
Abstract
Background
When a patient arrives in the emergency department following a stroke, a traumatic brain injury, or sudden cardiac arrest, there is no therapeutic drug available to help protect their jeopardized neurons. One crucial reason is that we have not identified the molecular mechanisms leading to electrical failure, neuronal swelling, and blood vessel constriction in newly injured gray matter. All three result from a process termed spreading depolarization (SD). Because we only partially understand SD, we lack molecular targets and biomarkers to help neurons survive after losing their blood flow and then undergoing recurrent SD.
Methods
In this review, we introduce SD as a single or recurring event, generated in gray matter following lost blood flow, which compromises the Na+/K+ pump. Electrical recovery from each SD event requires so much energy that neurons often die over minutes and hours following initial injury, independent of extracellular glutamate.
Results
We discuss how SD has been investigated with various pitfalls in numerous experimental preparations, how overtaxing the Na+/K+ ATPase elicits SD. Elevated K+ or glutamate are unlikely natural activators of SD. We then turn to the properties of SD itself, focusing on its initiation and propagation as well as on computer modeling.
Conclusions
Finally, we summarize points of consensus and contention among the authors as well as where SD research may be heading. In an accompanying review, we critique the role of the glutamate excitotoxicity theory, how it has shaped SD research, and its questionable importance to the study of early brain injury as compared with SD theory.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: Spreading Depolarizations and Their Clinical Importance
At the immediate onset of brain ischemia, spreading depolarization (SD) is the principal mechanism of electrochemical membrane failure and neuronal swelling in gray matter of the higher brain [1,2,3,4,5,6]. In this review, the “higher” brain includes all structures above the hypothalamus and brainstem. Within 1–2 min of severe ischemia, depletion of the available ATP pool leads to Na+/K+ pump failure with eruption of a front of cellular depolarization that propagates at 1–9 mm/minute throughout the ischemic tissue as well as into the surrounding penumbral and normal tissue [7,8,9,10,11]. This SD wave drives a sudden and active jump in neuronal membrane potential to near 0 mV over seconds (Fig. 1), in contrast to most other tissues of the body that slowly depolarize over many minutes in an energy crisis. However, SD does not arise only in ischemic tissue but can also be provoked by various noxious electrical, chemical, thermal, or mechanical disturbances of gray matter. Thus, SD is associated with a range of diseases and conditions, including migraine-associated aura, concussion [12], traumatic brain injury (TBI), subarachnoid hemorrhage, intracerebral hemorrhage, ischemic stroke, circulatory arrest (Fig. 2), brain death prior to circulatory collapse, and sudden unexplained death in epilepsy [3, 13,14,15,16,17,18]. When SD invades tissue that has not already been rendered electrically silent by the triggering pathology, SD induces a loss of spontaneous and evoked activity termed “spreading depression” [19].

Intracellular recordings from single rodent neurons undergoing SD in live brain slices. Neurons in the higher brain briskly undergo SD while most neurons in the lower brain respond slowly and then consistently recover. A, In response to 5-min 9.6 mM K+, a rat neocortical pyramidal cell depolarizes to − 73 mV then repolarizes in control aCSF. Exposure to 26 mM K+ elicits more depolarization with spiking and then spike inactivation, before a plateau of − 48 mV is reached. SD onset (arrow) is aborted just as control aCSF reaches the slice. A third K+ exposure again evokes firing and spike inactivation, reaching a plateau at − 48 mV. A steep depolarization then coincides with SD onset (arrow). Modified from [107]. B, Typical membrane potential changes (Vm) of a neocortical neuron (somatosensory cortex; mouse) shown in the upper trace with a simultaneous extracellular field potential recording (FP) acquired near the recorded neuron in the lower trace. After application of KCl in layer I/II of the neocortex, neurons in layer V depolarize abruptly during CSD as the negative DC shift initiates. The inset shows a brief burst of high-frequency population spikes recorded during the early DC deflection. Modified from [228]. C A rat hippocampal CA1 pyramidal neuron undergoes a terminal SD induced by 10 min of OGD. Typically, higher neurons in neocortex, thalamus, striatum and hippocampus reduce their firing during OGD before undergoing rapid SD to near-zero millivolts, with no recovery. D A ‘lower’ locus ceruleus neuron, like most neurons in hypothalamus and brainstem, only slowly depolarizes in response to OGD. As action potentials inactivate, the membrane potential slowly continues to near-zero millivolts (Weak SD). On return to control aCSF, the neuron slowly recovers, dramatically different from higher neurons post-OGD. C and D modified from [107]

Image sequence and the kinetics of the optical signal of spreading depolarization (SD) induced by cardiac arrest in the rat cerebral cortex. A,B The position of the closed cranial window and the parietal cortex exposed for imaging. C The propagation of anoxic terminal SD as visualized by a voltage‑sensitive dye (RH1838) (Farkas et al., 2008). Note the clear wave front and the rostro‑caudal direction of propagation of the terminal SD event. The scale bar in C1 represents 500 μm. D The variation of the optical signal at the two regions of interest (ROIs in C 1) reveals the evolution of terminal SD. The time delay between terminal SD onset at the two ROIs confirms the propagation of the event. The rate of propagation of this particular terminal SD event was 4.1 mm/min between ROI1 and ROI2. The DC potential trace shown with inverted polarity to match the optical signal traces indicates a terminal SD acquired with electrophysiology after cardiac arrest in another rat. Note correspondence of the optical and DC potential signals (unpublished data from the Farkas laboratory). E Six-contact Wyler electrode strip superimposed on a geometrically discretized whole human brain taken as MPRAGE magnetic resonance imaging sequence. F The process of brain death in the patient begins with the simultaneous decline of arterial pressure and spontaneous cortical activity (AC-ECoG; 0.5 Hz high-pass filter, subdural electrode). Cardiac arrest then triggers a spike in intracranial pressure, likely reflecting a cerebral vasodilatory response to the sharp decrease in cerebral perfusion pressure. Within 57 s of cardiac arrest, residual synaptic activity then ceases in a non-spreading depression (NSD) of activity. After a further 75 s, terminal SD develops as evidenced by the negative shift in DC-ECoG (no filter) that spreads from electrode 2 to electrode 1. The red asterisk marks the time delay between the two electrodes suggesting the propagation of anoxic terminal SD (ATSD). The cause of death was a hepatorenal syndrome after severe aneurysmal subarachnoid hemorrhage. G The other patient suffered extensive shear injury and basilar skull fractures as a result of a motor vehicle accident. Electrocorticography and partial pressure of brain oxygen were monitored through a bolt placed in the right frontal lobe. Approximately three hours before death, mean arterial pressure declined to <50 mm Hg. The traces then showed a further decline in arterial pressure with a lowering heart rate as spontaneous cortical activity (AC-ECoG) decreased progressively to a state of electrical silence (NSD). Anoxic terminal SD began 96 s later. Assuming an ideal linear spread, the time delay of 41 s between negative shifts of DC-ECoG would indicate a propagation velocity of 3.2 mm/min, since intraparenchymal electrodes were separated by 2.2 mm.
Stroke and SDs
In the ischemic core, neurons will die under a maintained depolarization that typically lasts 20–30 min or more [20,21,22,23,24,25,26]. However, if the ischemic core is reperfused within ~ 15 min, all neurons of the ischemic core will survive, even though the neurons have been persistently depolarized for about 15 min [23, 27]. In contrast, perfusion completely ceases after cardiac arrest, and so neurons start to die after about 5 min [28]. Importantly, there are also ischemia models that do not show an ischemic core because the reduction in cerebral blood flow is milder. In such in vivo models, there is no terminal SD, but typically a cluster of recurrent moderately prolonged SDs occurs superimposed on a relatively shallow negative ultraslow potential. Yet cell death also develops [29]. A similar pattern of clustered SDs is also seen when aerobic metabolism in astrocytes is selectively blocked by aconitase inhibitors, which is associated with cell death [30, 31]. Furthermore, this pattern is recorded in the ischemic penumbra in models of middle cerebral artery occlusion or photothrombosis in numerous studies. Clustered SD events pose a particularly high metabolic challenge for recovery.
The acute injury imposed by SD has been characterized as a “continuum,” determined by the degree of metabolic stress in the tissue where SD propagates [9, 32]. This concept can be illustrated in the middle cerebral artery occlusion model in which the first SD wave persists in the core, is moderately prolonged but reversible in the penumbra, and is short lasting in the surrounding well-perfused tissue where membrane potentials recover after 1–2 min [33, 34]. The SD duration and waveform change dramatically as it propagates from the core through the penumbra and into normal tissue. There is also a continuum with respect to neuronal injury in which core neurons rapidly begin to suffer necrotic death, whereas penumbral neurons can withstand several cycles of recurrent SD and recovery before dying. In the surrounding tissue, recovery follows each SD [3, 35, 36]. Also, the pharmacological properties of the SD event change along the SD continuum, as further detailed below.
The delayed nature of penumbral SDs presents a potential therapeutic window whereby their inhibition could improve neurological outcome [37]. These SDs appear to arise as a consequence of energy supply–demand mismatch [30, 38]. The cumulative effect of many secondary SDs is a progressive deterioration of metabolic status and lesion expansion. This occurs not only because of cytotoxic membrane failure, but also because of SD evoking microvascular constriction in injured tissue with impaired neurovascular coupling. Known as “spreading ischemia” [39], this inverse hemodynamic or initially vasoconstrictive response to SD promotes prolongation of the cellular depolarization and thus cell death [3, 10, 38, 40,41,42]. By contrast, the normal hemodynamic response to SD in naïve, normally perfused tissue consists of a predominantly hyperemic initial phase followed by mild long-lasting oligemia after tissue repolarization [43,44,45,46]. During this normal oligemic phase, neurovascular responses to hypercapnia and functional activation are transiently disturbed, but the tissue fully recovers [47, 48]. The hemodynamic response to SD often shows a continuum across tissue from an inverse ischemic response to an increasingly normal hyperemic response [49]. Toxic factors, for example, released after subarachnoid hemorrhage, may also disrupt neurovascular coupling [3]. Tissue regions displaying inverse hemodynamic responses to SDs can display impaired autoregulation, thereby increasing the risk of irreversible tissue damage [50]. Importantly, these concepts have all been clinically translated and validated by electrocorticographic monitoring of patients with severe acute brain injury [20, 51].
Beyond Stroke
In brain trauma as well as ischemic and hemorrhagic stroke, 50–90% of patients exhibit cortical SDs, and many show continuous, repetitive events lasting several days or even weeks after injury, with total counts of 50–100 or more. Even terminal SDs, those with no recovery, have been observed as the correlate of newly developing focal infarcts and of brain death at end of life [14, 15, 23, 52]. The full continuum from the normal hyperemic to the inverse ischemic response to SD has been found in patients with aneurysmal subarachnoid hemorrhage [23, 53, 54], TBI [50], and malignant hemispheric stroke [11].
The Na+/K+ ATPase is the main transporter regulating transmembrane cationic gradients. Its compromise leads to SD. The pump exchanges three cytosolic Na+ for two extracellular K+ via hydrolysis of adenosine triphosphate (ATP) [55]. As such, this transport is itself electrogenic, contributing several hyperpolarizing millivolts to the membrane potential. In mammalian gray matter, the Na+/K+ pump is responsible for ~ 50% of ATP hydrolysis [56]. Lack of blood oxygen and glucose inhibits ATP production, with pump failure evoking sudden SD, driven by the opening of a cryptic Na+/K+ current [57, 58]. Neurons in live slices undergo SD in response to oxygen–glucose deprivation (OGD) [59,60,61]. Also effective is severe hypoxia in an interface slice chamber [62, 63]. Similarly, SD is imaged and recorded in vivo under ischemia [64,65,66].
