
Abstract
Insulin-producing pancreatic neuroendocrine tumors (PanNETs)/insulinomas are generally considered to be indolent tumors with an excellent prognosis after complete resection. However, some insulinomas have a poor prognosis due to relapses and metastatic disease. Recently, studies in non-functional PanNETs indicated that behavior can be stratified according to alpha- and beta-cell differentiation, as defined by expression of the transcription factors ARX and PDX1, respectively. It is unknown whether similar mechanisms play a role in insulinomas. Therefore, we determined ARX and PDX1 expression in a cohort of 35 sporadic primary insulinomas and two liver metastases of inoperable primary insulinomas. In addition, WHO grade and loss of ATRX or DAXX were determined by immunohistochemistry, and alternative lengthening of telomeres (ALT) and CDKN2A status by fluorescence in situ hybridization. These findings were correlated with tumor characteristics and clinical follow-up data. In total, five out of 37 insulinoma patients developed metastatic disease. Metastatic insulinomas were all larger than 3 cm, whereas the indolent insulinomas were smaller (p value < 0.05). All three primary insulinomas that metastasized showed ARX expression, 2/3 showed ALT, and 1/3 had a homozygous deletion of CDKN2A as opposed to absence of ARX expression, ALT, or CDKN2A deletions in the 32 non-metastatic cases. The two liver metastases also showed ARX expression and ALT (2/2). The presence of ARX expression, which is usually absent in beta-cells, and genetic alterations not seen in indolent insulinomas strongly suggest a distinct tumorigenic mechanism in malignant insulinomas, with similarities to non-functional PanNETs. These observations may inform future follow-up strategies after insulinoma surgery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Insulinomas are the most common functional pancreatic neuroendocrine tumor (PanNET) type and are diagnosed by the triad of hypoglycemic symptoms, low blood glucose concentrations, and relief of symptoms after glucose administration. Most insulinomas are indolent tumors: there are few mitoses (low grade) and metastases are very rare [1, 2]. In contrast, 40–50% of non-functional PanNETs present with liver metastases at time of initial diagnosis [3, 4]. Surgery for insulinomas is primarily indicated to alleviate symptoms of hypoglycemia. Survival after surgery is not different from the general population [5, 6]. There are no insulinoma-specific international recommendations for follow-up after surgery [5, 7]. Nevertheless, about 10% of insulinoma patients develop metastases that are mostly present at time of initial diagnosis, but may sometimes develop years after resection of the primary tumor [8]. Median survival is less than 2 years in patients with metastatic insulinoma, similar to metastatic non-functional PanNET [9,10,11].
Because of the rarity of metastatic insulinomas, little is known about the mechanisms of tumorigenesis. Recent sequencing studies identified recurrent YY1 gene mutations in insulinomas; however, relapses or metastases were rarely reported in the studied cohorts [12,13,14,15]. There is a need to increase our understanding of insulinoma development, in particular of metastatic insulinomas. This will improve identification of patients at risk for recurrence, who may benefit from follow-up after surgery and thereby earlier detection and treatment of metastases.
Metastatic non-functional PanNETs are more common and have been characterized more extensively. Next-generation sequencing studies have demonstrated that sporadic PanNETs harbor relatively few gene mutations. The genes that are most frequently mutated are MEN1, ATRX, and DAXX [12, 16, 17]. Mutually exclusive mutations in ATRX or DAXX—coding for two chromatin-modifying proteins which form a histone chaperone complex—are associated with the alternative lengthening of telomere phenotype (ALT) [18]. Immunohistochemical loss of ATRX and DAXX can be used as a surrogate marker of inactivating mutations of ATRX or DAXX, respectively [18, 19]. The presence of either of these alterations is associated with recurrence and liver metastases [20,21,22,23,24,25]. In addition to ATRX or DAXX mutations and ALT, loss of ARID1A, loss of H3K36 trimethylation (H3K36me3) by SETD2 dysfunction, and CDKN2A deletions were also reported to be of prognostic value for non-functional PanNET [26]. A recent finding in non-functional PanNETs is that alpha- or beta-cell types-of-origin may predict clinical behavior [20, 27]. Beta-cell like non-functional PanNETs, marked by expression of the endocrine transcription factor PDX1, were generally indolent, while almost all relapses were observed in the group of alpha-cell like non-functional PanNETs marked by ARX expression. Furthermore, somatic mutations in ATRX, DAXX, and MEN1 and acquisition of ALT were observed more often in the alpha-type non-functional PanNETs [20, 27].
Whether the ARX and PDX1 transcription factors play a role in the clinical behavior of insulinomas is not known. We determined protein expression of ARX and PDX1 together with ATRX, DAXX, ARID1A, and H3K36me3 by immunohistochemistry, as well as ALT and CDKN2A deletions by fluorescence in situ hybridization (FISH), in a cohort of clinically defined sporadic insulinomas.
