
Abstract
Infection of mice with variants of mouse hepatitis virus, strain JHM (MHV-JHM), provide models of acute and chronic viral infection of the central nervous system (CNS). Through targeted recombination and reverse genetic manipulation, studies of infection with MHV-JHM variants have identified phenotypic differences and examined the effects of these differences on viral pathogenesis and anti-viral host immune responses. Studies employing recombinant viruses with a modified spike (S) glycoprotein of MHV-JHM have identified the S gene as a major determinant of neurovirulence. However, the association of S gene variation and neurovirulence with host ability to generate anti-viral CD8 T cell responses is not completely clear. Partially protective anti-viral immune responses may result in persistent infection and chronic demyelinating disease characterized by myelin removal from axons of the CNS and associated with dense macrophage/microglial infiltration. Demyelinating disease during MHV-JHM infection is immune-mediated, as mice that lack T lymphocytes fail to develop disease despite succumbing to encephalitis with high levels of infectious virus in the CNS. However, the presence of T lymphocytes or anti-viral antibody can induce disease in infected immunodeficient mice. The mechanisms by which these immune effectors induce demyelination share an ability to activate and recruit macrophages and microglia, thus increasing the putative role of these cells in myelin destruction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the absence of infection or injury, the resting state of the central nervous system (CNS) is highly resistant to peripheral immune cell infiltration and activation. Access to CNS tissues by peripheral blood leukocytes is prohibited by tight intercellular junctions and low expression of adhesion molecules by endothelial cells of the blood brain barrier (BBB). Within the CNS, immune activation is further diminished through downregulation of MHC molecules by resident CNS cells, and through constitutive expression of immunoregulatory molecules such as TGF-β. Furthermore, secreted factors which regulate normal brain function and development exhibit immunosuppressive activity. When CNS infection or injury occurs, these barriers to immune infiltration and activation are altered to allow access to circulating leukocytes and protective antibody. During CNS immune responses, however, a balance between immune activation and suppression is maintained. This conserves integrity and function of non-regenerative CNS tissue, while permitting immune responses against invading pathogens. Tilting the balance toward immune suppression could result in persistent or chronic infection, whereas excessive immune activation could result in autoimmunity or bystander destruction and loss of function of vital CNS tissue [1–3].
One pathogen that infects the CNS of mice and thus challenges the local balance between immune activation and suppression is Mouse Hepatitis Virus, strain JHM (MHV-JHM). Contrary to its name, MHV-JHM is a neurotropic, not a hepatotropic, single stranded RNA virus of the family coronaviridae. Human coronaviruses related to MHV-JHM cause a variety of infection and disease ranging from the common cold to severe acute respiratory syndrome (SARS) [1, 4]. Naïve susceptible mice infected with virulent variants of MHV-JHM develop lethal acute encephalitis. However, other variants of MHV-JHM provide tools for studying both acute and chronic infection of the CNS [5]. Studies of CNS infection with these variants have identified distinct mutations in virus structure, and have compared the effect of these structural differences on viral pathogenesis and host immune responses to infection. Differences between these variants are often reflected by changes in cell tropism, neurovirulence, and in the quality of host anti-viral immune responses in the infected CNS. In order to more closely compare virus structure and function with associated pathogenesis, additional MHV-JHM variants are generated through targeted recombination, with methods similar to those originally used to create MHV strain A59 recombinant viruses [6–9]. Initially, mutations or introduced coding sequences are generated by PCR from MHV-JHM, or from host genes encoded on plasmids, respectively. The site used for expression of non-MHV sequences is within gene 4, which is not essential for virus replication or infection [10, 11]. RNA is generated from the final linearized plasmid, containing the MHV-JHM S and gene 4 sequences, by in vitro transcription. Donor RNA is then transfected into cells infected with a homologous recipient virus. Desired recombinants are selected by host cell specificity, which is altered by donor/recipient recombination. In this manner, recombinant MHV have been generated for functional studies of MHV-JHM structural proteins, or for the production of immune effectors, and for functional studies of non-structural proteins of the human coronavirus SARS-CoV [12–14].
While acute infection is characterized by rapid virus spread and CNS inflammation, chronic disease during MHV-JHM infection is characterized by incomplete clearance of infectious virus and concomitant development of demyelinating disease [5]. MHV-JHM infected mice with demyelinating disease serve as a relevant animal model of the human autoimmune disease multiple sclerosis (MS) [15, 16]. As in MS, demyelination induced during MHV-JHM infection is characterized by macrophage infiltration into the white matter of the CNS, with subsequent destruction of the protective myelin sheath surrounding the axons of CNS neuronal cells. Demyelination may be induced in mice through persistent infection with attenuated MHV-JHM strains, or by partial protection from virulent MHV-JHM by MHV-specific antibody [5, 17, 18]. Demyelinating disease induced during MHV-JHM infection is partly immune mediated, as mice lacking the ability to generate T cell responses fail to develop demyelination, despite high viral loads and widespread inflammation in the CNS of infected mice [19, 20]. To examine this relationship between virus infection and demyelination, immunodeficient mice infected with MHV-JHM receive adoptively transferred enriched populations of T cells or other immune effectors [21, 22]. These models of MHV-JHM infection provide excellent tools for investigating both acute and chronic viral infection of the CNS. However, the factors that mediate the initiation of host anti-viral immune responses, virus-induced pathology, and the regulation of these events by the specialized local environment of the CNS, are only partly understood.