Despite the potential clinical relevance and the decades-long history of SD research, including recent comparative and molecular genetic approaches in insects [67,68,69], it is still unclear how compromise of the Na+/K+ pump links to SD generation. Essentially, we have not yet identified the molecular mechanisms underlying either the spread or the depolarization. The specific ionic channel type (or types) that opens to drive SD is not yet known, despite its hypothesized existence based on computer modeling [70] and its general characterization as a nonspecific Na+/K+ current [57, 58]. Voltage-gated Na+ channels also carry some inward current, and because Na+ influx predominates over K+ efflux, neurons depolarize. It is likely that the same conductance drives SD propagation through healthy, nonischemic tissues, as during migraine aura.
The purpose of this critique is to challenge and discuss several basic issues concerning the largely unknown molecular mechanisms that initiate, drive, and terminate SD. We will also highlight our issues of consensus and of contention regarding SD. Year after year, published reviews imply that the basic cellular mechanisms underlying acute neuronal death following brain ischemia are reasonably well established based on the glutamate excitotoxicity theory. In fact they are not [71]. The critical role of SD in acute brain damage has been consistently underestimated. Here we highlight areas of agreement and progress as well as those open to debate that require more research into SD. First, how failure of the Na+/K+ ATPase (the “pump”) leads to SD initiation is unclear, as well as how the SD event regenerates and propagates. Elevated levels of released extracellular K+ or of glutamate can help perpetuate SD propagation, but as argued below, neither appear to be a viable driver of SD within metabolically stressed gray matter. Second, the specific channel that opens to drive SD is not yet identified. Third, the processes that terminate SD and establish a period that is refractory to SD have not yet been fully determined. Recovery from SD requires, at minimum, a reactivation of the Na+/K+ ATP pump to reestablish lost ion concentration gradients. This itself will delay SD reignition [9, 72].
An important complicating aspect of SD research is that SD can be generated in isolated preparations using different forms of metabolic stress, as discussed in the next section.
SD and the Central Role of the Na+/K+ ATPase
What Does the Na+/K+ Pump Do?
The living animal cell has a high concentration of impermeable anions inside as well as a cell membrane permeable to K+ and less so to Na+. The pump actively removes Na+ and replenishes leaking K+. Both cations are similarly imbalanced, but K+ is near its electrochemical equilibrium because the negative resting potential opposes its concentration gradient. Thus, energy is associated primarily with the Na+ electrochemical gradient. The pump maintains these gradients and, in turn, cell volume [1].
Most treatments that initiate SD act to overwhelm or reduce the activity of the Na+/K+ ATPase. This is most obvious when using ouabain to specifically inhibit the pump, which induces SD in vivo and in slice preparations [29, 73,74,75,76,77]. Similarly, ischemia, OGD, or hypoxia evokes SD because the Na+/K+ pump stops working when ATP reserves are depleted. Gray matter cannot completely repolarize from SD until the ATP-dependent active transport of Na+ and K+ against their physiological gradients is reestablished. Thus, during SD, intracellular Na+ ([Na+]i ) increases from 10 to ~ 40 mM, whereas intracellular K+ ([K+]i ) decreases from 135 to ~ 105 mM [78]. By contrast, Na+ concentration outside ([Na+]o ) decreases from 145 to 60 mM and [K+]o increases from 3 to ~ 60 mM [79, 80]. In the presence of 3 mM ATP, the affinity for Na+ is about three times that of K+ at the cytoplasmic site of the Na+/K+ pump, whereas the affinity for K+ is about I00 times higher than that of Na+ on the extracellular site [81].
In the well-perfused neocortex, ATP levels fall by ~ 50% as nonischemic SD taxes the Na+/K+ ATPase [89]. By contrast, in the severely ischemic neocortex, ATP levels are already low, and resulting pump failure causes the SD event. The pivotal role of the Na+/K+ ATPase in clearing [K+]o becomes apparent when its normal function is impaired. Partial pharmacological inhibition of the Na+/K+ ATPase by using a low concentration of ouabain considerably slows K+ removal [76, 90, 92]. Whether the recovery of Na+/K+ ATPase transport is the sole mechanism terminating SD is unknown but unlikely.
Na+/K+ ATPase Isoforms and SD
The alpha, beta, and FXYD subunits form the Na+/K+ ATPase. Each have several isoforms, so many variants of the enzyme exist with differing properties [93]. Nevertheless, SD is a feature of insects that express only one alpha and one beta isoform and no FXYD subunit. Na+/K+ pump dysfunction also affects other linked transport processes, reducing, for example, the local extracellular/intracellular [Ca2+] gradient.
The alpha 1 isoform is expressed in all vertebrate cells. It has a high affinity for Na+ and so activates to extrude small Na+ increases during baseline neuronal activity, essentially serving a housekeeping function [94]. Compared with the alpha 3 isoform (below), it is much less efficient at handling Na+ removal during rapid discharge or during maintained depolarization. It is the most prevalent isoform in higher brain neurons, those most prone to SD, such as the neocortex, striatum, hippocampus, and thalamus [95]. So upregulating alpha 1 activity represents a target for protection or augmentation of pump function to combat ischemia. However, being expressed in every cell type, it is a less specific therapeutic target compared with alpha 2 and alpha 3 (below).
Alpha 2 is expressed in striated muscle and neural tissue, primarily astrocytes and other glia. The a2b2 complex is maximally active when [K+]o is high and the membrane potential is depolarized and so is efficient for clearing K+ after intense neuronal activity. The role of the alpha 2 isoform in SD is supported by the clinical and experimental observations that mutations in ATP1A2, the gene encoding alpha 2, induce familial hemiplegic migraine type 2. This is a Mendelian model of disease for SD and SD-induced spreading depression of activity, resulting in the patient percept of migraine aura [96]. Reduced alpha 2 expression leads to a propensity for SD because the astrocytic alpha 2 component is less efficient [9, 92]. In addition to the model in which aerobic metabolism in astrocytes is selectively blocked by aconitase inhibitors [30], familial hemiplegic migraine type 2 provides another example that SD, although a primarily neuronal phenomenon, can also arise as a consequence of impaired astrocyte function. A third example is provided by genetically modified mice in which astrocyte-driven inactivation of connexin 43 reduces astrocytic gap junction communication, leading to increased SD susceptibility and propagation [97].
Alpha 3 is found in the heart and is highly expressed in the brain, mainly in neuronal projections [98] and dendritic spines [94]. During intense neuronal activity, the local Na+ concentration in dendrites and spines increases from 15 mM to as high as 100 mM [99]. The required Na+ clearance that follows is mainly attributed to alpha 3 [100], which has a low sodium affinity [82, 101, 102] such that alpha 1 < alpha 2 < alpha 3. The alpha-3-containing pumps in neurons appear optimized for clearance of elevated Na+ concentration during high-intensity neuronal firing at depolarized levels. This has implications for resilience during SD. In support, the alpha 2 and alpha 3 isoforms are proposed to reduce vulnerability to ischemia compared with alpha 1 [56, 103]. Mined data from the Allen Brain Bank [104] indicate that the hypothalamus and the brainstem neurons express a greater proportion of alpha 3 to alpha 1 compared with the higher brain regions of the neocortex, thalamus, striatum, and hippocampus [105]. This may help explain how hypothalamic neurons [106] and brainstem neurons [95] can maintain firing, better resist complete depolarization, and better recover during OGD in contrast to neurons in higher brain regions, which immediately succumb to SD. Note that the hippocampus displays an intermediate proportion of alpha 3 expression yet is susceptible to SD and ischemic damage, so an elevated neuronal alpha 3 expression does not guarantee increased resistance to ischemia [105]. The lower projection neurons resist SD evoked by OGD or ouabain [107] or by high [K+]o [108], likely because alpha 3 more efficiently removes higher levels of intracellular Na+. Therefore, compared with the alpha 1 isoform, alpha 3 can, in theory, better handle large Na+ loads in electrically active cells and, by extension, during SD. Increasing alpha 3 expression in neurons might improve postischemia resilience and survival.
Elevating [K+]o and Pump Inhibition
Raising [K+]o is a common technique to induce SD (see Modes of SD induction), but how it activates SD is still unclear. At the level of the single cortical neuron, SD induced by raising [K+]o (Fig. 1A, B) is different from SD induced by OGD (Fig. 1C). From the Nernst equation for K+, high [K+]o directly depolarizes neurons, thereby activating voltage-sensitive Na+ channels and so promoting discharge. With further depolarization, action potentials inactivate. Graded K+ application (Fig. 1A) indicates that this passive depolarization is separate and distinct from the SD current in higher brain neurons (Fig. 1A, arrows). Presumably the Na+/K+ ATPase is compromised, activating the SD current, but this requires further research.
In contrast, hypoxia or OGD initially quiets firing by dampening synaptic input and briefly hyperpolarizing the neuron before SD current is activated (Fig. 1C) [95, 109]. In higher brain slices, the one-two punch of metabolic stress and SD means that the neuron has trouble recovering. In contrast (Fig. 1D), neurons from hypothalamus [106] and brainstem [95] undergo weak SD and more easily repolarize.
The normally functioning pump clears [Na+]i and [K+]o, both of which increase during neuronal firing. The pump also compensates for cell swelling. It essentially protects against SD onset. And yet the glutamate excitotoxicity theory ignores the Na+/K+ pump as a major player in the development of acute brain injury [71].
Variability in Experimental Preparations Used for Investigating SD
There are several variables which have complicated research aiming to reach a consensus on the mechanisms evoking and driving SD. See also a discussion of `false positives` that can mistakenly imply SD is blocked [71].
Terminology
One source of confusion in the literature is equating the terms ‘ischemia,’ ‘oxygen–glucose deprivation’ and ‘hypoxia.’ Although hypoxia is an important component of all three conditions, the three terms should be used precisely. ‘Ischemia’ should be reserved for in vivo models characterized by (1) lack of oxygen, (2) lack of glucose, and (3) lack of perfusion. Hypoxic-ischemic brain injury [110] is related to a frequent scenario in the clinic when the patient is deeply anesthetized and stops breathing, which requires intubation. Otherwise, the patient becomes hypoxic, affecting cerebral and cardiovascular function [15]. The hypoxia can evoke cardiac arrest, causing a double hit to the brain from initial hypoxia and subsequent global ischemia. Only models that precisely reproduce this clinical scenario should be called models of hypoxic-ischemic brain injury.
In addition, the term ‘peri-infarct depolarization’ (PID) is misleading and should be avoided [20, 32]. It is better to use the term ‘spreading depolarization’ and to refer to the SD continuum. PID implies being able to precisely differentiate ischemic regions of the core, penumbra, and surrounding healthy tissue. But ischemic lesion development follows a dynamic continuum in space and time. This has been argued in the aforementioned articles and will not be detailed here [38]. The term ‘PID’ supports the incorrect concept that SDs are the consequence of the infarct. In fact, SDs precede the development of the infarct and initiate the cascades leading to the infarct.
Another source of confusion is that SD may be accompanied by two different depression patterns. It can be preceded by nonspreading depression in severely energy-compromised tissue, or it can cause a spreading depression of activity in less ischemic or well-supplied tissue. If the first SD begins in the core area of focal ischemia, after a nonspreading depression of spontaneous activity has developed there about 2 min before, the SD can no longer induce a spreading depression of spontaneous activity there because there is no activity to depress. However, when the SD wave propagates through the penumbra, where spontaneous activity is still present, it leads to a spreading depression of that activity. In clinical recordings, it is important that spreading depression of activity is defined because SDs in electrically silent tissue are associated with worse patient outcome than SDs inducing depression of activity that spreads [111].
Also confusing, equating ischemia with hypoxia in the slice literature [112] is misleading because hypoxia-induced SD is not equivalent to OGD-induced SD. With hypoxia, there can be more robust recovery, and therefore putative neuroprotectants can appear more potent. Additionally, blocking glutamate receptors can inhibit the onset of hypoxic SD, whereas OGD usually only delays SD [71] (see Extracellular K+ promotes SD but is unlikely to be the natural SD activator).