Materials and Methods
Study Cohort
The study was approved by the University Medical Center (UMC) Utrecht Biobank Research Ethics Committee. Tissue-microarrays (TMAs) were constructed of primary sporadic insulinomas resected between 1991–2017 and 1997–2017 for the UMC Utrecht and Amsterdam UMC, respectively. Inherited cases (e.g., MEN1 syndrome) were excluded. Neuroendocrine tumor diagnosis was confirmed by an experienced gastrointestinal pathologist (LAAB). Three 0.6-mm cores per tumor were randomly taken from annotated tumor areas in formalin-fixed paraffin-embedded (FFPE) blocks. In case of multiple tumors, the largest tumor was used. Biopsies of insulinoma liver metastases were identified by a search in the UMC Utrecht pathology archive.
Information on age, sex, multifocality, surgery type, surgery date, tumor size, location, grade, resection margin, and lymph nodes was collected from pathology reports. If possible, macroscopic tumor size was used. Free margins were interpreted as R0, also if the distance was less than 1 mm from the resection margin. Medical records were reviewed to collect information on the functional status of the tumor, presence of genetic syndromes, and follow-up. Events of tumor relapse (local recurrence, liver metastases, or other metastases) were either histologically proven or diagnosed by the treating clinician. The first radiological evidence of proven relapse was used as event time point. Follow-up time is counted from date of surgery until described events, death, or was censored at the last visit to a relevant hospital clinician (surgery, internal medicine, endocrinology, gastroenterology, or oncology) or most recent clinic visit. For overall survival, any cause of death and the most recent clinic visit were used. Relapse was defined as any distant metastasis (liver or other location) or local recurrence. Relapse-free, distant-free, and liver metastases-free survival were censored at last visit to a relevant hospital clinician.
Immunohistochemistry
Four-micrometer sections of FFPE tissue were cleared at 60 °C and deparaffinized in xylene. Endogenous peroxidase was blocked by immersion in 0.6% H2O2 (7210, Merck, Kenilworth, USA) in methanol for 15 min. Antigen retrieval was performed by cooking slides in a 10 mM citrate (pH 6) or 10/1 mM Tris/EDTA (pH 9) solution for 20 min. Nonspecific binding was reduced by with Protein Block Serum Free (X0909, Dako, Santa Clara, United States of America). Antibodies were diluted in normal antibody diluent (Immunologic, Duiven, The Netherlands) and applied on the slides (Table 1). After incubation of post antibody blocking solution for 15 min (Immunologic), the secondary antibody Poly-HRP-goat anti Mouse/Rabbit IgG (cat. no. VWRKDPVB110HRP, Immunologic) was incubated for 30 min. Peroxidase activity was detected by DAB (D5637, Sigma, St. Louis, USA) or Bright-DAB (cat. no. VWRKBS04–110, Immunologic) as chromogen for 8 min. After all incubation steps, except the protein block, slides were washed with PBS-Tween-20 0.1% four times. Slides were counterstained with hematoxylin and mounted with Pertex (Histolab, Askim, Sweden).
Scoring was performed by at least two independent researchers (WMH, WS, LAAB), blinded for each other’s results and clinical information. Disagreements were resolved by consensus.
For ARX and PDX1, negative protein expression in tumor tissue was defined as weak nuclear staining in < 50% of cells or strong nuclear staining in < 10% of cells. Positive expression was defined as weak nuclear staining in > 50% of cells or intermediate/strong nuclear staining > 10% of cells [27]. For insulin and glucagon, cytoplasmic staining of > 10% of cells was considered positive expression for the respective peptide hormone. Normal islets, containing a mix of cells expressing or not expressing the respective peptide hormone, were used as positive and negative controls, respectively. If < 10% of cells had expression, cases were called scattered. DAXX, ATRX, and ARID1A were considered negative if < 5% of cells had positive nuclear staining and if there was non-tumoral tissue present with positive nuclear staining serving as internal control, e.g., islets of Langerhans, stromal cells, endothelial cells, or lymphocytes [26, 28]. All negative cases in the TMA were also stained on whole sections to confirm the results. For H3K36me3 loss, a cut-off of 30% of cells was used [26, 29]. Negative cases without a positive internal control were non-informative. All cytoplasmic staining was ignored.
Ki67 labeling index (LI) was counted in at least 2000 cells by digital image analysis with Sectra (PACS, Sectra AB, Linköping, Sweden), as previously described [30]. Digital counts were confirmed by visual assessment. PanNETs were graded by the 2017 WHO criteria (Ki67 G1 < 3%, G2 3 to 20%, G3 > 20%) [31]. If the pathology report also mentioned tumor grade based on Ki67 or mitoses per 10 HPF, the highest grade was used for further analysis as the location of tumor cores not always represents the most proliferative region.