Virulence and antiviral immune responses in acute infection of mice with variants of MHV-JHM
Susceptible mice infected intracerebrally with the MHV-JHM variant MHV.SD (also termed MHV-4) develop fatal acute encephalitis with a 50% mortality after inoculation with 1 plaque forming unit (pfu) [9]. In highly virulent variants of MHV-JHM, intranasal inoculation also results in uniformly fatal encephalitis in susceptible mice. The resulting pathology is generally attributed to sequence variation in the spike (S) glycoprotein. The contributions of non-S versus S genes to enhanced MHV.SD neurovirulence were determined by infection with recombinant viruses comprised of either the non-S genes of the less virulent MHV strain A59 with the S gene of neurovirulent MHV.SD, or the non-S genes of MHV-JHM with the S gene of MHV-A59 [9, 23–25]. These studies demonstrate that recombinant viruses expressing the MHV.SD S protein are more neurovirulent than MHV-A59 S glycoprotein expressing viruses.
In addition to differences in mortality between MHV.SD and MHV-A59 infected mice, host immune responses also differ. Mice infected with MHV.SD exhibit a prolonged innate response characterized by IFN-β production in the CNS beyond 5 days post infection with decreased IL-12p40 and IFN-γ transcription [24]. In MHV-A59 infected mice, IFN-β transcripts decrease after 5 days post infection, with increasing IFN-γ mRNA coincident with increasing T cell infiltration. Proinflammatory cytokines and chemokines such as IL-1, IL-6, MIP-1α, MIP-1β, and MIP-2 are upregulated in the CNS of MHV.SD mice in comparison to relatively low CNS expression during MHV-A59 infection. Adaptive immunity is suppressed during MHV.SD infection. Antigen specificity of adaptive immune responses of both CD4 and CD8 T cells are determined by recognition of defined immunodominant and subdominant epitopes within MHV strains [26, 27]. The immunodominant CD8 T cell epitope recognized in MHV-JHM infected C57BL/6 mice is presented on the H-2Db class I molecule and is located in the MHV-JHM spike glycoprotein from (amino acids 510–518 (S510)). An H-2Kb CD8 T cell epitope is located in the S protein of both MHV-JHM and MHV-A59 spanning amino acids 598–605 (S598). MHV-A59 infected mice mount a robust CD8 T cell response to the shared S598 epitope, while MHV.SD infected mice mount a weak response to both S598 and S510 epitopes [23, 24]. Total T cell infiltration (antigen specific and non-specific) is also reduced in the CNS of MHV.SD infected mice. Consistent with increased inflammation in virulent MHV.SD infection, mononuclear cells expressing CD11b and FcγRI/III are significantly increased in the CNS of MHV.SD infected mice. These results demonstrate that MHV.SD infection of the CNS results in a greater inflammatory response and a weakened CD8 T cell response when compared to MHV-A59.
To evaluate the contribution of the spike (S) glycoprotein to the pathogenesis of MHV.SD infection, targeted recombination and reverse genetic techniques were used by Phillips et al. to generate MHV.SD and MHV-A59 viruses that express the S glycoprotein of the other strain [9]. Rempel et al. further characterized the immune responses to these viruses, using recombinant MHV-A59 viruses with the S glycoprotein of either MHV-A59 (WTR13) or MHV.SD (S4R22) [25]. Infection of mice with S4R22 results in increased virulence compared to the MHV-A59 wild type recombinant WTR13. However, this increased virulence is not accompanied by a suppressed CD8 T cell response or prolonged IFN-β production, which were observed in the highly virulent MHV.SD infection. Increased virulence in S4R22 infection is associated with increased CNS macrophage infiltration and increased MIP-1α and MIP-1β transcription, while infection with either S4R22 or WTR13 results in a robust CD8 T cell response and increased CNS IFN-γ transcription. These results were repeated and confirmed by Iacono et al., who further explored the role of background (non-S) genes by generating an MHV-JHM virus that expressed the MHV-A59 S glycoprotein. Mice infected with this virus (SA59/RJHM) display an attenuated neurovirulence compared to wild type recombinant MHV-A59 (rA59). However, these mice mount a diminished antigen specific CD8 T cell response to infection similar to the suppressed response to rJHM infection. From these results, the authors concluded that although the S glycoprotein determines the neurovirulence of MHV strains, the background genes determine the extent of the CD8 T cell response.
This conclusion conflicts with results obtained from infection of mice with a slightly attenuated, yet closely related MHV-JHM variant, MHV.IA [13]. Similar to the neurovirulent MHV.SD, 50% of mice infected intracerebrally with 1 pfu of MHV.IA succumb to fatal encephalitis. However, in contrast to the weak response to MHV.SD infection, MHV.IA infected mice mount a robust MHV specific CD8 T cell response [28]. Furthermore, attenuation in neurovirulence in mice infected with MHV.IA is attributed solely to a single amino acid change in the spike glycoprotein [13]. As with studies comparing MHV.SD and MHV-A59 virulence, examining the association of the MHV S glycoprotein with neurovirulence in MHV.IA and MHV.SD infection was performed through generation of recombinant rJHM.IA virus via targeted recombination. With this strategy, a background rJHM.IA virus expressing the spike glycoprotein of MHV.SD was generated (rJHM.SD). Comparison of rJHM.IA and rJHM.SD infection revealed that the MHV.SD S expressed by rJHM.IA increases neurovirulence with accompanied increases in viral titers and lateral spread throughout the CNS of infected mice. Furthermore, the spike glycoprotein of rJHM.SD was shown to mediate increased receptor independent spread in infection of tissue culture cells lacking the carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1, the cellular receptor for MHV-JHM entry). Sequencing of these viruses indicated that the structures of the spike proteins of rJHM.IA and rJHM.SD differ by four amino acids. Targeted mutation of the S protein of rJHM.IA resulting in a single change in the amino acid 310 from a serine to a glycine generated a virus (rJIA.S310G) that exhibited increased neurovirulence in infected mice when compared to rJHM.IA infection.