Modes of SD Induction
There are several experimental slice techniques to induce SD, each varying considerably in their level of metabolic stress that compromises the Na+/K+ pump. These stressors include (in reducing order of metabolic load) the following: chemical ischemia (metabolic inhibitors combined with OGD) [113, 114], blockade of mitochondrial respiration [115], OGD alone [59, 60, 88, 116], exposure to the pump inhibitor ouabain [73, 75], hyperthermia [117], and hypoxia in interface slices (i.e., oxygen removed with reduced glucose) [7, 118]. Also, severe hypoglycemia induces SD in vivo [119]. Finally veratridine, which blocks Na+ channel closure (inactivation), causing abnormally high and sustained Na+ influx, is another inducer of SD [120].
There are less metabolically demanding techniques to evoke SD in vivo, usually involving raising [K+]o. These include prolonged elevation of [K+]o at mildly increased concentrations [121,122,123] and prolonged elevation of [K+]o at higher concentrations [124, 125]. Additionally, SD can be evoked focally by local K+ ejection in vivo [13, 123, 126, 127] or in slices [128]. Also in slices, brief elevation of bath [K+]o allows induction of SD (Fig. 1A, B) repeatedly with recovery [88, 129, 130]. It is also important to note that raising [K+]o for a sufficiently long time will produce a terminal SD [131]. This eventually results in neuronal death but takes longer than does ischemia [72, 132]. Focal induction of SD in vivo is also possible optogenetically [47], by using pin prick [133], or by using strong electrical stimulation [23, 134]. The latter is far more intense than even the most pathological brain activity and is associated with histopathological evidence of a small lesion [23].
Highly unphysiologically hypoosmotic media can promote SD in slices [135]. This is likely the result of greatly elevated excitability and synchronicity involving combined increases in the strength of synaptic transmission [136], elevated field effects [137], and intrinsic neuronal excitability [138], as reviewed in [1]. An acute decrease in plasma osmolality in intact mammals will quickly elicit seizure activity [139, 140]. However, SD associated with dilute plasma osmolality, even at pathophysiological levels, has not been reported in patients.
We conclude that SD is a highly conserved response by the brain’s gray matter that can be evoked by a plethora of stressors. And the properties of SD varies as it propagates further away from the site of action of the stressor [9, 32]. This continuum complicates research on SD and on neuroprotection in general.
Experimental Temperature
The metabolic stressors used to induce SD have been tested in vitro at different temperatures ranging from 28 to 37 °C. Higher temperature imposes a greater metabolic challenge, leading to earlier failure of the Na+/K+ pump and thus earlier SD onset. In rodent brain slices, temperatures > 40 °C promote SD onset [141]. However, similar to the condition of high [K+]O [76], previous data in the immature hippocampus suggest that this is not necessarily due to a lack of ATP [142] but rather overwhelming of the working pump’s transport capacity. On the other hand, lower temperature within the 28–37 °C range is neuroprotective partly because it reduces metabolic demand for ATP, thereby lowering propensity for SD [46, 60, 143]. So a drug or other treatment suspected of inhibiting SD onset, say at 32 °C, will appear more effective than at 37 °C. Therefore, it is more physiologically relevant to test potential SD inhibitors near body temperature, particularly with respect to simulated ischemia.
Regarding in vivo studies, TBI patients with hourly core temperatures of > 38.4 °C have a 63% chance of depolarization, compared with only 21% in patients with temperatures of ≤ 38.4 °C [144]. This is consistent with the finding that induced hyperthermia increases SD frequency following focal cerebral ischemia in the rat [143]. Higher temperature pushes the balance of energy toward deficit. Additionally, hyperthermia increased SD frequency in the comatose patient, and SD events themselves increased brain temperature but not body temperature [145]. Local brain temperature was also elevated during subarachnoid hemorrhage in patients who showed a terminal SD cluster, whereas delayed cerebral infarction developed at the recording site [23]. Notably, SD itself will increase brain temperature: SD releases ~ 90% of Gibbs free energy (i.e., the energy associated with a chemical reaction that can be used to do work), which is normally contained in the ion concentration gradients across the neuronal membranes. This energy is then converted to heat [125].
Age of the Experimental Animal
A second likely source of variability is that investigations in rodents in vivo and in brain slices have been performed across a substantial age range. Advancing age and ischemia appear to elevate the thresholds of SD both in vivo [146] and in brain slices [147, 148]. On the other hand, neonatal brains do not seem to support SD [149]. However, OGD-induced SD is already found by P5 in rats [150]. Milder SD stimuli may fail until postnatal day 10 and continue to fail occasionally up until day 20 [151]. However, SD susceptibility of the tissue then rapidly reaches its maximum at 16–30 days of age and slowly decreases again thereafter [147, 148, 152]. So in general, age correlates negatively with SD propagation [153].
The CNS Region Under Study
A third variation is that SD has been studied using slices from several brain regions, including the neocortex, hippocampus, striatum, thalamus/hypothalamus, brainstem, and cerebellum, in addition to the excised retina. First, the mouse primary sensory cortex is more susceptible to SD than other neocortical regions exposed to anoxia, a trend also seen in patients with TBI [8].
Next, gray matter in the hypothalamus and the brainstem are significantly more resistant to OGD-evoked SD than in higher brain regions [107, 154] as shown in Fig. 1D. In fact, it is not possible to elicit full-blown SD using high K+ artificial cerebrospinal fluid (aCSF) in slices of the hypothalamus or brainstem [108, 155]. The reason is still unclear, but this SD resistance helps explain why the hypothalamus and brainstem suffer less damage following global ischemia, possibly promoting the emergence of the persistent vegetative state.
As another example of regional susceptibility, AMPAR activation of Purkinje cells in cerebellar slices appear to have a greater role in SD generation [155] than in other brain regions. Severe ischemia is a potent stimulus of SD in the cerebellum [156,157,158]. However, selective neuronal necrosis of the highly vulnerable Purkinje cells could also result from decreases in blood flow and tissue partial pressure of oxygen when ischemia was too mild to cause an SD [159]. Under identical conditions in the neocortex, selective neuronal necrosis was only observed in the hypoperfused cortex when either the mild ischemia triggered at least one SD or an SD was chemically triggered at a distance and invaded the hypoperfused zone [29, 160]. This means that Purkinje cells may have the lowest ischemic threshold for cell death in the CNS.
Regarding spinal cord gray matter, George Somjen wrote that it was “immune” to SD ([63], p. 260) but that under “special conditions,” SD could be evoked in a minority of slices [162]. However, that study used infant mice under hypoxia, and so SD would be more difficult to elicit compared with adult slices of the spinal cord using OGD (adult slices of spinal cord are exceedingly difficult to keep viable). It was later shown that the dorsal and ventral horns clearly undergo SD in response to ischemia both in vivo [163] and in response to OGD using spinal cord slices [164]. It has been suggested that, like higher gray matter, this SD susceptibility reflects greater vulnerability of the spinal cord to contusion injury (similar to higher brain) as compared with the better protected brainstem [107].
Submerged Versus Interfaced Brain Slices
Compared with being submerged, slices held at a gas–liquid interface display a higher baseline excitability for several reasons. They acquire a compacted extracellular space caused by tissue flattening imposed by high surface tension at the gas/fluid boundary [165]. Thus, ephaptic interactions (field effects) are increased, promoting local synchronous discharge among neuronal populations particularly in laminated cortical gray matter [166]. Whether field effects can promote SD onset is debatable. However, released glutamate and K+ levels are more concentrated within the compacted tissue, further increasing electrophysiological excitability compared with submerged slices. Glucose is more slowly replenished because the slice is only perfused from below, so these slices are likely somewhat hypoglycemic when made anoxic. Anoxic interface slices show good recovery from SD when the slices are reoxygenated after a couple of minutes. This contrasts with OGD in submerged slices, which is more a model for global ischemia after cardiac arrest without resuscitation attempts.
Two disadvantages to interface slices are the slow diffusion of drugs into and out of the preparation and the difficulty in acquiring imaging data when objective lens submersion is required, as with multiphoton laser microscopy [2, 167]. George Somjen’s laboratory commonly used interface slices combined with severe hypoxia to induce SD [168, 169]. The interface preparation allowed rapid restoration of oxygen, leading to near-complete recovery of slice health and enabling the induction of repeated SD events in a single slice. Hypoxia alone is not sufficient to induce SD in submerged slices [170]. Slices that are made hypoxic are used to simulate clinical situations that produce hypoxemia, such as apnea, asphyxia, respiratory failure (i.e., acute respiratory distress syndrome), drowning, and high altitude, showing similar modifications of SD as observed under hypoxia in vivo [15], whereas slices deprived of oxygen and glucose (OGD) simulate the condition of severe ischemia.
In both hypoxic interface slices and submerged slices exposed to OGD, normal action potential-evoked synaptic release fails a minute or more prior to SD onset [161]. In in vivo models of ischemia, hypoxic interface slices and OGD paradigms in submerged slices demonstrate that synaptic failure occurs rapidly and precedes anoxic SD [95, 118, 171, 172]. In the case of hypoxia, failure of synaptic transmission can persist over hours [173]. The evoked field excitatory postsynaptic potential is lost about 2 min before SD onset in neocortical slices exposed to OGD [59, 174, 175]. This early electrical inactivity that precedes SD in severe energy compromise is associated with a hyperpolarization of neurons both in slices and in vivo [61, 80, 176]. Spontaneous release of small quanta of neurotransmitters, such as gamma minobutryic acid (GABA) and glutamate, is also recorded [118]. Also, adenosine release may inhibit synaptic transmission even ahead of the transmitter pool of glutamate becoming exhausted [115].
Kunkler and Kraig [177] found that SD could be initiated only rarely in organotypic slices submerged under normal saline superfusion. Acute slice models of hypoxia, and particularly of OGD, simulate severe energy deficiency, whereas brief K+ elevation simulates milder stress. In all cases, the result are dramatic SD events that are easily recorded or imaged. Slices are also advantageous for investigating whether an SD-related mechanism is still present in the absence of intact blood circulation.
The Channel Type(s) Initiating and Driving SD
We have yet to pinpoint a major source of inward current driving SD. The main conductance driving SD is known to be a dual Na+ influx/K+ efflux [58], but the actual channel that opens is not yet identified. Computer models often rely on a persistent inward voltage-sensitive Na+ current (see Issues of Contention Among the Authors Regarding SD), but these channels are of low density on neurons and drive only minor depolarization. SD proceeds in the presence of voltage-sensitive Na+ channel blockers [71]. Müller and Somjen [169] showed that when tetrodotoxin (TTX, which blocks action potentials) was added to selective n-methyl-d-aspartate (NMDA) and non-NMDA glutamate antagonists, SD was prevented in half the hypoxic slices. When SD did occur, it was delayed, yet neurons depolarized to the same extent as in normal solution. The SD-related sudden decrease in [Na+]o was depressed by only 19% in the presence of all three drugs, indicating that Na+ flows into neurons other than through ionotropic glutamate receptors and TTX-sensitive Na+ channels, presumably through an, as yet, unidentified nonspecific Na+/K+ channel [58].