Fluorescence In Situ Hybridization
After deparaffinization in xylene, 4 μm FFPE sections for CDKN2A/CEN9 FISH were pre-treated in 0.2 N HCL for 20 min, cooked in a 10 mM citrate buffer (pH 6) for 20 min and washed in PBS. Slides were then digested in proteinase K buffer for 10 min at 37 °C (5 μM Tris–HCL, 1μM EDTA, 1 μM NaCl, 10 mg/L Proteinase K), washed with PBS and dried. Ten microliters of CDKN2A/CEN9 probe mix (CDKN2A/CEN 9 Dual Color probe, Zytolight, Bremerhaven, Germany) was applied per slide. Slides were denatured at 78 °C for 5 min and cooled on ice for 5 min. Hybridization was performed in a ThermoBrite (Abbott Laboratories, Chicago, IL) at 37 °C overnight. After removing coverslips, slides were washed in washing buffer (WB) 1 (0.4× SCC, 0.5% NP-40, 73 °C), WB 2 (2× SCC, 0.1% NP-40, room temperature), WB 3 (2× SCC, room temperature), and PBS, for 2, 1, 5 min, and 20 s respectively. Nuclei were counterstained and mounted with Vectashield with DAPI (H-1200, Vector laboratories, Amsterdam, The Netherlands).
Slides for telomere/centromere FISH were cooked in 10 mM citrate buffer (pH 6) for 20 min, washed in dH2O and dried. Probes (TelC-Cy3 F1002 PNA 180723PL-01, Cent-FITC, F3013 172865, Panagene, Daejeon, Republic of Korea) were diluted in hybridization mix (50% deionized Formamide, 50% SCC 4×, 5% Dextran sulphate, Tween-20 0.5%) at a 400 nM concentration and applied on the slides. After 5 min denaturation at 84 °C, slides were cooled on ice for 5 min before hybridizing at 37 °C overnight. After removing coverslips, slides were washed in two cycles of 1× WB (70% Formamide, 30% dH2O, 10 mM Tris, 15 min) and 3× PBS (2 min each time). Nuclei were counterstained with DAPI in PBS 2 μg/ml (Sigma-Aldrich, D9542), and coverslips were mounted with Vectashield (H-1000, Vector laboratories).
Slides were stored at 4 °C before imaging, and viewed with a Leica DM5500 B using appropriate excitation and emission filters. Images were made at 100× magnification with a Z stack of 14 steps in Leica application Suite X (Leica Microsystems, Rijswijk, The Netherlands).
The number of CDKN2A gene probe and centromere probe signals were counted in at least 50 tumor cells for each case (WMH, WS). At least nine photographs of tumor tissue (confirmed on H&E) were made for counting of cells. Only intact non-overlapping nuclei with at least one centromere probe were counted. Multiple signals separated ≤ 1 signal distance were counted as one. If no signals were observed (gene and centromere) in the tumor and surrounding stromal cells or if there was too much background, cases were called non-informative. Homozygous loss was defined as at least 20% of counted cells lacking CDKN2A probe signals with at least one CEP9 probe. Hemizygous deletion of CDKN2A was concluded if 45% of counted cells had one CDKN2A probe and two CEP9 probes; monosomy of chromosome 9 (which can be considered hemizygous loss) if 15% of cells had one CDKN2A probe and one CEP9 probe. Cut-off values were based on previous literature [26, 32]. If results were discordant, additional photos were made and at least 100 cells were counted (WMH).
ALT positivity was defined as ultra-bright, intra-nuclear telomere FISH signals, 10× the signal intensity of cumulative single telomere sum intensities in normal stromal/endothelial cells, which are present in more than 1% of cells [18, 33]. The percentage of ALT cells > 1% was determined on × 20 magnification by visual assessment (WMH) in areas of tumor tissue (annotated in H&E slide), with a cut-off of more than 20 ultra-bright foci per tumor core (max 2000 cells). If less than 1% by visual assessment, all ultra-bright foci were counted. All ultra-bright foci were confirmed at × 100 magnification. Using the same laser/microscope settings, representative ultra-bright foci near stromal cells were photographed and digital grayscale TIFF images exported for each fluorophore channel. Telomere signals were quantified using Telometer (a free custom software ImageJ plug-in, downloaded from demarzolab.pathology.jhmi.edu/telometer). Rolling ball radius was set at the maximal telomeric signal diameter, which was measured per photograph. Signals were separated by the draw function if necessary. Centromere signals were used as positive hybridization efficiency control and to confirm specific probe binding.
Statistics
Data was managed and statistical tests were performed in SPSS version 25 (IBM Nederland, Amsterdam, The Netherlands). The Kaplan Meier method was used to plot the effect of variables on events occurring over time, and significance was assessed by the log-rank test. The Χ2 or Fishers exact test was used when comparing categorical data between groups. Follow-up time was calculated using a reverse Kaplan Meier for relapse-free survival and observation time was calculated from date of surgery until last visit to a relevant hospital clinician, irrespective of events. P values < 0.05 were considered significant. Data was visualized in R version 3.5.1 using packages Beeswarm and Survplot.