Although these studies have identified structural determinants of virulence, the importance of the spike protein in the generation of contrasting immune responses to MHV.SD and MHV.IA infection is not fully understood. Prolonged production of IFN-β in the CNS of MHV.SD infected mice may provide one clue [24, 25]. Stimulation of IFN-β production in the CNS through toll like receptor 3 (TLR3) suppresses EAE [29]. In certain conditions, IFN-β induces the upregulation of numerous effectors that ultimately limit T cell responses. In cultured CNS cells, IFN-β production limits the capacity of antigen presenting cells to activate T cells [30]. In addition, mice deficient in IFN-β have increased antigen-specific CD8 T cell responses to peptide or DNA vaccination, with decreased IL-10 producing T regulatory cells (Treg) [31]. Both Treg cells and IL-10 have been implicated in regulating immune responses to viral infection as well as autoimmunity [32–34]. By controlling the magnitude of anti-viral immune responses, the activity of Treg cells and IL-10 effectively limit immune-mediated pathology while providing a potential opening to chronic viral infection. Determining the source of IFN-β in the CNS of MHV.SD infected mice is important for understanding the mechanism of MHV.SD mediated immune suppression. Neurons are a major source of type I IFN in mice infected with Theiler’s murine encephalitis virus (TMEV) [35]. Furthermore, neurons inhibit T cell responses and ameliorate disease in the CNS of mice with experimental autoimmune encephalomyelitis (EAE), via conversion of encephalitogenic T cells into regulatory T cells [36]. Determining the source of type I IFN and important associated factors in MHV.SD mediated immune suppression are the focus of current investigation.
In addition to providing a model for examining structural components and determinants of virulence, targeted recombination of MHV-JHM has provided insight into the role of immunodominant epitopes in both CD4 and CD8 T cell responses to infection. Persistent infection with viruses like HIV-1 or Hepatitis B or C viruses may select viral mutants that evade the host CD8 T cell response [37]. These mutations are commonly selected in the immunodominant CD8 epitopes recognized by a large portion of virus-specific cytotoxic T cells, thus directly diminishing their ability to clear viral infection. These CD8 T cell epitope, or CTL escape, mutations also occur during MHV-JHM infection, allowing for viral persistence and chronic demyelinating disease [38]. MHV-JHM CTL escape mutations are selected in the immunodominant S510, but not the subdominant S598 epitope. Through targeted recombination, a second high avidity CD8 T cell immunodominant epitope from lymphocytic choriomeningitis virus (LCMV gp33) [39] was added to recombinant JHM [40]. In the presence of both S510 and gp33 high avidity epitopes, demyelinating disease associated with CTL escape is prevented. In contrast to the persistent infection associated with elimination of the immunodominant MHV-JHM CD8 T cell epitope, mice infected with recombinant MHV-JHM with a single mutation in the immunodominant CD4 T cell epitope M133-147 (rJ.MY135Q) exhibit milder disease with no mortality [41]. Virus is ultimately cleared in rJ.MY135Q-infected mice and antigen specific CD8 T cell responses are equivalent to those detected in mice infected with wild type recombinant JHM. The absence of disease in rJ.MY135Q-infected mice is not attributed to decreases in either TNF-α or to increased Th2 cytokine production in the CNS. These results are striking, particularly because numerous studies have reported decreased viral clearance and increased mortality in the absence of a CD4 T cell response [19, 42–44]. Future studies will be aimed at further characterizing the roles of these specific epitopes in MHV-JHM pathogenesis, the role of other epitopes such as the subdominant Kb S598 epitope in CTL escape selection, and the mechanism by which the loss of the immunodominant CD4 epitope attenuates disease in infected animals.
In addition to studies of acute MHV-JHM infection using the MHV.SD and MHV.IA variants, infection of mice with an attenuated variant of MHV.SD, MHV-J2.2-v1, provides a model for studying acute and chronic infection of mice with subsequent development of demyelinating disease [5, 45]. J2.2v-1 differs from MHV.SD in structure by a single amino acid in the MHV S glycoprotein. Early responses to J2.2v-1 infection are similar to those in MHV-JHM-infected mice. CNS inflammation allows breakdown of the BBB, permitting peripheral blood neutrophil and monocyte infiltration. Concomitant with a T cell response, infectious virus is mostly cleared from the CNS by 2 weeks post-infection [46], but may remain detectable by low levels of viral RNA well after viral clearance [47]. However, macrophage recruitment into areas of white matter within the CNS continues in the absence of infectious virus, and mice develop plaques of demyelination in associated areas of macrophage infiltration. Thus, infection of mice with MHV-J2.2v-1 provides a model for the study of persistent viral infection and chronic demyelination.