Given the lack of viable candidates, it is probable that the initial SD and the ensuing penumbral SDs are driven by the opening of a still unidentified channel [178, 179] that is normally inactive in healthy, nonischemic gray matter [178]. It could become modified from an existing channel that then can conduct larger molecules under ischemic conditions, a so-called megachannel, as discussed by Anderson et al. [59]. Fujiwara et al. in 1992 [180] originally showed that carboxyfluorescein (MW 376 Da) leaks rapidly from blebbing CA1 neurons during 5 to 7 min of OGD, leading Tanaka et al. [181] in 1999 to predict “the formation of micro-pores in the small blebs may generate the rapid depolarization (i.e., SD) because it is voltage-independent and is due to a nonselective increase in permeability to all participating ions, which probably occurs only in pathological conditions.” Calcein (MW 623 Da) and SR-101 (MW 607 Da) also leak on exposure to OGD [182]. Pannexin 1 had been a megachannel candidate because it opens after SD onset, providing a further conduit for the flux of ions during SD [182]. Nevertheless, OGD-induced SD still proceeds and causes acute neuronal damage in the presence of the pannexin 1 antagonist carbenoxolone [174]. In vivo, neither SDs in normal tissue nor SDs induced by moderate ischemia are inhibited by carbenoxolone [183, 184]. Moreover probenecid, another pannexin 1 antagonist, does not inhibit hypoxia-induced SD [118] or OGD-induced SD [185]. Typically, the story is less clear for K+-evoked SD, in which selective pharmacological inhibition of the P2X7 channel does not affect SD but inhibiting of the P2X7–PANX1 pore complex reduces SD frequency and duration [186].
Alternately, an open SD channel might arise as a conversion from what is normally a transporter molecule. Membrane patch- clamp studies provide elegant evidence that the marine poison palytoxin transforms the Na+/K+ ATPase into an open channel in cardiac myocytes and human embryonic kidney (HEK) 293 cells [187, 188], mammalian erythrocytes [189], and frog oocytes [190]. In a similar fashion, maitotoxin binds to the plasma membrane Ca2+ ATPase and turns it into a Ca2+-permeable ion channel [191]. Conversion from pump to channel is not surprising given the evolutionary link between transporters and channels, in which mutations can convert one to the other [192,193,194]. Perhaps metabolic stress can do the same. Recent evidence indicates that an SD-like inward current is generated by the Na+/K+-ATPase conversion from transporter to channel in the presence of palytoxin. Andrew et al. [195] proposed that this toxin might mimic a released molecule that binds to and opens the pump, generating SD. Compromise of the Na+/K+ pump clearly initiates SD, but whether ischemia can convert it to an open channel is an important question.
Extracellular K+ Promotes SD But is Unlikely to be the Natural SD Activator
The mechanism of SD propagation is controversial, although it apparently follows a nonlinear reaction/diffusion process [196,197,198,199]. That is, neurons (or possibly other brain cells) release a neuroactive substance that diffuses to adjacent cells, where it triggers in neurons a depolarizing, self-propagating, regenerative process. Essentially, SD is induced at every spot in the tissue reached by the SD wave. Among candidates for a humoral factor, K+ and glutamate are commonly considered. Glutamate was discussed as unlikely [71]. Regarding K+, there are also significant arguments against it being the key mediator of SD propagation.
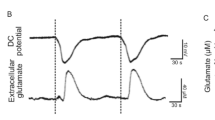
First, at a given point in gray matter, [K+]o does not begin to significantly increase before the negative DC shift is underway [79, 115, 121, 125]. Yet the concept is so influential that the steep rise in [K+]o can be found illustrated ahead of the negative DC shift [200]. Note that under severe hypoxia or ischemia, or if the experimenter applies K+, a minor increase in [K+]o arises prior to SD (Fig. 1B).
Second, the magnitude of [K+]o necessary for SD ignition casts doubt that diffusion of [K+]o is sufficiently high enough to sequentially drive SD initiation as it propagates. At the SD front, where neuronal depolarization is just under way, measured [K+]o is barely above baseline, so it cannot be continually reinitiating SD as the front propagates. The same rationale holds for glutamate as an SD initiator.
Third, it is difficult to explain how tissue with a maintained and elevated [K+]o of ~ 15 mM (the approximate minimum to evoke SD) can maintain well-configured and regular SD events, as observed both in vivo and in brain slices.
Why does flooding the slice with physiological levels of bath-applied glutamate (0.1–1.0 mM) or of NMDA or kainate fail to induce an organized spreading event, whereas elevating [K+]o induces spread? Both released K+ and released glutamate will initially be buffered by glial reuptake mechanisms that will be quickly swamped, leading to depolarization of the entire field of neurons. Yet an SD is evoked by bath-applied K+ but not bath-applied glutamate. One explanation is that elevated [K+]o directly decreases pump activity, whereas massive glutamate receptor activation simply depolarizes every neuron at once, later leading to pump overload. We have hypothesized that pump compromise is a prerequisite for generating a propagating event (see SD and the central role of the Na +/K + ATPase). Clearly, the way to fully refute the K+ hypothesis is to chemically identify a factor that is released by gray matter in response to metabolic stress which then becomes elevated just prior to SD onset.
There is a consensus that released K+ helps drive SD propagation. Hypoxia- or ischemia-evoked SD is preceded by a slow increase in [K+]o [29, 80, 109]. This may contribute to the ignition of SD in the energy-deficient brain region because, as shown by the Nernst equation for K+, high [K+]o directly depolarizes neurons and astrocytes. In neurons, this will open V-sensitive Na+ channels [201] as well as a small persistent neuronal Na+ conductance [202]. Also contributing to the depolarization, high [K+]o reduces glutamate uptake by the excitatory amino acid transporters [203, 204]. But the main driver of SD is the Na+/K+ conductance [58] through an, as yet, unidentified channel type.
Hints of a Released Activator of SD that is Neither Glutamate Nor K+
The slow migration of SD suggests that a chemical activator could be released by stressed gray matter extracellularly just ahead of the SD front. The initiator has often been assumed to be released K+ [205], but as noted with glutamate, there is the cause/effect issue as well as other problems [71]. One hypothesis is that an as yet unidentified SD activator (SDa) may be released by gray matter, acting to inhibit the pump. That an SDa might both initiate and promote SD regeneration is an intriguing idea. Martins-Ferreira et al. [206] found that the medium bathing isolated retinas undergoing light-evoked SD could trigger SD in another otherwise untreated retina. Moreover, Kunkler and Kraig [177] found that SD could be initiated only rarely in organotypic slices submerged under normal saline superfusion. However, in an interface chamber where saline exposure was restricted to the bottom surface of the cultures, SD could be initiated by electrical stimulation. They suggested that a soluble substance released to the interstitial space was essential for SD. Pomper et al. [207], also using hippocampal slice cultures, found that repetitive SD-like events at intervals of 10–15 min increased in duration and decreased in threshold for induction by electrical stimulation. The propensity for SD, however, does not increase after daily SD induction for 1–2 weeks [208], so there is no apparent cumulative effect over hours and days. Theoretically, an ideal SDa might potently inhibit the pump but quickly inactivate once the metabolic stress was removed. As several research groups have hypothesized an SDa, perhaps it reflects a dissatisfaction with K+ or glutamate fulfilling this role.
An alternate scenario is that an intracellularly transmitted SDa might pass through gap junctions between astrocytes, which are tightly electrically coupled, and adult neurons likewise might display enough coupling to support a similar spread among neurons. For instance, the positive field spikes that herald SD in the hippocampus were interpreted as return currents for negative spikes in adjacent neurons using a transcellular path for current [209]. However, blockers of gap junctions are not specific and have inconsistent effects on SD (p. 312–3 in 268). For example, neither carbenoxolone nor probenecid delayed or blocked OGD–SD in brain slices [118, 183, 210]. SDs induced by moderate ischemia in vivo were not inhibited by carbenoxolone either. Octanol and other long-chain alcohols inhibit gap junctions and SDs [211], but they are not specific [184]. For example, octanol blocks voltage-dependent sodium currents [212], potentiates GABAA receptors [213], and inhibits N-methly-d-aspartate receptors (NMDARs ) [213, 214]. Nevertheless, it remains a possibility that a small (< 1,000 Da) signaling molecule could move through open connexons of the astrocytic syncytium to activate and/or promote propagation of SD. Mouse models knocking out connexons have proven inconclusive because of the possibility of developmental compensation.
Computer Modeling of SD
The first computational model for SD was proposed by Alan Hodgkin and published in a 1963 article by Bernice Grafstein [205]. For the 4 decades that followed, computational models were more concerned with the propagation of SD than its initiation and the biophysical mechanism behind it. These models mostly followed three different theories about SD propagation: (1) K+ diffusion through extracellular space [196], (2) glutamate release from intracellular stores and diffusion through extracellular space [215], and (3) diffusion of K+ through gap junctions between neurons [216]. The model by Hodgkin and Grafstein was based on the K+ diffusion hypothesis [205], in which the governing equations included K+ diffusion and two phenomenological terms representing the release of K+ from neuronal intracellular space and recovery (clearance) by unspecified pathways. A study in 1978 [218] generalized this formalism to allow for Na+, Cl−, K+, and Ca2+ concentrations in multiple compartments.
Although most of these models give a reasonable estimate for the velocity and other basic features of SD waves, they present a simplified macroscopic view of several complex underlying microscopic mechanisms. These models were mostly inspired by the similarities between the neuronal action potential and the SD wave. Both are considered all-or-none events, in which the trajectory takes its due course controlled by inward and outward currents that are activated over different time scales. Thus, like action potential, an SD event can be represented by a simple FitzHugh–Nagumo-like formalism. That is,
and
where C and D represent the concentration and diffusion coefficient of K+ and glutamate, respectively. The nonlinear term F(C,R) models the release and recovery (uptake) of K+ or glutamate, and R is the recovery variable with linear dependence on C and R (G(C,R)). Istim is the stimulus igniting the SD wave, such as a local increase in K+ or glutamate level. However, the actual mechanism causing this stimulus is never specified. Furthermore, the release and recovery terms are often phenomenological in nature and might not necessarily represent the underlying biophysical processes.
More sophisticated models have evolved over the past 20 years but make several assumptions that may or may not be correct. First, most SD models generally assume that a persistent voltage-dependent Na+ conductance is the primary conductance driving SD. Being voltage-sensitive, they should start inactivating at ~ − 30 mV, so it is unclear how neurons are driven to 0 mV during SD. Alternately in Somjen’s laboratory, Czeh et al. [57] used TTX and other blockers to demonstrate a nonspecific Na+/K+ conductance driving the neurons to 0 mV during SD. However, a channel candidate was lacking at that time, so the Somjen laboratory turned to modeling an activated persistent Na+ current and an NMDA-controlled current [219]. In pyramidal neurons, persistent Na+ channels exist at low density on cell bodies and dendrites (compared with the axonal initial segment) [176], making those channels an unlikely source of the formidable SD current. Elevating [K+]o could enhance the persistent Na+ current [202], so it probably supports SD. Less conventional scenarios, such as the opening of a megachannel or conversion of a transporter to a channel, have not yet been modeled. In a model mimicking OGD conditions, neuronal membrane potential reaches 0 mV when the Na+/K+ pump is completely blocked [220]. However, for that scenario, ATP levels would have to be near 0, which might not be reached in the tissue. Modeling how an SD event terminates has also been challenging. Presumably with return of oxygen and glucose, the initially open channels close. Also, K+ uptake by glia and return of pump function aid in repolarization. Models may also include activation of a K+ current analogous to the K+ rectifier, although there is little evidence for such a current from neuronal recordings. Release of adenosine or other, as yet, unidentified messengers that inhibit SD may occur.
Issues of Consensus Regarding SD Among the Authors
SD evoked by ischemia of focal stroke, cardiac arrest, subarachnoid hemorrhage, or TBI is itself a major contributor to the neuronal injury that follows. But the preeminence of glutamate excitotoxicity is continually reinforced by new review articles particularly on stroke that do not question the excitotoxicity theory, do not provide new experimental support, and do not consider the important role of SD in ischemic brain damage as reviewed in [71]. Meanwhile, recent reviews on other neurodegenerative diseases (Alzheimer`s disease, Huntington`s disease, amyotrophic lateral sclerosis, traumatic brain injury (AD, HD, ALS, TBI) have abandoned glutamate excitotoxicity as causal [71]. We concur that more data are required to further support causality between SD and injury and certainly between glutamate release and long-term brain injury.