Results
The cohort consisted of 35 primary insulinomas. The clinical characteristics of the patients are listed in Table 2. All cases were confirmed to be clinically functioning insulinomas by the treating hospital clinician. Three patients developed liver metastases during follow-up, one of whom also had local recurrence and tumor depositions around the uterine appendages. While all metastastic primary cases initially presented with symptomatic hypoglycemia, only two out of three cases had hypoglycemic events at relapse by liver metastasis (Table 3). Overall survival could not be analyzed as there were no deaths during follow-up. The metastatic primary insulinomas were larger than the indolent primary insulinomas, but this did not reach statistical significance (mean 32 indolent insulinomas 1.48 cm, SD 0.45, mean 3 metastatic insulinomas 7.20 cm, SD 3.12, t test p = 0.086; Fig. 1). In addition, biopsies of liver metastases of two patients with inoperable primary insulinoma were available (Table 3), as well as a biopsy of a corresponding liver metastasis of one of the primary insulinomas in the cohort (patient 2, Table 3). When the sizes of the primary tumors of the inoperable insulinoma patients were included, the five metastatic insulinomas were significantly larger than the indolent cases (mean 1.48 cm, SD 0.45, metastatic mean 6.16 cm, SD 2.63, t test P = 0.016). A size cut-off of 3 cm perfectly separated all metastatic from indolent insulinomas.

ARX expression, tumor size and liver metastases-free survival in primary insulinomas. a–c Positive ARX expression in the three metastatic primary insulinomas (patient 1, 2, 3) and no ARX expression in a non-metastatic insulinoma (d). Photos of immunohistochemistry, white-balanced, with 50-μm scale bar. e Tumor size plot. Cases with liver metastases in the size plot are circled (green), WHO grade is given. f Kaplan Meier of liver metastases-free survival for ARX expression in the primary insulinoma cohort. P value was calculated with the log-rank test
Positive or scattered insulin expression was observed in all insulinomas. Positive or scattered glucagon expression was seen in about 60% of insulinomas. PDX1 expression was observed in 34/35 primary insulinomas (97%), and 2/3 liver metastases; in contrast, only 3/35 (9%) of primary insulinomas were ARX positive, while all liver metastases were ARX positive (Fig. 1, Tables 2 and 3). All ARX positive primary insulinomas developed metastases during follow-up. The ARX-positive percentage of cells varied between 10 and 90% of cells (Fig. 1, Table 3), and PDX1 expression was intermediate to strong in 70–100% of cells for all positive cases. Two (of 3) metastasizing primary insulinomas were multifocal (two tumors per patient). To exclude the possibility of the smaller tumor being the insulinoma and the larger tumor (tested in the TMA) possibly being an ARX-positive non-functional PanNET, whole slides of all multifocal tumors were stained for ARX, PDX1, and insulin. ARX, PDX1, and insulin expression was identical between tumors of the same patient. Thus, strong ARX expression in more than 10% of cells identified metastatic insulinomas with a 100% sensitivity and specificity. Of note, one of the metastatic insulinomas showed areas with < 10% ARX positive cells with scattered positive cells when assessing the whole slide (Patient 1, Fig. 1c). Although no obvious heterogeneity was seen between different tumor cores when scoring for ARX and PDX1, it is conceivable that the number of ARX-positive cells was underestimated using TMA cores for other cases. We therefore sought to further confirm our results by reviewing all ARX-negative insulinomas by digital image analyses on the TMA similar to the Ki67 count [30]. In all these cases, expression was observed in far less than 10% of cells, most often absent or in less than 1% of cells.
Of 33 insulinomas interpretable by telomere FISH, two cases (6%) had ALT which both developed liver metastases during follow up (Fig. 1; Table 3). All tested liver metastasis biopsies had ALT. All insulinomas (n = 34) with tumor tissue present in the TMAs had retained ATRX expression, only one case had heterogenic DAXX loss (also ALT positive). Insulin, glucagon, ARID1A, H3K36me3 IHC, and CDKN2A FISH were only tested on 31 cases, as for the last four insulinoma cases, no unstained TMA slides were available. All cases (n = 30) with tumor tissue present in the TMA had retained H3K36me3 and ARID1A expression. One case with homozygous CDKN2A deletion and one case with monosomal CDKN2A were observed (of 21 interpretable cases; Table 3). The case with homozygous CDKN2A loss developed liver metastases. The metastatic primary insulinomas were grade 1 (2/3) or grade 2 (1/3), and both metastasis biopsies were grade 2 (Ki67 labeling index, Table 3).