Viral clearance and demyelinating disease in persistent MHV-JHM infection
MS is a debilitating human disease with a worldwide distribution, and is characterized by immune-mediated destruction of the myelin sheaths surrounding neuronal axons and, in some cases, degeneration of the axons themselves [15, 16]. MS patients can exhibit disease with several different, yet potentially overlapping, clinical and pathological profiles. Due to this diversity, the etiology of MS is likely diverse as well. Although not completely understood, both genetic and environmental factors play a role in disease development and progression. MS patients in remission often experience relapses after common viral infections, indicating an environmental component may be necessary to trigger disease in susceptible individuals.
Due to the multiple factors that promote and affect MS in humans, numerous animals models of demyelinating disease have been developed to examine particular aspects of its pathogenesis [15]. The well-established model experimental EAE examines immune responses to myelin antigens in rodents. Viral demyelination induced during infections with Semliki Forest virus (SFV), Theiler’s murine encephalomyelitis virus (TMEV), or MHV-JHM provide models of pathogen associated demyelinating disease [15, 48].
A hallmark of MS and associated animal models of the disease is the presence of infiltrating macrophages and resident microglia in demyelinating plaques located in the white matter of the CNS. Both cell types are able to phagocytose myelin, and therefore are potential contributors to autoimmune tissue destruction. Specific contributions of macrophages and microglia to demyelinating disease within each animal model are less clear. In models of demyelinating disease in which CNS inflammation results in a breakdown of the BBB, other peripheral blood leukocytes are also present in the CNS. Myelin-specific T cells are present in EAE lesions, while during MHV-JHM infection, T cells are predominantly specific for viral antigens. B cells play a role in demyelinating disease, as the presence of myelin specific antibody exacerbates disease in EAE. Professional antigen presenting cells such as DCs are also present in the CNS in mice in both the EAE and TMEV models, and are able to prime naïve T cells in situ [49].
Demyelination during MHV-JHM infection has been studied in several contexts. One particular model relies on infection of mice partially protected by nursing on MHV-JHM immune dams (suckling mouse model) [18]. In this scenario, maternal MHV-specific antibody facilitates partial viral clearance. Thus, these mice survive the acute infection, but subsequently develop demyelinating disease accompanied by clinical signs of hind limb paresis. In this model, development of chronic disease is associated with mutations in the S510 CD8 T cell epitope [38]. These CTL escape mutations allow for viral persistence and chronic demyelination. Emergence of CTL escape in MHV-JHM infected animals is strain dependent but MHC independent, as C57BL/6 mice allow CTL escape while Balb/c or Balb/b mice do not [50]. Subsequent studies indicate that this difference is due to increased endogenous anti-viral antibody production, specifically due to an increase in the amount of antibody secreting, or plasma cells, in the CNS of Balb mice. Despite the clear link between prevention of CTL escape and anti-viral antibody production in the CNS of MHV-JHM infected mice, important questions remain. One important facet of CTL escape is the selection of mutations in the immunodominant Db S510, but not the subdominant Kb S598 epitope. An explanation for this selection is likely found in the differences between functional avidity of the two epitopes, with the S510 epitope exhibiting higher functional avidity than the subdominant S598 epitope [26]. Current studies are aimed at examining these differences by alteration of epitope avidity in recombinant MHV-JHM.
A second model of demyelinating disease during MHV-JHM infection involves infection of adult mice with the attenuated MHV-J.2.2v-1 variant [45]. Although generally resistant to intranasal infection, C57BL/6 mice infected intracerebrally with J2.2v-1 develop mild acute encephalitis, followed by viral clearance mediated by an anti-viral T cell response. Demyelination with clinical signs of hindlimb weakness occurs during the process of virus clearance. As in other animal models of demyelinating disease, plaques of demyelination in J2.2v-1 infection are characterized by dense macrophage/microglial infiltration [15]. Due to their similar function and surface marker expression, specific contributions of macrophages and microglia to demyelination during MHV-JHM infection are not well understood. Chemical depletion of blood borne macrophages prior to infection does not affect disease, suggesting that microglia and/or perivascular macrophages are sufficient for demyelination to occur [51].
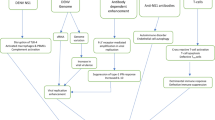
Induction of demyelinating disease in J2.2v-1 infected mice is usually T cell or antibody mediated, as mice lacking the recombinase activating gene 1 (RAG1) or severe combined immunodeficient (SCID) mice do not clear virus and ultimately succumb to encephalitis with little or no demyelination [20, 52, 53]. Infection of RAG1-/- or SCID mice, which completely lack T or B lymphocytes, provides a basis to determine the role of adaptive immune components in the development of demyelinating disease (Table 1). J2.2v-1 infected RAG1-/- mice that receive adoptively transferred splenocytes from immunocompetent MHV-JHM immunized mice regain the demyelinating phenotype [21]. Depletion of both CD4 and CD8 T cells from the donor splenocyte population abrogates demyelination, while depletion of a single population does not. However, demyelination mediated by each T cell population is characterized by a distinct disease profile. Infected RAG1-/- mice that receive CD4 T cell-enriched splenocytes develop severe acute encephalitis with moderate amounts of demyelination. Mice that receive CD8 T cell-enriched splenocytes exhibit less encephalitis and a prolonged disease course that is characterized by high levels of demyelination. Furthermore, CD8, but not CD4, mediated demyelination and associated macrophage infiltration into the spinal cord white matter is significantly reduced when donor mice lack the ability to produce IFN-γ [54, 55]. Perforin, an essential component of CTL cytolytic activity, is not required. How IFN-γ produced by CD8 T cells contributes to demyelination in MHV-JHM-infected mice is unclear. Potent activation of macrophages/microglia by IFN-γ likely plays a role in cell recruitment and demyelination. For example, IFN-γ treatment of macrophages results in increased production of nitric oxide and increased phagocytosis. Since IFN-γ is required for clearance of J2.2v-1 from oligodendroglia [56], it is possible that IFN-γ produced by CD8 T cells as part of the anti-viral response results in increased activation and recruitment of myelin-destroying macrophages/microglia. IFN-γ also plays a role in autoimmune destruction of myelin, by enhancing CD8 T cell-mediated EAE [57]. Interestingly, IFN-γ produced in the CNS in mice with EAE induces upregulation of CCL2 (MCP-1) [58], a macrophage recruiting chemokine that also promotes infiltration into the CNS during MHV-JHM infection [59]. Furthermore, in a CD8 T-cell mediated model of spontaneous demyelination utilizing transgenic mice that constitutively express the costimulatory ligand CD86 on microglial cells within the CNS, IFN-γ receptor deficient mice exhibited no disease [60]. These studies suggest that IFN-γ responsiveness by macrophages/microglia may be critical for CD8 T cell-mediated demyelination in MHV-J2.2v-1 infected mice.