-
Experimental variables when using acute brain slices to study SD can confound results. OGD-evoked SD is not equivalent to severe hypoxia, which is less metabolically demanding. We argue for more standardized parameters when testing ischemia-like stress in brain slices.
-
The molecular mechanisms leading to SD initiation are still unclear. Moreover, how SD regeneration drives propagation is poorly understood. Elevated levels of released extracellular K+ or of glutamate can help promote SD propagation, but neither is a credible candidate as the biological initiator/perpetuator of SD. Both do play a role in K+-evoked and optogenetically evoked SD. Alternately, there are hints that one or more small molecules may be released from gray matter during ischemia to initiate and then promote SD regeneration.
-
The channel type (or types) that then opens to drive SD is still unidentified but is known to conduct both Na+ and K+, which explains why neurons depolarize to near 0 mV. It would be useful to modify current computer models of SD in this regard because TTX studies show that opening of the voltage-dependent Na+ channel initially helps drive depolarization but then it inactivates between − 30 and − 20 mV.
-
To make meaningful predictions and help resolve some of these issues, computer models must transition from the phenomenological approach, in which the focus is usually on reproducing a single experiment, to a biophysical paradigm, in which the model can explain all observations about SD. The formalism of key factors related to SD, such as the kinetics of the Na+/K+ pump, glutamate homeostasis, the spatiotemporal dynamics of extracellular K+, and the related pathways, has to be based on biophysical facts. Furthermore, the glial network, vasculature, inhibitory networks, and GABA homeostasis should be essential components of SD models because they all have roles in SD.
-
Susceptibility to SD varies among brain regions. For example hypothalamic and brainstem nuclei of the adult rodent do not support strong SD [95, 107] unless chemically depolarized [221] or preconditioned with high K+ [222]. This SD resistance can help account for hypothalamic and brainstem survival post ischemia in patients in a persistent vegetative state. The increased susceptibility of the immature brainstem to SD has been advanced as a possible explanation for sudden unexplained death in epilepsy [13, 16].
-
There is also variability in SD susceptibility across neocortical gray matter [8, 197, 223, 224]. Again, SD is not simply a default of mammalian gray matter. Differences in SD susceptibility can be clinically relevant. One underlying reason may be differential expression of Na+/K+ pump isoforms across brain regions as well as across their constituent neuronal populations.
Issues of Contention Among the Authors Regarding SD
-
How does elevated [K+]o generate experimental SD? Elevated [K+]o activates the pump at 5–8 mM, at which point pump efficiency peaks. However, once it approaches 15 mM, SD is evoked, which implicates suppression of pump activity at that point. How Na+/K+ pump transport declines in the presence of elevated [K+]o is an important issue that requires further experimentation to clarify.
-
There is indirect evidence that an endogenously generated activator of SD, neither glutamate nor K+, is released by metabolically stressed brain tissue. Experiments can be devised to test this idea. The specific molecular mechanisms underlying SD initiation, propagation, and arrest have not yet been identified. Computer modeling of the release and diffusion of a theoretical SDa and the resulting opening of nonspecific Na+/K+ channels are interesting questions for future study. Brain metabolic stress leading to release of a molecule that initiates cell depolarization could represent the long sought-after local “oxygen detector” that represents a sentinel of hypoxic threat, as suggested by Somjen in 2004 (p. 328 in [161]).
-
We know that processes are activated over seconds and minutes to terminate SD and establish a period refractory to renewed SD. It is unclear if the kinetics of pump reactivation alone can fully account for SD termination and the refractory period, which can last hours. Alternately, SD itself might lead to the release of one or more chemical messengers that inhibit the reonset of SD over minutes and possibly hours. For example, adenosine is a glutamatergic synaptic inhibitor, and its release is correlated with SD. As SD duration increases, more adenosine is released [225].
-
It is unclear if SD initiation, propagation, and duration each represent separate phenomena driven by distinct molecular and electrophysiological mechanisms. The simplest scenario is that all three aspects are essentially versions of a single process whereby depolarizing channels open and stay open until the Na+/K+ pump can at least partly recover. Some authors believe that the process is more complicated.
-
Reperfusion injury is a potential mechanism for lesion expansion, although the pathophysiology is unclear. Kloner et al. [226] recently reviewed the no-reflow phenomenon, suggesting that poor reperfusion in the brain may involve SD.
-
“Brain tsunami” has been adopted as a colloquial term for SDs, illustrating their wave-like and deadly nature (https://www.charite-academy.de/). Conversely, whether SDs could be beneficial for survival in the surrounding normal gray matter of mammals has been debated [227]. Such benefits could include upregulation of growth factors, termination of seizure discharge, stress response proteins, and inflammatory mediators. These preconditioning actions could promote plasticity and regeneration and might reduce the vascular steal effect on ischemic zones through the physiological oligemia [3]. Perhaps also, SD is a way that compromised brain tissue signals distress to neighboring regions. Certainly, there are still many fascinating aspects of SD yet to be discovered.
References
Hellas JA, Andrew RD. Neuronal swelling: a non-osmotic consequence of spreading depolarization. Neurocrit Care. 2021; (in press).
Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA. Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex. 2007;17:787–802.
Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–47.
Dreier JP, Reiffurth C. Exploitation of the spreading depolarization-induced cytotoxic edema for high-resolution, 3D mapping of its heterogeneous propagation paths. Proc Natl Acad Sci U S A. 2017;114:2112–4.
Kirov SA, Fomitcheva IV, Sword J. Rapid neuronal ultrastructure disruption and recovery during spreading depolarization-induced cytotoxic edema. Cereb Cortex. 2020;5517–31.
Mestre H, Du T, Sweeney AM, Liu G, Samson AJ, Peng W, et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science. 2020;367.
Aitken PG, Borgdorff AJ, Juta AJA, Kiehart DP, Somjen GG, Wadman WJ. Volume changes induced by osmotic stress in freshly isolated rat hippocampal neurons. Pflugers Arch J Physiol. 1998;436:991–8.
Bogdanov VB, Middleton NA, Theriot JJ, Parker PD, Abdullah OM, Ju YS, et al. Susceptibility of primary sensory cortex to spreading depolarizations. J Neurosci. 2016;
Dreier JP, Reiffurth C. The stroke-migraine depolarization continuum. Neuron. 2015;86:902–22. https://doi.org/10.1016/j.neuron.2015.04.004.
Strong AJ, Anderson PJ, Watts HR, Virley DJ, Lloyd A, Irving EA, et al. Peri-infarct depolarizations lead to loss of perfusion in ischaemic gyrencephalic cerebral cortex. Brain. 2007;130:995–1008.
Woitzik J, Hecht N, Pinczolits A, Sandow N, Major S, Winkler MKL, et al. Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology. 2013;80:1095–102.
Bouley J, Chung DY, Ayata C, Brown RH, Henninger N. Cortical spreading depression denotes concussion injury. J Neurotrauma. 2019;36:1008–17.
Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med. 2015;7:282ra46.
Dreier JP, Major S, Foreman B, Winkler MKL, Kang EJ, Milakara D, et al. Terminal spreading depolarization and electrical silence in death of human cerebral cortex. Ann Neurol. 2018;83.
Dreier JP, Major S, Lemale CL, Kola V, Reiffurth C, Schoknecht K, et al. Correlates of spreading depolarization, spreading depression, and negative ultraslow potential in epidural versus subdural electrocorticography. Front Neurosci. 2019;13.
Loonen ICM, Jansen NA, Cain SM, Schenke M, Voskuyl RA, Yung AC, et al. Brainstem spreading depolarization and cortical dynamics during fatal seizures in Cacna1a S218L mice. Brain. 2019;142:412–25.
Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:17–35.
Soldozy S, Sharifi KA, Desai B, Giraldo D, Yeghyayan M, Liu L, et al. Cortical spreading depression in the setting of traumatic brain injury. World Neurosurg. 2020;134:50–7.
Leao AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:359–90.
Dreier JP, Fabricius M, Ayata C, Sakowitz OW, William Shuttleworth C, Dohmen C, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J Cereb Blood Flow Metab. 2017;37.
Heiss W, Rosner G. Functional recovery of cortical neurons as related to degree and duration of ischemia. Ann Neurol. 1983;14:294–301.
Kaminogo M, Suyama K, Ichikura A, Onizuka M, Shibata S. Anoxic depolarization determines ischemic brain injury. Neurol Res. 1998;20:343–8.
Lückl J, Lemale CL, Kola V, Horst V, Khojasteh U, Oliveira-Ferreira AI, et al. The negative ultraslow potential, electrophysiological correlate of infarction in the human cortex. Brain. 2018;141:1734–52.
Memezawa H, Smith ML, Siesjo BK. Penumbral tissues salvaged by reperfusion following middle cerebral artery occlusion in rats. Stroke. 1992;23:552–9.
Pignataro G, Simon RP, Boison D. Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. J Cereb Blood Flow Metab. 2007;27:1–5.
Shen Q, Ren H, Cheng H, Fisher M, Duong TQ. Functional, perfusion and diffusion MRI of acute focal ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:1265–79.
Nozari A, Dilekoz E, Sukhotinsky I, Stein T, Eikermann-Haerter K, Liu C, et al. Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol. 2010;67:221–9.
Ayad M, Verity MA, Rubinstein EH. Lidocaine delays cortical ischemic depolarization: relationship to electrophysiologic recovery and neuropathology. J Neurosurg Anesthesiol. 1994;6:98–110.
Dreier JP, Kleeberg J, Petzold G, Priller J, Windmüller O, Orzechowski H-D, et al. Endothelin-1 potently induces Leão’s cortical spreading depression in vivo in the rat: a model for an endothelial trigger of migrainous aura? Brain. 2002;125:102–12.
Largo C, Cuevas P, Somjen GG, Martín Del Río R, Herreras O. The effect of depressing glial function in rat brain in situ on ion homeostasis, synaptic transmission, and neuron survival. J Neurosci. 1996;16:1219–29.
Menyhárt Á, Frank R, Farkas AE, Süle Z, Varga VÉ, Nyúl-Tóth Á, et al. Malignant astrocyte swelling and impaired glutamate clearance drive the expansion of injurious spreading depolarization foci. J Cereb Blood Flow Metab. 2021:271678X211040056. https://doi.org/10.1177/0271678X211040056.
Hartings JA, Shuttleworth CW, Kirov SA, Ayata C, Hinzman JM, Foreman B, et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leao’s legacy. J Cereb Blood Flow Metab. 2017;37:1571–94.
Sawant-Pokam PM, Suryavanshi P, Mendez JM, Dudek FE, Brennan KC. Mechanisms of neuronal silencing after cortical spreading depression. Cereb Cortex. 2017;27:1311–25.
Sugaya E, Takato MNY. Neuronal and glial activity during spreading depression in cerebral cortex of cat. J Neurophysiol. 1975;38:822–41.
Charriaut-Marlangue C, Margaill I, Represa A, Popovici T, Plotkine M, Ben-Ari Y. Apoptosis and necrosis after reversible focal ischemia: An in situ DNA fragmentation analysis. J Cereb Blood Flow Metab. 1996;16:186–94.
Nedergaard M, Hansen AJ. Spreading depression is not associated with neuronal injury in the normal brain. Brain Res. 1988;449:395–8.
Hartings JA, Rolli ML, Lu X-CM, Tortella FC. Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection. J Neurosci. 2003;23:11602–10.
von Bornstädt D, Houben T, Seidel JL, Zheng Y, Dilekoz E, Qin T, et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron. 2015;85:1117–31.
Dreier JP, Körner K, Ebert N, Görner A, Rubin I, Back T, et al. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab. 1998;18:978–90.