Discussion
All metastatic insulinomas in this cohort were larger than 3 cm. Strikingly, all metastatic insulinoma lesions showed ARX expression, which was not observed in any of the indolent primary insulinomas. Four out of five (80%) metastatic insulinomas had ALT—not reported before in insulinomas—while none of the indolent insulinomas showed ALT. Interestingly, the two recent studies that identified ARX as marker for PanNET relapse after surgery, also included non-metastatic insulinomas: Cejas et al. found no ARX immunohistochemical expression in 17 primary insulinomas [27] and Chan et al. reported ARX mRNA expression (alpha signature) in one out of three insulinomas. None of the insulinomas in these studies metastasized, but remarkably, the ARX-expressing case in the study of Chan et al. was also ATRX mutated and very large (8 cm), while all the negative cases were small (< 2cm) and ATRX, DAXX, MEN1 wild type [20]. These results are in line with our observation that there is an association between size, ALT, and ARX expression in insulinomas. To our knowledge, ALT has not been described before in any insulinoma. Retention of ATRX/DAXX protein expression in one of the insulinomas with ALT may be explained by non-truncating mutations, translocations, or other underlying mutations causing the ALT phenotype [34]. In the literature, presence of somatic ATRX and DAXX mutations or protein loss detected by immunohistochemistry is uncommon in sporadic insulinoma—in contrast to non-functional PanNET [12,13,14,15, 26]. A recent large whole-genome sequencing study definitively established that insulinomas and non-functional PanNETs have distinct genetic underpinnings, and recurrent copy number variations together with ATRX and DAXX mutations are a characteristic feature of non-functional PanNETs [12]. However, such alterations might be more prevalent in metastatic insulinomas, of which only few are present in the cohorts previously reported in the literature. Interestingly, in a cohort with multiple rare malignant/metastatic insulinomas, a high number of chromosomal aberrations was strongly associated with metastases [35]. As ATRX/DAXX mutations and ALT correlate with copy number variations and chromosomal instability [21, 36], this may be a reflection of ATRX/DAXX mutations that were not tested at that time. Loss of the tumor suppressor CDKN2A has been reported once before in a malignant insulinoma [37] and was recently described as a marker of malignant behavior in non-functional PanNET [26].
Several studies have reported large insulinomas to be malignant more often, and the late symptomatology suggests relatively low or acquired insulin production (Fig. 2) [1, 38, 39]. The ARX transcription factor is not expected to be expressed in insulinomas [40], as it is not expressed in pancreatic beta-cells [41, 42]. In contrast, around 50–60% of non-functional sporadic PanNETs express ARX [27]. Although focal nesidioblastosis was considered as explanation for ARX expression, it was deemed highly unlikely due to the characteristic tumor morphology, random peptide hormone expression, and the presence of metastases [43].

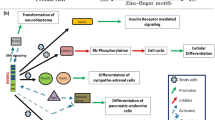
Tumorigenic mechanisms in clinically defined insulinomas. Hypothetic distinct pathways of tumorigenesis in clinical insulinomas based on previously published data in combination with current findings [1, 12, 26, 35]. 1 Typical small insulinomas characterized by recurrent YY1 mutations (25%), neutral, or amplified chromosomal copy numbers, and endocrine transcription factor expression consistent with normal beta-cell differentiation (PDX1+/ARX−). 2 Large insulinomas with distinct tumorigenic mechanisms often seen in non-functional PanNETs and endocrine transcription factor expression inconsistent with normal beta-cell differentiation (ARX+)
Acquired insulin production could be the result of transdifferentiation of ARX positive non-functional PanNETs or (subclinical) glucagonomas. The latter phenomenon has been shown in mice [44, 45], and a recent report described a human non-functional PanNET that progressed into a metastatic insulinoma with liver metastases over the course of 2 years [46]. Importantly, all malignant cases in this study had hypoglycemic symptoms at presentation, which resolved after surgery of the primary tumor. All but one metachronous liver metastases (patient 2) gave new episodes of symptomatic hypoglycemia when diagnosed. Interestingly, in patient 2, both the primary and the corresponding liver metastasis only had few PDX1-positive cells (potential insulin producing cells), so perhaps the solitary liver metastasis (3 cm) did not grow to a size in which a low percentage of cells could have caused symptoms.
There are several strengths of this study. The assay to assess expression of the nuclear transcription factor ARX is very robust. Whole slides that were stained show no variable expression due to fixation, and pancreatic islets, enteroendocrine duodenal/antrum cells serve as positive internal controls. Although several other markers have been proposed based on molecular analyses, protein, or mRNA expression, this is the first study to show a clear association of one single immunohistochemical marker with malignant behavior in insulinomas [35, 47, 48].
The small cohort size and number of metastatic cases is a limitation of this study, caused by the fact that metastatic insulinomas are very rare. Validation of our findings in other cohorts would be of important value. For the limitations inherent to cut-off scoring systems and TMAs, we have tried to minimize their effect by scoring blinded by two independent observers, confirming key percentages by digital analysis, and staining whole slides to confirm immunohistochemical expression when necessary.