Although transfer of CD4 or CD8 T cells induces demyelination in RAG1-/- mice, J2.2 infected athymic nude mice, also lacking CD4 or CD8 T cells, develop demyelination in the absence of adoptive cell transfer [19]. However, nude mice lack only conventional αβ T cells, but still retain functional γδ T cells. Depletion of γδ T cells from infected nude mice significantly reduces demyelination [61]. Demyelination in infected mice lacking the TCRβ gene, which also lack conventional αβ T cells, provides further proof that γδ T cells can mediate demyelination [62]. Furthermore, antibody depletion of IFN-γ in J2.2 infected TCRβ-/- mice significantly reduces demyelination, showing that IFN-γ is critical for demyelination induced by γδ T cells, as it is in CD8+ T cell-mediated myelin destruction. In addition to the requirement for IFN-γ, recognition of NKG2D is also important for γδ T cell mediated demyelination [62]. Antibody blockade of NKG2D, a classic activating receptor of NK cells also expressed on a subset of mouse γδ T cells, abrogates demyelination in TCRβ deficient mice. Therefore, γδ T cells are capable of mediating demyelination during viral infection through the action of two critical effector molecules. In contrast, the role of γδ T cells in EAE is still controversial. Depletion of γδ T cells in EAE results in either milder or more severe disease, depending on whether cells are depleted early or late in the inflammatory process [63–67].
In addition to cell-mediated demyelination, anti-MHV antibody is also capable of induction of disease. MHV-J2.2v-1 infected RAG1-/- mice treated with anti-MHV antibodies develop demyelinating disease with associated white matter infiltration of macrophages/microglia [22]. Although the precise role of humoral immunity in MS is not completely understood, B cells and antibody production are believed to be involved in myelin destruction [68–70]. Oligoclonally expanded B cells and high levels of immunoglobulin are detected in the cerebrospinal fluid of MS patients [71], some of which are directed against myelin components, or against common viruses such as Epstein–Barr [72] or Varicella-Zoster [73]. Antibody mediated demyelination during MHV-J2.2v-1 infection requires the activating Fcγ receptors I and III, since anti-MHV antibody treated infected RAG1-/- mice deficient in these receptors fail to develop disease [22]. Furthermore, the complement pathway may play a role in antibody-mediated demyelination, as depletion of complement by treatment of mice with cobra venom factor (CVF) results in a significant decrease in demyelination. FcγR and complement may function together to activate macrophages with subsequent demyelination. Adoptively transferred antibody is detectable in the CNS of infected RAG1-/- mice, supporting the idea of direct interaction with activating Fcγ receptors on macrophages. In the EAE model of demyelination, mice lacking the macrophage activating FcγRI/III exhibit attenuated disease, while mice lacking the inhibitory FcγRII display increased disease [74]. Furthermore, C3 derived complement products play a critical role in the pathogenesis of EAE [75]. In addition to liver production of serum components of complement, many resident CNS cells are capable of producing complement proteins. Through the classical pathway, these proteins may be activated on the surface of antibody bound MHV infected cells or in MHV/antibody immune complexes, thus enhancing the recruitment and effector functions of CNS macrophages/microglia. Of note, RAG1-/- mice also deficient in C3, the central component of the complement pathway, develop antibody-mediated demyelination (Templeton and Perlman, unpublished). Contrasting results between C3 deficiency and CVF depletion of complement are not surprising, since CVF exhibits considerable toxicity [76]. Furthermore, recent evidence indicates that C5a may be produced independently of C3, providing a possibility that complement products downstream of C3 may play a role in demyelination in MHV-JHM infected C3-/-RAG1-/- mice [77]. Current studies are focused on examination of the role of complement in CNS disease and in induction of immune responses to MHV-JHM infection in both immunocompetent and immunodeficient mice.