Bere Z, Obrenovitch TP, Bari F, Farkas E. Ischemia-induced depolarizations and associated hemodynamic responses in incomplete global forebrain ischemia in rats. Neuroscience. 2014;260.
Dreier JP, Ebert N, Priller J, Megow D, Lindauer U, Klee R, et al. Products of hemolysis in the subarachnoid space inducing spreading ischemia in the cortex and focal necrosis in rats: a model for delayed ischemic neurological deficits after subarachnoid hemorrhage? J Neurosurg. 2000;93:658–66.
Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, Ayata C. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab. 2006;26:1018–30.
Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev. 2015;95:953–93.
Chang JC, Shook LL, Biag J, Nguyen EN, Toga AW, Charles AC, et al. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain. 2010;133:996–1012.
Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab. 2009;29:1517–27.
Takaoka S, Pearlstein R, Warner D. Hypothermia reduces the propensity of cortical tissue to propagate direct current depolarizations in the rat. Neurosci Lett. 1996;218:25–8.
Böhm M, Chung DY, Gómez CA, Qin T, Takizawa T, Sadeghian H, et al. Neurovascular coupling during optogenetic functional activation: local and remote stimulus-response characteristics, and uncoupling by spreading depression. J Cereb Blood Flow Metab. 2020;40:808–22.
Wahl M, Lauritzen M, Schilling L. Change of cerebrovascular reactivity after cortical spreading depression in cats and rats. Brain Res. 1987;411:72–80.
Bere Z, Obrenovitch TP, Kozák G, Bari F, Farkas E. Imaging reveals the focal area of spreading depolarizations and a variety of hemodynamic responses in a rat microembolic stroke model. J Cereb Blood Flow Metab. 2014;34:1695–705.
Hinzman JM, Andaluz N, Shutter L, Okonkwo DO, Pahl C, Strong AJ, et al. Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain. 2014;2960–72.
Lauritzen M, Strong AJ. ‘Spreading depression of Leão’ and its emerging relevance to acute brain injury in humans. J Cereb Blood Flow Metab. 2017;
Carlson AP, Abbas M, Alunday RL, Qeadan F, Shuttleworth CW. Spreading depolarization in acute brain injury inhibited by ketamine: a prospective, randomized, multiple crossover trial. J Neurosurg. 2018;1–7.
Bosche B, Graf R, Ernestus RI, Dohmen C, Reithmeier T, Brinker G, et al. Recurrent spreading depolarizations after subarachnoid hemorrhage decreases oxygen availability in human cerebral cortex. Ann Neurol. 2010;67:607–17.
Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132:1866–81.
Gottron MA, Lo DC. The Na+/K+-ATPase as a drug target for ischemic stroke. 2009; https://doi.org/10.1007/978-1-60761-280-3
Dobretsov M, Stimers JR. Neuronal function and alpha3 isoform of the Na/K-ATPase. Front Biosci. 2005;10:2373–96.
Czeh G, Aitken PG, Somjen GG. Membrane currents in CA1 pyramidal cells during spreading depression (SD) and SD-like hypoxic depolarization. Brain Res. 1993;632:195–208.
Czéh G, Aitken PG, Somjen GG. Whole-cell membrane current and membrane resistance during hypoxic spreading depression. NeuroReport. 1992;3:197–200.
Anderson TR, Jarvis CR, Biedermann AJ, Molnar C, Andrew RD. Blocking the anoxic depolarization protects without functional compromise following simulated stroke in cortical brain slices. J Neurophysiol. 2005;93:963–79.
Joshi I, Andrew RD. Imaging anoxic depolarization during ischemia-like conditions in the mouse hemi-brain slice. J Neurophysiol. 2001;85:414–24.
Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol. 1997;78:891–902.
Gerich FJ, Hepp S, Probst I, Müller M. Mitochondrial inhibition prior to oxygen-withdrawal facilitates the occurrence of hypoxia-induced spreading depression in rat hippocampal slices. J Neurophysiol. 2006;96:492–504.
Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–96.
Murphy TH, Li P, Betts K, Liu R. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci. 2008;28:1756–72.
Petzold GC, Windmüller O, Haack S, Major S, Buchheim K, Megow D, et al. Increased extracellular K+ concentration reduces the efficacy of N-methyl-D-aspartate receptor antagonists to block spreading depression-like depolarizations and spreading ischemia. Stroke. 2005;36:1270–7.
Risher WC, Ard D, Yuan J, Kirov SA. Recurrent spontaneous spreading depolarizations facilitate acute dendritic injury in the ischemic penumbra. J Neurosci. 2010;30:9859–68.
Robertson RM, Dawson-Scully KD, Andrew RD. Neural shutdown under stress: an evolutionary perspective on spreading depolarization. J Neurophysiol. 2020;123:885–95.
Spong KE, Andrew RD, Robertson RM. Mechanisms of spreading depolarization in vertebrate and insect central nervous systems. J Neurophysiol. 2016;116:jn.00352.2016.
Spong KE, Dreier JP, Robertson RM. A new direction for spreading depolarization: investigation in the fly brain. Channels. 2017;11:97–8.
Makarova J, Ibarz JM, Canals S, Herreras O. A steady-state model of spreading depression predicts the importance of an unknown conductance in specific dendritic domains. Biophys J. 2007;92:4216–32.
Andrew RD, Farkas E, Hartings JA, Brennan KC, Herreras O, Muller M, et al. Questioning glutamate excitotoxicity in acute brain damage: the importance of spreading depolarization. Neurocrit Care. https://doi.org/10.1007/s12028-021-01429-4.
Dreier JP, Victorov IV, Petzold GC, Major S, Windmüller O, Fernández-Klett F, et al. Electrochemical failure of the brain cortex is more deleterious when it is accompanied by low perfusion. Stroke. 2013;44.
Balestrino M, Young J, Aitken P. Block of (Na+, K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices. Brain Res. 1999;838:37–44.
Basarsky TA, Duffy SN, Andrew RD, MacVicar BA. Imaging spreading depression and associated intracellular calcium waves in brain slices. J Neurosci. 1998;18:7189–99.
Jarvis CR, Anderson TR, Andrew RD. Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex (New York, NY 1991). 2001;11:249–59.
Major S, Petzold GC, Reiffurth C, Windmüller O, Foddis M, Lindauer U, et al. A role of the sodium pump in spreading ischemia in rats. J Cereb Blood Flow Metab. 2017;37.
Obeidat AS, Andrew RD. Spreading depression determines acute cellular damage in the hippocampal slice during oxygen/glucose deprivation. Eur J Neurosci. 1998;10:3451–61.
Hübel N, Ullah G. Anions govern cell volume: a case study of relative astrocytic and neuronal swelling in spreading depolarization. PLoS ONE. 2016;11:e0147060.
Hansen J, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand. 1981;113:437–45.
Müller M, Somjen GG. Na(+) and K(+) concentrations, extra- and intracellular voltages, and the effect of TTX in hypoxic rat hippocampal slices. J Neurophysiol. 2000;83:735–45.
Skou JC, Esmann M. The Na, K-ATPase. J Bioenerg Biomembr. 1992;24:249–61.
Crambert G, Hasler U, Beggah AT, Yu C, Modyanov NN, Horisberger JD, et al. Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J Biol Chem. 2000;275:1976–86.
Hasler U, Greasley PJ, von Heijne G, Geering K. Determinants of topogenesis and glycosylation of type II membrane proteins. Analysis of Na, K-ATPase beta 1 AND beta 3 subunits by glycosylation mapping. J Biol Chem. 2000;275:29011–22.
Horisberger J-D, Kharoubi-Hess S. Functional differences between alpha subunit isoforms of the rat Na, K-ATPase expressed in Xenopus oocytes. J Physiol. 2002;539:669–80.
Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, et al. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia. 2014;62:608–22.
Armstrong CM. The Na/K pump, Cl ion, and osmotic stabilization of cells. Proc Natl Acad Sci U S A. 2003;100:6257–62.
Brown CE, Wong C, Murphy TH. Rapid morphologic plasticity of peri-infarct dendritic spines after focal ischemic stroke. Stroke. 2008;39:1286–91.
Risher WC, Andrew RD, Kirov SA. Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia. 2009;57:207–21.
Mies G, Paschen W. Regional changes of blood flow, glucose, and ATP content determined on brain sections during a single passage of spreading depression in rat brain cortex. Exp Neurol. 1984;84:249–58.
D’Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J Neurophysiol. 2002;87:87–102.
Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol. 2000;522(Pt 3):427–42.
Reiffurth C, Alam M, Zahedi-Khorasani M, Major S, Dreier JP. Na+/K+-ATPase α isoform deficiency results in distinct spreading depolarization phenotypes. J Cereb Blood Flow Metab. 2020;40:622–38.
Clausen MV, Hilbers F, Poulsen H. The structure and function of the Na, K-ATPase isoforms in health and disease. Front Physiol. 2017;8:1–16.
Blom H, Bernhem K, Brismar H. Sodium pump organization in dendritic spines. Neurophotonics. 2016;3:041803.
Brisson CD, Hsieh YT, Kim D, Jin AY, Andrew RD. Brainstem neurons survive the identical ischemic stress that kills higher neurons: insight to the persistent vegetative state. PLoS ONE. 2014;9:e96585.
Leo L, Gherardini L, Barone V, de Fusco M, Pietrobon D, Pizzorusso T, et al. Increased susceptibility to cortical spreading depression in the mouse model of Familial hemiplegic migraine type 2. PLoS Genet. 2011;7.
Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Döring B, et al. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–76.
Böttger J, Margulies DS, Horn P, Thomale UW, Podlipsky I, Shapira-Lichter I, et al. A software tool for interactive exploration of intrinsic functional connectivity opens new perspectives for brain surgery. Acta Neurochir. 2011;1561–72.
Rose CR, Konnerth A. NMDA receptor-mediated na+ signals in spines and dendrites. J Neurosci. 2001;21:4207–14.
Azarias G, Kruusmagi M, Connor S, Akkuratov EE, Liu X-L, Lyons D, et al. A specific and essential role for Na, K-ATPase 3 in neurons co-expressing 1 and 3. J Biol Chem. 2013;288:2734–43.
Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–50.
Zahler R, Zhang ZT, Manor M, Boron WF. Sodium kinetics of Na, K-ATPase α isoforms in intact transfected HeLa cells. J Gen Physiol. 1997;110:201–13.
Blanco G. Na, K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin Nephrol. 2005;25:292–303.
Shen EH, Overly CC, Jones AR. The Allen Human Brain Atlas: comprehensive gene expression mapping of the human brain. Trends Neurosci. 2012;35:711–4.
Lowry CA, Golod ME, Andrew RD, Bennett BM. Expression of neuronal Na+/K+-ATPase α subunit isoforms in the mouse brain following genetically programmed or behaviourally-induced oxidative stress. Neuroscience. 2020;442:202–15.
Brisson CD, Lukewich MK, Andrew RD. A distinct boundary between the higher brain’s susceptibility to ischemia and the lower brain’s resistance. PLoS One. 2013;8.
Andrew RD. The persistent vegetative state: evidence that the lower brain survives because its neurons intrinsically resist ischemia. In: Monti MM, Sannita WG, editors. Brain funct responsiveness disord conscious. Springer: Berlin; 2016. p. 1–207.
Andrew RD, Hsieh Y-T, Brisson CD. Spreading depolarization triggered by elevated potassium is weak or absent in the rodent lower brain. J Cereb Blood Flow Metab. 2016;0271678X16657344.
Erdemli G, Xu YZ, Krnjević K. Potassium conductance causing hyperpolarization of CA1 hippocampal neurons during hypoxia. J Neurophysiol. 1998;80:2378–90.
Sekhon MS, Ainslie PN, Griesdale DE. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: a “two-hit” model. Crit Care. 2017;21:1–10.