To conclude, large tumor size (> 3 cm) was confirmed to be a strong marker of metastatic behavior in insulinomas. In addition, we found that large metastatic insulinomas are driven by tumorigenic mechanisms often seen in non-functional PanNETs, but not in small indolent insulinomas. Furthermore, we demonstrate that ARX—which is normally not present in beta-cells—is expressed in a subset of insulinomas and is associated with large tumor size, ALT, and metastatic disease. In contrast, ARX expression was absent in any of the small indolent insulinomas in this study or the previous literature. We hypothesize that cellular differentiation and tumorigenic mechanisms more closely related to non-functional PanNETs are important for the development of malignant insulinoma. Our findings with regard to rare metastatic insulinomas may be of value to personalize follow-up and treatment strategies in the future.
References
Camara-de-Souza AB, Toyoshima MTK, Giannella ML, Freire DS, Camacho CP, Lourenco DM, Jr., Rocha MS, Bacchella T, Jureidini R, Machado MCC, Almeida MQ, Pereira MAA (2018) Insulinoma: A retrospective study analyzing the differences between benign and malignant tumors. Pancreatology 18 (3):298–303. https://doi.org/10.1016/j.pan.2018.01.009
Nikfarjam M, Warshaw AL, Axelrod L, Deshpande V, Thayer SP, Ferrone CR, Fernandez-del Castillo C (2008) Improved contemporary surgical management of insulinomas: a 25-year experience at the Massachusetts General Hospital. Ann Surg 247 (1):165–172. https://doi.org/10.1097/SLA.0b013e31815792ed
Keutgen XM, Schadde E, Pommier RF, Halfdanarson TR, Howe JR, Kebebew E (2018) Metastatic neuroendocrine tumors of the gastrointestinal tract and pancreas: A surgeon’s plea to centering attention on the liver. Semin Oncol 45 (4):232–235. https://doi.org/10.1053/j.seminoncol.2018.07.002
Man D, Wu J, Shen Z, Zhu X (2018) Prognosis of patients with neuroendocrine tumor: a SEER database analysis. Cancer Manag Res 10:5629–5638. https://doi.org/10.2147/CMAR.S174907
Falconi M, Eriksson B, Kaltsas G, Bartsch DK, Capdevila J, Caplin M, Kos-Kudla B, Kwekkeboom D, Rindi G, Kloppel G, Reed N, Kianmanesh R, Jensen RT, Vienna Consensus Conference p (2016) ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 103 (2):153–171. https://doi.org/10.1159/000443171
Service FJ, McMahon MM, O'Brien PC, Ballard DJ (1991) Functioning insulinoma--incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc 66 (7):711–719. https://doi.org/10.1016/s0025-6196(12)62083-7
Kunz PL, Reidy-Lagunes D, Anthony LB, Bertino EM, Brendtro K, Chan JA, Chen H, Jensen RT, Kim MK, Klimstra DS, Kulke MH, Liu EH, Metz DC, Phan AT, Sippel RS, Strosberg JR, Yao JC, North American Neuroendocrine Tumor S (2013) Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas 42 (4):557–577. https://doi.org/10.1097/MPA.0b013e31828e34a4
Hirshberg B, Cochran C, Skarulis MC, Libutti SK, Alexander HR, Wood BJ, Chang R, Kleiner DE, Gorden P (2005) Malignant insulinoma: spectrum of unusual clinical features. Cancer 104 (2):264–272. https://doi.org/10.1002/cncr.21179
Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, Shih T, Yao JC (2017) Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol 3 (10):1335–1342. https://doi.org/10.1001/jamaoncol.2017.0589
Yao JC, Eisner MP, Leary C, Dagohoy C, Phan A, Rashid A, Hassan M, Evans DB (2007) Population-based study of islet cell carcinoma. Ann Surg Oncol 14 (12):3492–3500. https://doi.org/10.1245/s10434-007-9566-6
Keutgen XM, Nilubol N, Kebebew E (2016) Malignant-functioning neuroendocrine tumors of the pancreas: A survival analysis. Surgery 159 (5):1382–1389. https://doi.org/10.1016/j.surg.2015.11.010
