeckhoven C (2008) Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet 24:186–194
Cruts M, Gijselinck I, van der Zee J et al (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924
Finch N, Baker M, Crook R et al (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132:583–591
Finch N, Carrasquillo MM, Baker M et al (2011) TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76:467–474
Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G (2008) Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71:1235–1239
Gijselinck I, Engelborghs S, Maes G et al (2010) Identification of 2 loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol 67:606–616
Harold D, Abraham R, Hollingworth P et al (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41:1088–1093
Lambert JC, Heath S, Even G et al (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41:1094–1099
Le Ber I, Camuzat A, Berger E et al (2009) Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology 72:1669–1676
Luty AA, Kwok JB, Thompson EM et al (2008) Pedigree with frontotemporal lobar degeneration–motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol 8:32
Morita M, Al-Chalabi A, Andersen PM et al (2006) A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66:839–844
Nuytemans K, Rademakers R, Theuns J et al (2008) Founder mutation p.R1441C in the leucine-rich repeat kinase 2 gene in Belgian Parkinson's disease patients. Eur J Hum Genet 16:471–479
Rollinson S, Mead S, Snowden J et al (2011) Frontotemporal lobar degeneration genome wide association study replication confirms a risk locus shared with amyotrophic lateral sclerosis. Neurobiol Aging 32:758
Simon-Sanchez J, Schulte C, Bras JM et al (2009) Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41:1308–1312
Sleegers K, Brouwers N, Van Damme P et al (2009) Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 65:603–609
Sleegers K, Brouwers N, Van Broeckhoven C (2010) Role of progranulin as a biomarker for Alzheimer's disease. Biom Med 4:37–50
Valdmanis PN, Kabashi E, Dion PA, Rouleau GA (2008) ALS predisposition modifiers: knock NOX, who's there? SOD1 mice still are. Eur J Hum Genet 16:140–142
Van Deerlin VM, Sleiman PM, Martinez-Lage M et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239
van der Zee J, Van Langenhove T, Kleinberger G et al (2011) TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134:808–815
van Es MA, Veldink JH, Saris CG et al (2009) Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet 41:1083–1087
Vance C, Al CA, Ruddy D et al (2006) Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain 129:868–876
Vass R, Ashbridge E, Geser F et al (2011) Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol 121:373–380
Acknowledgments
Research in the authors' research group was funded in part by the Interuniversity Attraction Poles (IAP) program P6/43 of the Belgian Federal Science Policy office; the Foundation for Alzheimer Research (SAO/FRMA); the Association for Frontotemporal Dementias (AFTD); the Medical Foundation Queen Elisabeth; a Methusalem excellence grant of the Flemish Government; the Research Foundation Flanders (FWO); the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-V); and the Special Research Fund (BOF) of the University of Antwerp, Belgium. The FWO provided a postdoctoral fellowship to [J.v.d.Z.].
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van der Zee, J., Van Broeckhoven, C. TMEM106B a Novel Risk Factor for Frontotemporal Lobar Degeneration. J Mol Neurosci 45, 516–521 (2011). https://doi.org/10.1007/s12031-011-9555-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9555-x