Hartings JA, Andaluz N, Bullock MR, Hinzman JM, Mathern B, Pahl C, et al. Prognostic value of spreading depolarizations in patients with severe traumatic brain injury. JAMA Neurol. 2020;77:489–99.
Thompson RJ. Pannexin channels and ischaemia. J Physiol. 2015;593:3463–70.
Allen NJ, Rossi DJ, Attwell D. Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices. J Neurosci. 2004;24:3837–49.
Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–21.
Canals S, Makarova I, López-Aguado L, Largo C, Ibarz JM, Herreras O. Longitudinal depolarization gradients along the somatodendritic axis of CA1 pyramidal cells: a novel feature of spreading depression. J Neurophysiol. 2005;94:943–51.
Obeidat AS, Jarvis CR, Andrew RD. Glutamate does not mediate acute neuronal damage after spreading depression induced by O2/glucose deprivation in the hippocampal slice. J Cereb Blood Flow Metab. 2000;20:412–22.
Wu J, Fisher RS. Hyperthermic spreading depressions in the immature rat hippocampal slice. J Neurophysiol. 2000;84:1355–60.
Revah O, Lasser-Katz E, Fleidervish IA, Gutnick MJ. The earliest neuronal responses to hypoxia in the neocortical circuit are glutamate-dependent. Neurobiol Dis. 2016;95:158–67.
Astrup J, Norberg K. Potassium activity in cerebral cortex in rats during progressive severe hypoglycemia. Brain Res. 1976;103:418–23.
Ashton D, Willems R, Wynants J, Van Reempts J, Marrannes R, Clincke G. Altered Na+-channel function as an in vitro model of the ischemic penumbra: action of lubeluzole and other neuroprotective drugs. Brain Res. 1997;745:210–21.
Herreras O, Somjen GG. Analysis of potential shifts associated with recurrent spreading depression and prolonged unstable spreading depression induced by microdialysis of elevated K+ in hippocampus of anesthetized rats. Brain Res. 1993;610:283–94.
Maslarova A, Alam M, Reiffurth C, Lapilover E, Gorji A, Dreier JP. Chronically epileptic human and rat neocortex display a similar resistance against spreading depolarization in vitro. Stroke. 2011;42:2917–22.
Reiffurth C, Alam M, Zahedi-Khorasani M, Major S, Dreier JP. Na+/K+-ATPase α isoform deficiency results in distinct spreading depolarization phenotypes. J Cereb Blood Flow Metab. 2019;40:622–38.
Ayata C, Jin H, Kudo C, Dalkara T, Moskowitz MA. Suppression of cortical spreading depression in migraine prophylaxis. Ann Neurol. 2006;59:652–61.
Dreier JP, Isele T, Reiffurth C, Offenhauser N, Kirov SA, Dahlem MA, et al. Is spreading depolarization characterized by an abrupt, massive release of gibbs free energy from the human brain cortex? Neuroscientist. 2013;19:25–42.
Aiba I, Carlson AP, Sheline CT, Shuttleworth CW. Synaptic release and extracellular actions of zn 2+ limit propagation of spreading depression and related events in vitro and in vivo. J Neurophysiol. 2012;107:1032–41.
Takano T, Tian GF, Peng W, Lou N, Lovatt D, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;47:754–62.
Steffensen AB, Sword J, Croom D, Kirov SA, MacAulay N. Chloride cotransporters as a molecular mechanism underlying spreading depolarization-induced dendritic beading. J Neurosci. 2015;35:12172–87.
Anderson TR, Andrew RD. Spreading depression: imaging and blockade in the rat neocortical brain slice. J Neurophysiol. 2002;88:2713–25.
Zhou N, Rungta RL, Malik A, Han H, Wu DC, MacVicar B. Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression. J Cereb Blood Flow Metab. 2013;33.
Herreras O, Somjen GG. Effects of prolonged elevation of potassium on hippocampus of anesthetized rats. Brain Res. 1993;617:194–204.
Muramatsu H, Karikó K, Welsh FA. Induction of tolerance to focal ischemia in rat brain: dissociation between cortical lesioning and spreading depression. J Cereb Blood Flow Metab. 2004;24:1167–71.
Verhaegen MJJ, Todd MM, Hindman BJ, Warner DS. Cerebral autoregulation during moderate hypothermia in rats. Stroke. 1993;24:407–14.
Van Den Maagdenberg AMJM, Pietrobon D, Pizzorusso T, Kaja S, Broos LAM, Cesetti T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–10.
Chebabo SR, Hester MA, Jing J, Aitken PG, Somjen GG. Interstitial space, electrical resistance and ion concentrations during hypotonia of rat hippocampal slices. J Physiol. 1995;487:685–97.
Rosen AS, Andrew RD. Osmotic effects upon excitability in rat neocortical slices. Neuroscience. 1990;38:579–90.
Ballyk BA, Quackenbush SJ, Andrew RD. Osmotic effects on the CA1 neuronal population in hippocampal slices with special reference to glucose. J Neurophysiol. 1991;65:1055–66.
Azouz R, Alroy G, Yaari Y. Modulation of endogenous firing patterns by osmolarity in rat hippocampal neurones. J Physiol. 1997;502:175–87.
Andrew RD. Seizure and acute osmotic change: clinical and neurophysiological aspects. J Neurol Sci. 1991;101:7–18.
Andrew RD, Fagan M, Ballyk BA, Rosen AS. Seizure susceptibility and the osmotic state. Brain Res. 1989;498:175–80.
Ollen-Bittle N, Lee K, Fisher M, Gagolewicz P, Simon D, Oleschuk R, Jin A AR. Investigating a potential activator of spreading depolarization released by stressed gray matter. Can Assoc Neurosci. 2019.
Wu J, Takeo T, Wakui M, Ellsworth K, Fisher RS. Intracellular energy failure does not underlie hyperthermic spreading depressions in immature rat hippocampal slice. Brain Res. 2003;987:240–3.
Chen Q, Chopp M, Bodzin G, Chen H. Temperature modulation of cerebral depolarization during focal cerebral ischemia in rats: correlation with ischemic injury. J Cereb Blood Flow Metab. 1993;13:389–94.
Hartings JA, Strong AJ, Fabricius M, Manning A, Bhatia R, Dreier JP, et al. Spreading depolarizations and late secondary insults after traumatic brain injury. J Neurotrauma. 2009;26:1857–66.
Schiefecker AJ, Kofler M, Gaasch M, Beer R, Unterberger I, Pfausler B, et al. Brain temperature but not core temperature increases during spreading depolarizations in patients with spontaneous intracerebral hemorrhage. J Cereb Blood Flow Metab. 2018;38:549–58.
Hertelendy P, Menyhárt Á, Makra P, Süle Z, Kiss T, Tóth G, et al. Advancing age and ischemia elevate the electric threshold to elicit spreading depolarization in the cerebral cortex of young adult rats. J Cereb Blood Flow Metab. 2017;37:1763–75.
Hablitz JJ, Heinemann U. Alterations in the microenvironment during spreading depression associated with epileptiform activity in the immature neocortex. Dev Brain Res 1989;46:243–52.
Maslarova A, Alam M, Reiffurth C, Lapilover E, Gorji A, Dreier JP. Chronically epileptic human and rat neocortex display a similar resistance against spreading depolarization in vitro. Stroke 2011;42:2917–22.
Schade JP. Maturational aspects of EEG and of spreading depression in rabbit. J Neurophysiol 1959;22:245–57.
Dzhala V, Khalilov I, Ben-Ari Y, Khazipov R. Seizures accelerate anoxia-induced neuronal death in the neonatal rat hippocampus. Ann Neurol 2000;48:632–40.
Bures J. The ontogenetic development of steady potential differences in the cerebral cortex in animals. Electroencephalogr Clin Neurophysiol 1957;9:121–130
Hertelendy P, Menyhárt Á, Makra P, Süle Z, Kiss T, Tóth G, Ivánkovits-Kiss O, Bari F, Farkas E. Advancing age and ischemia elevate the electric threshold to elicit spreading depolarization in the cerebral cortex of young adult rats. J. Cereb. Blood Flow Metab. (2017). https://doi.org/10.1177/0271678X16643735.
Guedes RCA, Abadie-Guedes R. Brain aging and electrophysiological signaling: Revisiting the spreading depression model. Front Aging Neurosci 2019;11.
Richter F, Rupprecht S, Lehmenkühler A, Schaible H-G. Spreading depression can be elicited in brain stem of immature but not adult rats. J Neurophysiol. 2003;90:2163–70.
Hamann M, Rossi DJ, Mohr C, Andrade AL, Attwell D. The electrical response of cerebellar Purkinje neurons to simulated ischaemia. Brain. 2005;128:2408–20.
Fifkova E, Bures J, Koshtoyants OK, Krivanek J, Weiss T. Leao’s spreading depression in the cerebellum of rat. Experientia. 1961;17:572–3.
Kraig RP, Ferreira-Filho CR, Nicholson C. Alkaline and acid transients in cerebellar microenvironment. J Neurophysiol. 1983;49:831–50.
Nicholson C, Bruggencate GT, Steinberg R, Stöckle H. Calcium modulation in brain extracellular microenvironment demonstrated with ion-selective micropipette. Proc Natl Acad Sci USA. 1977;74:1287–90.
Oliveira-Ferreira AI, Major S, Przesdzing I, Kang EJ, Dreier JP. Spreading depolarizations in the rat endothelin-1 model of focal cerebellar ischemia. J Cereb Blood Flow Metab. 2020;40:1274–89.
Ferreira A, Milakara D, Alam M, Jorks DVL, Major S, Hartings J, et al. Experimental and preliminary clinical evidence of an ischemic zone with prolonged negative DC shifts surrounded by a normally perfused tissue belt with persistent electrocorticographic depression. J Cereb Blood Flow Metab. 2010;30:1504–19.
Somjen GG. Ions in the brain, seizures, and stroke. Oxford: Oxford University Press; 2004.
Czeh G, Somjen GG. Hypoxic failure of synaptic transmission in the isolated spinal cord, and the effects of divalent cations. Brain Res. 1990;527:224–33.
Gorji A, Zahn PK, Pogatzki EM, Speckmann EJ. Spinal and cortical spreading depression enhance spinal cord activity. Neurobiol Dis. 2004;15:70–9.
Nohda K, Nakatsuka T, Takeda D, Miyazaki N, Nishi H, Sonobe H, et al. Selective vulnerability to ischemia in the rat spinal cord: a comparison between ventral and dorsal horn neurons. Spine (Phila Pa 1976). 2007;32:1060–6.
Schuchmann S, Meierkord H, Stenkamp K, Breustedt J, Windmüller O, Heinemann U, et al. Synaptic and nonsynaptic ictogenesis occurs at different temperatures in submerged and interface rat brain slices. J Neurophysiol. 2002;87:2929–35.
Dudek FE, Yasumura T, Rash JE. “Non-synaptic” mechanisms in seizures and epileptogenesis. Cell Biol Int. 1998;22:793–805.
Enger R, Tang W, Vindedal GF, Jensen V, Helm PJ, Sprengel R, et al. Dynamics of ionic shifts in cortical spreading depression. Cereb Cortex. 2015;25.
Fischer M, Reuter J, Gerich FJ, Hildebrandt B, Hägele S, Katschinski D, et al. Enhanced hypoxia susceptibility in hippocampal slices from a mouse model of Rett syndrome. J Neurophysiol. 2009;101:1016–32.
Müller M, Somjen GG. Na(+) dependence and the role of glutamate receptors and Na(+) channels in ion fluxes during hypoxia of rat hippocampal slices. J Neurophysiol. 2000;84:1869–80.
Croning MDR, Haddad GG. Comparison of brain slice chamber designs for investigations of oxygen deprivation in vitro. J Neurosci Methods. 1998;81:103–11.
Hofmeijer J, van Putten MJAM. Ischemic cerebral damage: an appraisal of synaptic failure. Stroke. 2012;43:607–15.