Inflammatory chemokines such as the macrophage chemoattractant protein CCL2 also play a role in demyelinating disease. Expression of CCL2 during EAE follows T cell entry into the CNS [78], and may be induced by T cell-associated increases in IFN-γ [58]. CCL2 and the CCL2 receptor CCR2 are both important for recruitment of immune cells and in increased pathogenesis in EAE [79]. CCL2/CCR2 also promote monocyte recruitment and viral clearance during MHV-JHM infection [59, 80]. Introduction of the mouse CCL2 gene into a recombinant J2.2 (rJ2.2.CCL2) virus results in an infectious virus capable of producing secreted CCL2 in infected cells in vitro [12]. RAG1-/- mice infected with rJ2.2.CCL2 develop demyelinating disease, whereas RAG1-/- mice infected with a control virus that lacks a functional CCL2 protein (rJ2.2.ΔCCL2) do not. Therefore, T cells or anti-MHV antibody are not required for demyelination in MHV-JHM-infected mice. Rather, it is likely that factors induced by adaptive immune responses result in the recruitment and/or activation of macrophages/microglia into white matter areas of the CNS. Therefore, these cells are the final effectors in the demyelinating process and any intervention that induces their migration and activation will likely result in demyelination. Further studies involve introduction of other chemoattractants into recombinant MHV-JHM, to evaluate their role in demyelinating disease, cell recruitment, generation of immune responses, and clearance of infectious virus in MHV-JHM-infected mice.
Summary/conclusions
Studies of MHV-JHM infection of mice have provided insights into the relationship of virus phenotype to CNS neurovirulence and anti-viral immune responses. The differences in infection with variants of MHV-JHM allow for examination of pathogenic mechanisms associated with both acute and chronic CNS disease. Recombinant virus technology has been employed to study both loss and gain of function in MHV-JHM structure, as well as the effects of host immune factors on anti-viral immune responses. Using these techniques, the S gene has been identified as a primary determinant of neurovirulence in MHV-JHM infection [9, 13, 25, 79]. However, the role that virus phenotype and neurovirulence plays in the hosts’ ability to generate anti-viral immune responses is less clear. Some MHV-JHM variants, like MHV.IA or MHV-J2.2v-1. elicit robust anti-viral CD8 T cell responses in infected mice, while, in contrast, the highly neurovirulent MHV.SD does not [23, 24, 28]. Mice which survive acute MHV-JHM infection develop demyelinating disease with associated macrophage/microglial infiltration [5]. Many of the factors that mediate demyelination are capable of activating and/or recruiting macrophages (Table 1), suggesting a pathogenic role for macrophage recruitment via adaptive immune responses. Future studies will aim to further understand how changes in MHV-JHM gene expression affect the pathogenesis of acute infection and demyelinating disease in the CNS of infected mice.
References
Bergmann CC, Lane TE, Stohlman SA. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol 2006;4:121–32.
Griffin DE. Immune responses to RNA-virus infections of the CNS. Nat Rev Immunol 2003;3:493–502.
Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia 2001;36:118–24.
Perlman S, Dandekar AA. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol 2005;5:917–27.
Stohlman SA, Bergmann CC, Perlman S. Mouse hepatitis virus. In: Ahmed R, Chen I, editors. Persistent viral infections. John Wiley & Sons, Ltd.; 1998. p. 537–557.
Fischer F, Stegen C, Koetzner CA, Masters PS. Analysis of a recombinant mouse hepatitis virus expressing a foreign gene reveals a novel aspect of coronavirus transcription. J Virol 1997;71:5148–60.
Kuo L, Godeke GJ, Raamsman MJ, Masters PS, Rottier PJ. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J Virol 2000;74:1393–406.
Masters PS. Reverse genetics of the largest RNA viruses. Adv Virus Res 1999;53:245–64.
Phillips JJ, Chua MM, Lavi E, Weiss SR. Pathogenesis of chimeric MHV4/MHV-A59 recombinant viruses: the murine coronavirus spike protein is a major determinant of neurovirulence. J Virol 1999;73:7752–60.
Ontiveros E, Kuo L, Masters PS, Perlman S. Inactivation of expression of gene 4 of mouse hepatitis virus strain JHM does not affect virulence in the murine CNS. Virology 2001;290:230–8.
Yokomori K, Lai MMC. Mouse hepatitis virus S RNA sequence reveals that nonstructural protein ns4 and ns5a are not essential for murine coronavirus replication. J Virol 1991;65:5605–8.
Kim TS, Perlman S. Viral expression of CCL2 is sufficient to induce demyelination in RAG1-/- mice infected with a neurotropic coronavirus. J Virol 2005;79:7113–20.
Ontiveros E, Kim TS, Gallagher TM, Perlman S. Enhanced virulence mediated by the murine coronavirus, mouse hepatitis virus Strain JHM, Is associated with a glycine at residue 310 of the spike glycoprotein. J Virol 2003;77:10260–9.
Pewe L, Zhou H, Netland J, Tangadu C, Olivares H, Shi L, et al. A severe acute respiratory syndrome-associated coronavirus-specific protein enhances virulence of an attenuated murine coronavirus. J Virol 2005;79:11335–42.
Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 2002;3:291–301.
Sospedra M, Martin R. Immunology of multiple sclerosis. Ann Rev Immunol 2005;23:683–747.
Buchmeier MJ, Lewicki HA, Talbot PJ, Knobler RL. Murine hepatitis virus-4 (strain JHM)-induced neurologic disease is modulated in vivo by monoclonal antibody. Virology 1984;132:261–70.
Perlman S, Schelper R, Bolger E, Ries D. Late onset, symptomatic, demyelinating encephalomyelitis in mice infected with MHV-JHM in the presence of maternal antibody. Microb Pathog 1987;2:185–94.