Hong X, Qiao S, Li F, Wang W, Jiang R, Wu H, Chen H, Liu L, Peng J, Wang J, Jia C, Liang X, Dai H, Jiang J, Zhang T, Liao Q, Dai M, Cong L, Han X, Guo D, Liang Z, Li D, Zheng Z, Ye C, Li S, Zhao Y, Wu K, Wu W (2019) Whole-genome sequencing reveals distinct genetic bases for insulinomas and non-functional pancreatic neuroendocrine tumours: leading to a new classification system. Gut. https://doi.org/10.1136/gutjnl-2018-317233
Wang H, Bender A, Wang P, Karakose E, Inabnet WB, Libutti SK, Arnold A, Lambertini L, Stang M, Chen H, Kasai Y, Mahajan M, Kinoshita Y, Fernandez-Ranvier G, Becker TC, Takane KK, Walker LA, Saul S, Chen R, Scott DK, Ferrer J, Antipin Y, Donovan M, Uzilov AV, Reva B, Schadt EE, Losic B, Argmann C, Stewart AF (2017) Insights into beta cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat Commun 8 (1):767. https://doi.org/10.1038/s41467-017-00992-9
Cao Y, Gao Z, Li L, Jiang X, Shan A, Cai J, Peng Y, Li Y, Jiang X, Huang X, Wang J, Wei Q, Qin G, Zhao J, Jin X, Liu L, Li Y, Wang W, Wang J, Ning G (2013) Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat Commun 4:2810. https://doi.org/10.1038/ncomms3810
Lichtenauer UD, Di Dalmazi G, Slater EP, Wieland T, Kuebart A, Schmittfull A, Schwarzmayr T, Diener S, Wiese D, Thasler WE, Reincke M, Meitinger T, Schott M, Fassnacht M, Bartsch DK, Strom TM, Beuschlein F (2015) Frequency and clinical correlates of somatic Ying Yang 1 mutations in sporadic insulinomas. J Clin Endocrinol Metab 100 (5):E776–E782. https://doi.org/10.1210/jc.2015-1100
Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, Velculescu VE, Diaz LA, Jr., Vogelstein B, Kinzler KW, Hruban RH, Papadopoulos N (2011) DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331 (6021):1199–1203. https://doi.org/10.1126/science.1200609
Scarpa A, Chang DK, Nones K, Corbo V, Patch A-M, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A, Rusev B, Scardoni M, Antonello D, Barbi S, Sikora KO, Cingarlini S, Vicentini C, McKay S, Quinn MCJ, Bruxner TJC, Christ AN, Harliwong I, Idrisoglu S, McLean S, Nourse C, Nourbakhsh E, Wilson PJ, Anderson MJ, Fink JL, Newell F, Waddell N, Holmes O, Kazakoff SH, Leonard C, Wood S, Xu Q, Nagaraj SH, Amato E, Dalai I, Bersani S, Cataldo I, Dei Tos AP, Capelli P, Davì MV, Landoni L, Malpaga A, Miotto M, Whitehall VLJ, Leggett BA, Harris JL, Harris J, Jones MD, Humphris J, Chantrill LA, Chin V, Nagrial AM, Pajic M, Scarlett CJ, Pinho A, Rooman I, Toon C, Wu J, Pinese M, Cowley M, Barbour A, Mawson A, Humphrey ES, Colvin EK, Chou A, Lovell JA, Jamieson NB, Duthie F, Gingras M-C, Fisher WE, Dagg RA, Lau LMS, Lee M, Pickett HA, Reddel RR, Samra JS, Kench JG, Merrett ND, Epari K, Nguyen NQ, Zeps N, Falconi M, Simbolo M, Butturini G, Van Buren G, Partelli S, Fassan M, Initiative APCG, Khanna KK, Gill AJ, Wheeler DA, Gibbs RA, Musgrove EA, Bassi C, Tortora G, Pederzoli P, Pearson JV, Waddell N, Biankin AV, Grimmond SM (2017) Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 543 (7643):65–71
Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, He Y, Yan H, Bigner DD, Oba-Shinjo SM, Marie SK, Riggins GJ, Kinzler KW, Vogelstein B, Hruban RH, Maitra A, Papadopoulos N, Meeker AK (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333 (6041):425. https://doi.org/10.1126/science.1207313
Hechtman JF, Klimstra DS, Nanjangud G, Frosina D, Shia J, Jungbluth AA (2019) Performance of DAXX Immunohistochemistry as a Screen for DAXX Mutations in Pancreatic Neuroendocrine Tumors. Pancreas 48 (3):396–399. https://doi.org/10.1097/MPA.0000000000001256
Chan CS, Laddha SV, Lewis PW, Koletsky MS, Robzyk K, Da Silva E, Torres PJ, Untch BR, Li J, Bose P, Chan TA, Klimstra DS, Allis CD, Tang LH (2018) ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat Commun 9 (1):4158. https://doi.org/10.1038/s41467-018-06498-2
Marinoni I, Kurrer AS, Vassella E, Dettmer M, Rudolph T, Banz V, Hunger F, Pasquinelli S, Speel EJ, Perren A (2014) Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 146 (2):453–460 e455. https://doi.org/10.1053/j.gastro.2013.10.020