Leao AAP. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol. 1947;10:409–14.
Hofmeijer J, Mulder ATB, Farinha AC, van Putten MJAM, le Feber J. Mild hypoxia affects synaptic connectivity in cultured neuronal networks. Brain Res. 2014;1557:180–9.
Douglas HA, Callaway JK, Sword J, Kirov SA, Andrew RD. Potent inhibition of anoxic depolarization by the sodium channel blocker dibucaine. J Neurophysiol. 2011;105:1482–94.
Kim JH, Park YK, Kim JH, Kwon TH, Chung HS. Transient recovery of synaptic transmission is related to rapid energy depletion during hypoxia. Neurosci Lett. 2006;400:1–6.
Fleidervish IA, Gebhardt C, Astman N, Gutnick MJ, Heinemann U. Enhanced spontaneous transmitter release is the earliest consequence of neocortical hypoxia that can explain the disruption of normal circuit function. J Neurosci. 2001;21:4600–8.
Kunkler PE, Kraig RP. Calcium waves precede electrophysiological changes of spreading depression in hippocampal organ cultures. J Neurosci. 1998;18:3416–25.
Andrew RD. The dogma of glutamate excitotoxicity as a major cause of stroke damage: an alternate target. Abstr 60010 2004 Neurosci Meet Planner San Diego, CA Soc Neurosci 2004 Online.
Herreras O, Makarova J. Mechanisms of the negative potential associated with Leão’s spreading depolarization: a history of brain electrogenesis. J Cereb Blood Flow Metab. 2020;40:1934–52.
Fujiwara N, Abe T, Endoh H, Warashina A, Shimoji K. Changes in intracellular pH of mouse hippocampal slices responding to hypoxia and/or glucose depletion. Brain Res. 1992;572:335–9.
Tanaka E, Yamamoto S, Inokuchi H, Isagai T, Higashi H. Membrane dysfunction induced by in vitro ischemia in rat hippocampal CA1 pyramidal neurons. J Neurophysiol. 1999;81:1872–80.
Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–7.
Világi I, Klapka N, Luhmann HJ. Optical recording of spreading depression in rat neocortical slices. Brain Res. 2001;898:288–96.
Petzold GC, Dreier JP. Spreading depolarization evoked by endothelin-1 is inhibited by octanol but not by carbenoxolone. Brain Hemorrhages. 2020;
Madry C, Haglerød C, Attwell D. The role of pannexin hemichannels in the anoxic depolarization of hippocampal pyramidal cells. Brain. 2010;133:3755–63.
Chen S-P, Qin T, Seidel JL, Zheng Y, Eikermann M, Ferrari MD, et al. Inhibition of the P2X7–PANX1 complex suppresses spreading depolarization and neuroinflammation. Brain. 2017;
Artigas P, Gadsby DC. Large diameter of palytoxin-induced Na/K pump channels and modulation of palytoxin interaction by Na/K pump ligands. J Gen Physiol. 2004;123:357–76.
Gadsby DC, Takeuchi A, Artigas P, Reyes N. Review. Peering into an ATPase ion pump with single-channel recordings. Philos Trans R Soc Lond B Biol Sci. 2009;364:229–38.
Kim SY, Marx KA, Wu CH. Involvement of the Na, K-ATPase in the induction of ion channels by palytoxin. Naunyn-Schmiedeberg’s Arch Pharmacol. 1995;351:542–54.
Wang X, Horisberger JD. Palytoxin effects through interaction with the Na,K-ATPase in Xenopus oocyte. FEBS Lett. 1997;409:391–5.
Sinkins WG, Estacion M, Prasad V, Goel M, Shull GE, Kunze DL, et al. Maitotoxin converts the plasmalemmal Ca 2+ pump into a Ca 2+ -permeable nonselective cation channel. Am J Physiol Physiol. 2009;297:C1533–43.
Durell SR, Hao Y, Nakamura T, Bakker EP, Guy HR. Evolutionary relationship between K(+) channels and symporters. Biophys J. 1999;77:775–88.
Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 1992;8:67–113.
Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006;440:484–9. https://doi.org/10.1038/nature04713.
Andrew RD, Britton R, McQueen SA, Kim D, Jin AY. Could the failing Na+/K+ pump convert to a channel during stroke? Soc Neurosci Abstr. 337.07/T10. 2013.
Grafstein B. Mechanism of spreading spreading depression. J Neurophysiol. 1956;19:154–71.
Kaufmann D, Theriot JJ, Zyuzin J, Service CA, Chang JC, Tang YT, et al. Heterogeneous incidence and propagation of spreading depolarizations. J Cereb Blood Flow Metab. 2017;
Milakara D, Grozea C, Dahlem M, Major S, Winkler MKL, Lückl J, et al. Simulation of spreading depolarization trajectories in cerebral cortex: correlation of velocity and susceptibility in patients with aneurysmal subarachnoid hemorrhage. NeuroImage Clin. 2017;16:524–38.
Zandt BJ, ten Haken B, van Putten MJAM. Diffusing substances during spreading depolarization: analytical expressions for propagation speed, triggering, and concentration time courses. J Neurosci. 2013;33:5915–23.
Pietrobon D, Moskowitz M. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci. 2014;15:379–93.
Takahashi S, Shibata M, Fukuuchi Y. Role of sodium ion influx in depolarization-induced neuronal cell death by high KCl or veratridine. Eur J Pharmacol. 1999;372:297–304.
Somjen GG, Müller M. Potassium-induced enhancement of persistent inward current in hippocampal neurons in isolation and in tissue slices. Brain Res. 2000;885:102–10.
Divito CB, Underhill SM. Excitatory amino acid transporters: roles in glutamatergic neurotransmission. Neurochem Int. 2014;73:172–80.
Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–6.
Grafstein B. Neuronal release of potassium during spreading depression. In: Brazier MAB, editor. Brain Funct, vol. I. Berkeley: University of California Press; 1963.
Martins-Ferreira H, De Oliveira CG, Stuchiner CJ, Rodrigues PS. Liberation of chemical factors during spreading depression in isolated retina. J Neurophysiol. 1974;37:785–91.
Pomper JK, Haack S, Petzold GC, Buchheim K, Gabriel S, Hoffmann U, et al. Repetitive spreading depression-like events result in cell damage in juvenile hippocampal slice cultures maintained in normoxia. J Neurophysiol. 2006;95:355–68.
Sukhotinsky I, Dilekoz E, Wang Y, Qin T, Eikermann-Haerter K, Waeber C, et al. Chronic daily cortical spreading depressions suppress spreading depression susceptibility. Cephalalgia. 2011;31:1601–8.
Herreras O, Largo C, Ibarz JM, Somjen GG, Del Río RM. Role of neuronal synchronizing mechanisms in the propagation of spreading depression in the in vivo hippocampus. J Neurosci. 1994;14:7087–98.
Gagolewicz P, Tressider K, Andrew RD. Still unidentified: the channel driving spreading depression during ischemia. Soc Neurosci Abstr. 2016;2016-S-148.
Largo C, Tombaugh GC, Aitken PG, Herreras O, Somjen GG. Heptanol but not fluoroacetate prevents the propagation of spreading depression in rat hippocampal slices. J Neurophysiol. 1997;77:9–16.
Nelson WL, Makielski JC. Block of sodium current by heptanol in voltage-clamped canine cardiac Purkinje cells. Circ Res. 1991;68:977–83.
Dildy-Mayfield JE, Mihic SJ, Liu Y, Deitrich RA, Harris RA. Actions of long chain alcohols on GABAA and glutamate receptors: relation to in vivo effects. Br J Pharmacol. 1996;118:378–84.
McLarnon JG, Wong JHP, Sawyer D, Baimbridge KG. The actions of intermediate and long-chain n-alkanols on unitary NMDA currents in hippocampal neurons. Can J Physiol Pharmacol. 1991;69:1422–7.
van Harreveld A. Compounds in brain extracts causing spreading depression of cerebral cortical activity and contraction of crustacean muscle. J Neurochem. 1959;3:300–15.
Herrerras O. Letter to the Editor. Electrical prodromals of spreading depression void Grafstein’s potassium hypothesis. J Neurophysiol. 2005;94:3656.
Shapiro BE. Osmotic forces and gap junctions in spreading depression: a computational model. J Comput Neurosci. 2001;10:99–120.
Tuckwell HC, Miura RM. A mathematical model for spreading cortical depression. Biophys J. 1978;23:257–76.
Kager H, Wadman WJ, Somjen GG. Conditions for the triggering of spreading depression studied with computer simulations. J Neurophysiol. 2002;88:2700–12.
Hübel N, Hosseini-Zare MS, Žiburkus J, Ullah G. The role of glutamate in neuronal ion homeostasis: a case study of spreading depolarization. PLoS Comput Biol. 2017;13.
Richter F, Bauer R, Ebersberger A, Lehmenkuhler A, Schaible HG. Enhanced neuronal excitability in adult rat brainstem causes widespread repetitive brainstem depolarizations with cardiovascular consequences. J Cereb Blood Flow Metab. 2012;32:1535–45.
Kron M, Zimmermann JL, Dutschmann M, Funke F, Müller M. Altered responses of MeCP2-deficient mouse brain stem to severe hypoxia. J Neurophysiol. 2011;105:3067–79.
Farkas E, Bari F, Obrenovitch TP. Multi-modal imaging of anoxic depolarization and hemodynamic changes induced by cardiac arrest in the rat cerebral cortex. Neuroimage. 2010;51:734–42.
Juzekaeva E, Gainutdinov A, Mukhtarov M, Khazipov R. Reappraisal of anoxic spreading depolarization as a terminal event during oxygen–glucose deprivation in brain slices in vitro. Sci Rep. 2020;10:1–13.
Lindquist BE, Shuttleworth CW. Spreading depolarization-induced adenosine accumulation reflects metabolic status in vitro and in vivo. J Cereb Blood Flow Metab. 2014;34.
Kloner RA, King KS, Harrington MG. No-reflow phenomenon in the heart and brain. Am J Physiol Hear Circ Physiol. 2018;315:H550–62.
Shuttleworth CW, Andrew RD, Akbari Y, Ayata C, Balu R, Brennan KC, et al. Which spreading depolarizations are deleterious to brain tissue? Neurocrit Care. 2020;32:317–22.
Ghadiri MK, Kozian M, Ghaffarian N, Stummer W, Kazemi H, Speckmann EJ, et al. Sequential changes in neuronal activity in single neocortical neurons after spreading depression. Cephalalgia. 2012;32:116–24.
Funding
This work was supported by grants from the Heart and Stroke Foundation of Canada and the National Science and EngineeringResearch Council of Canada to RDA, an NIH grant (NS106901) to CWS, a National Research, Development and Innovation Office of Hungary grant (K1343777) and EU Horizon 2020 research and innovation program (739953) to EF and from DFG Deutsche Forschungsgemeinschaft (German Research Council) (DFG DR 323/5-1), DFG DR 323/10-1, and BMBF Bundesministerium fuer Bildung und Forschung (Era-Net Neuron EBio2, with funds from BMBF 01EW2004) to JPD.
Author information
Authors and Affiliations
Contributions
Much of the article was written by R. David Andrew and Jens P. Dreier, with important contributions, feedback, editorial comments, and corrections from each of the other authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest to disclose.
Ethical Approval/Informed Consent
This article also adheres to all ethical guidelines outlined by Neurocritical Care for review articles and aims to provide a balanced discussion of an issue of clinical relevance to the journal.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the collection “Spreading Cortical Depolarization”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Andrew, R.D., Hartings, J.A., Ayata, C. et al. The Critical Role of Spreading Depolarizations in Early Brain Injury: Consensus and Contention. Neurocrit Care 37 (Suppl 1), 83–101 (2022). https://doi.org/10.1007/s12028-021-01431-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-021-01431-w