Houtman JJ, Fleming JO. Dissociation of demyelination and viral clearance in congenitally immunodeficient mice infected with murine coronavirus JHM. J Neurovirol 1996;2:101–10.
Wang F, Stohlman SA, Fleming JO. Demyelination induced by murine hepatitis virus JHM strain (MHV-4) is immunologically mediated. J Neuroimmunol 1990;30:31–41.
Wu GF, Dandekar AA, Pewe L, Perlman S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J Immunol 2000;165:2278–86.
Kim TS, Perlman S. Virus-specific antibody, in the absence of T cells, mediates demyelination in mice infected with a neurotropic coronavirus. Am J Pathol 2005;166:801–9.
Iacono KT, Kazi L, Weiss SR. Both spike and background genes contribute to murine coronavirus neurovirulence. J Virol 2006;80:6834–43.
Rempel JD, Murray SJ, Meisner J, Buchmeier MJ. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology 2004;318:381–92.
Rempel JD, Murray SJ, Meisner J, Buchmeier MJ. Mouse hepatitis virus neurovirulence: evidence of a linkage between S glycoprotein expression and immunopathology. Virology 2004;318:45–54.
Castro RF, Perlman S. CD8+ T cell epitopes within the surface glycoprotein of a neurotropic coronavirus and correlation with pathogenicity. J Virol 1995;69:8127–31.
Xue S, Jaszewski A, Perlman S. Identification of a CD4+ T cell epitope within the M protein of a neurotropic coronavirus. Virology 1995;208:173–9.
Pewe L, Heard SB, Bergmann CC, Dailey MO, Perlman S. Selection of CTL escape mutants in mice infected with a neurotropic coronavirus: Quantitative estimate of TCR diversity in the infected CNS. J Immunol 1999;163:6106–13.
Touil T, Fitzgerald D, Zhang GX, Rostami A, Gran B. Cutting Edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J Immunol 2006;177:7505–9.
Teige I, Liu Y, Issazadeh-Navikas S. IFN-beta inhibits T cell activation capacity of central nervous system APCs. J Immunol 2006;177:3542–53.
Dikopoulos N, Bertoletti A, Kroger A, Hauser H, Schirmbeck R, Reimann J. Type I IFN negatively regulates CD8+ T cell responses through IL-10-producing CD4+ T regulatory 1 cells. J Immunol 2005;174:99–109.
Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med 2006;203:2461–72.
Liu XS, Xu Y, Hardy L, Khammanivong V, Zhao W, Fernando GJ, et al. IL-10 mediates suppression of the CD8 T cell IFN-gamma response to a novel viral epitope in a primed host. J Immunol 2003;171:4765–72.
Rouse BT, Sarangi PP, Suvas S. Regulatory T cells in virus infections. Immunol Rev 2006;212:272–86.
Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, et al. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U S A 2006;103:7835–40.
Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med 2006;12:518–25.
Ploegh HL. Viral strategies of immune evasion. Science 1998;280:248–53.
Pewe L, Wu G, Barnett EM, Castro R, Perlman S. Cytotoxic T cell-resistant variants are selected in a virus-induced demyelinating disease. Immunity 1996;5:253–62.
Hudrisier D, Oldstone MB, Gairin JE. The signal sequence of lymphocytic choriomeningitis virus contains an immunodominant cytotoxic T cell epitope that is restricted by both H-2D(b) and H-2K(b) molecules. Virology 1997;234:62–73.
Kim TS, Perlman S. Protection against CTL escape and clinical disease in a murine model of virus persistence. J Immunol 2003;171:2006–13.
Anghelina D, Pewe L, Perlman S. Pathogenic role for virus-specific CD4 T cells in mice with coronavirus-induced acute encephalitis. Am J Pathol 2006;1:209–22.
Liu T, Chambers TJ. Yellow fever virus encephalitis: properties of the brain-associated T-cell response during virus clearance in normal and gamma interferon-deficient mice and requirement for CD4+ lymphocytes. J Virol 2001;75:2107–18.
Murali-Krishna K, Ravi V, Manjunath R. Protection of adult but not newborn mice against lethal intracerebral challenge with Japanese encephalitis virus by adoptively transferred virus-specific cytotoxic T lymphocytes: requirement for L3T4+ T cells. J Gen Virol 1996;77(Pt 4):705–14.
Weidinger G, Henning G, ter Meulen V, Niewiesk S. Inhibition of major histocompatibility complex class II-dependent antigen presentation by neutralization of gamma interferon leads to breakdown of resistance against measles virus-induced encephalitis. J Virol 2001;75:3059–65.
Fleming JO, Trousdale MD, Bradbury J, Stohlman SA, Weiner LP. Experimental demyelination induced by coronavirus JHM (MHV-4): molecular identification of a viral determinant of paralytic disease. Microb Pathog 1987;3:9–20.
Fleming JO, Trousdale MD, El-Zaatari F, Stohlman SA, Weiner LP. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J Virol 1986;58:869–75.
Fleming JO, Adami C, Pooley J, Glomb J, Stecker E, Fazal F, et al. Mutations associated with viral sequences isolated from mice persistently infected with MHV-JHM. Adv Exp Med Biol 1995;380:591–5.
Fazakerley JK, Buchmeier MJ. Pathogenesis of virus-induced demyelination. Adv Virus Res 1993;42:249–324.
McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med 2005;11:335–9.
Dandekar AA, Jacobsen G, Waldschmidt TJ, Perlman S. Antibody-mediated protection against cytotoxic T-cell escape in coronavirus-induced demyelination. J Virol 2003;77:11867–74.
Xue S, Sun N, van Rooijen N, Perlman S. Depletion of blood-borne macrophages does not reduce demyelination in mice infected with a neurotropic coronavirus. J Virol 1999;73:6327–34.
Houtman JJ, Fleming JO. Pathogenesis of mouse hepatitis virus-induced demyelination. J Neurovirol 1996;2:361–76.
Wu GF, Perlman S. Macrophage infiltration, but not apoptosis, is correlated with immune-mediated demyelination following murine infection with a neurotropic coronavirus. J Virol 1999;73:8771–80.
Pewe L, Haring J, Perlman S. CD4 T-cell-mediated demyelination is increased in the absence of gamma interferon in mice infected with mouse hepatitis virus. J Virol 2002;76:7329–33.
Pewe LL, Perlman S. CD8 T cell-mediated demyelination is IFN-γ dependent in mice infected with a neurotropic coronavirus. J Immunol 2002;168:1547–51.
Parra B, Hinton D, Marten N, Bergmann C, Lin MT, Yang CS, Stohlman SA. IFN-γ(is required for viral clearance from central nervous system oligodendroglia. J Immunol 1999;162:1641–47.
Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med 2001;194:669–76.
Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol 2000;164:2759–68.
Held KS, Chen BP, Kuziel WA, Rollins BJ, Lane TE. Differential roles of CCL2 and CCR2 in host defense to coronavirus infection. Virology 2004;329:251–60.
Brisebois M, Zehntner SP, Estrada J, Owens T, Fournier S. A pathogenic role for CD8+ T cells in a spontaneous model of demyelinating disease. J Immunol 2006;177:2403–11.
Dandekar AA, Perlman S. Virus-induced demyelination in nude mice is mediated by gamma delta T cells. Am J Pathol 2002;161:1255–63.
Dandekar AA, O’Malley K, Perlman S. Important roles for gamma interferon and NKG2D in γδ T cell-induced demyelination in TCRß-deficient mice infected with a coronavirus. J Virol 2005;79:9388–96.
Kobayashi Y, Kawai K, Ito K, Honda H, Sobue G, Yoshikai Y. Aggravation of murine experimental allergic encephalomyelitis by administration of T-cell receptor gammadelta-specific antibody. J Neuroimmunol 1997;73:169–74.
Odyniec A, Szczepanik M, Mycko MP, Stasiolek M, Raine CS, Selmaj KW. Gammadelta T cells enhance the expression of experimental autoimmune encephalomyelitis by promoting antigen presentation and IL-12 production. J Immunol 2004;173:682–94.
Ponomarev ED, Novikova M, Yassai M, Szczepanik M, Gorski J, Dittel BN. Gamma delta T cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J Immunol 2004;173:1587–95.
Rajan AJ, Gao YL, Raine CS, Brosnan CF. A pathogenic role for γδ T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J Immunol 1996;157:941–9.
Spahn TW, Issazadah S, Salvin AJ, Weiner HL. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33–35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR δ chain gene. Eur J Immunol 1999;29:4060–71.
Archelos JJ, Storch MK, Hartung HP. The role of B cells and autoantibodies in multiple sclerosis. Ann Neurol 2000;47:694–706.
Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–17.
Wingerchuk DM, Lucchinetti CF, Noseworthy JH. Multiple sclerosis: current pathophysiological concepts. Lab Invest 2001;81:263–81.
Owens GP, Ritchie AM, Burgoon MP, Williamson RA, Corboy JR, Gilden DH. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol 2003;171:2725–33.
Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Multiple sclerosis and Epstein-Barr virus. JAMA 2003;289:1533–6.
Burgoon MP, Hammack BN, Owens GP, Maybach AL, Eikelenboom MJ, Gilden DH. Oligoclonal immunoglobulins in cerebrospinal fluid during varicella zoster virus (VZV) vasculopathy are directed against VZV. Ann Neurol 2003;54:459–63.
Abdul-Majid KB, Stefferl A, Bourquin C, Lassmann H, Linington C, Olsson T, et al. Fc receptors are critical for autoimmune inflammatory damage to the central nervous system in experimental autoimmune encephalomyelitis. Scand J Immunol 2002;55:70–81.
Barnum SR, Szalai AJ. Complement and demyelinating disease: no MAC needed? Brain research 2006;52:58–68.
Nataf S, Carroll SL, Wetsel RA, Szalai AJ, Barnum SR. Attenuation of experimental autoimmune demyelination in complement-deficient mice. J Immunol 2000;165:5867–73.
Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 2006;12:682–7.
Glabinski AR, Tani M, Tuohy VK, Tuthill RJ, Ransohoff RM. Central nervous system chemokine mRNA accumulation follows initial leukocyte entry at the onset of acute murine experimental autoimmune encephalomyelitis. Brain Behav Immun 1995;9:315–30.
Izikson L, Klein RS, Luster AD, Weiner HL. Targeting monocyte recruitment in CNS autoimmune disease. Clin Immunol 2002;103:125–31.
Chen BP, Kuziel WA, Lane TE. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol 2001;167:4585–92.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Templeton, S.P., Perlman, S. Pathogenesis of acute and chronic central nervous system infection with variants of mouse hepatitis virus, strain JHM. Immunol Res 39, 160–172 (2007). https://doi.org/10.1007/s12026-007-0079-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-007-0079-y