Singhi AD, Liu TC, Roncaioli JL, Cao D, Zeh HJ, Zureikat AH, Tsung A, Marsh JW, Lee KK, Hogg ME, Bahary N, Brand RE, McGrath KM, Slivka A, Cressman KL, Fuhrer K, O'Sullivan RJ (2017) Alternative Lengthening of Telomeres and Loss of DAXX/ATRX Expression Predicts Metastatic Disease and Poor Survival in Patients with Pancreatic Neuroendocrine Tumors. Clin Cancer Res 23 (2):600–609. https://doi.org/10.1158/1078-0432.CCR-16-1113
Kim JY, Brosnan-Cashman JA, An S, Kim SJ, Song KB, Kim MS, Kim MJ, Hwang DW, Meeker AK, Yu E, Kim SC, Hruban RH, Heaphy CM, Hong SM (2017) Alternative Lengthening of Telomeres in Primary Pancreatic Neuroendocrine Tumors Is Associated with Aggressive Clinical Behavior and Poor Survival. Clin Cancer Res 23 (6):1598–1606. https://doi.org/10.1158/1078-0432.CCR-16-1147
Chou A, Itchins M, de Reuver PR, Arena J, Clarkson A, Sheen A, Sioson L, Cheung V, Perren A, Nahm C, Mittal A, Samra JS, Pajic M, Gill AJ (2018) ATRX loss is an independent predictor of poor survival in pancreatic neuroendocrine tumors. Hum Pathol 82:249–257. https://doi.org/10.1016/j.humpath.2018.07.032
Park JK, Paik WH, Lee K, Ryu JK, Lee SH, Kim YT (2017) DAXX/ATRX and MEN1 genes are strong prognostic markers in pancreatic neuroendocrine tumors. Oncotarget 8 (30):49796-49806. https://doi.org/10.18632/oncotarget.17964
Roy S, LaFramboise WA, Liu T-C, Cao D, Luvison A, Miller C, Lyons MA, O’Sullivan RJ, Zureikat AH, Hogg ME, Tsung A, Lee KK, Bahary N, Brand RE, Chennat JS, Fasanella KE, McGrath K, Nikiforova MN, Papachristou GI, Slivka A, Zeh HJ, Singhi AD (2018) Loss of Chromatin-Remodeling Proteins and/or CDKN2A Associates With Metastasis of Pancreatic Neuroendocrine Tumors and Reduced Patient Survival Times. Gastroenterology 154 (8):2060-2063.e2068
Cejas P, Drier Y, Dreijerink KMA, Brosens LAA, Deshpande V, Epstein CB, Conemans EB, Morsink FHM, Graham MK, Valk GD, Vriens MR, Castillo CF, Ferrone CR, Adar T, Bowden M, Whitton HJ, Da Silva A, Font-Tello A, Long HW, Gaskell E, Shoresh N, Heaphy CM, Sicinska E, Kulke MH, Chung DC, Bernstein BE, Shivdasani RA (2019) Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat Med 25 (8):1260–1265. https://doi.org/10.1038/s41591-019-0493-4
de Wilde RF, Heaphy CM, Maitra A, Meeker AK, Edil BH, Wolfgang CL, Ellison TA, Schulick RD, Molenaar IQ, Valk GD, Vriens MR, Borel Rinkes IH, Offerhaus GJ, Hruban RH, Matsukuma KE (2012) Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol 25 (7):1033–1039. https://doi.org/10.1038/modpathol.2012.53
Ho TH, Park IY, Zhao H, Tong P, Champion MD, Yan H, Monzon FA, Hoang A, Tamboli P, Parker AS, Joseph RW, Qiao W, Dykema K, Tannir NM, Castle EP, Nunez-Nateras R, Teh BT, Wang J, Walker CL, Hung MC, Jonasch E (2016) High-resolution profiling of histone h3 lysine 36 trimethylation in metastatic renal cell carcinoma. Oncogene 35 (12):1565–1574. https://doi.org/10.1038/onc.2015.221
Conemans EB, Brosens LAA, Raicu-Ionita GM, Pieterman CRC, de Herder WW, Dekkers OM, Hermus AR, van der Horst-Schrivers AN, Bisschop PH, Havekes B, Drent ML, Timmers HTM, Offerhaus GJ, Valk GD, Vriens MR (2017) Prognostic value of WHO grade in pancreatic neuro-endocrine tumors in Multiple Endocrine Neoplasia type 1: Results from the DutchMEN1 Study Group. Pancreatology 17 (5):766–772. https://doi.org/10.1016/j.pan.2017.07.196
Inzani F, Petrone G, Rindi G (2018) The New World Health Organization Classification for Pancreatic Neuroendocrine Neoplasia. Endocrinol Metab Clin North Am 47 (3):463–470. https://doi.org/10.1016/j.ecl.2018.04.008
Chung CT, Santos Gda C, Hwang DM, Ludkovski O, Pintilie M, Squire JA, Tsao MS (2010) FISH assay development for the detection of p16/CDKN2A deletion in malignant pleural mesothelioma. J Clin Pathol 63 (7):630–634. https://doi.org/10.1136/jcp.2010.076794
Hackeng WM, Morsink FHM, Moons LMG, Heaphy CM, Offerhaus GJA, Dreijerink KMA, Brosens LAA (2019) Assessment of ARX expression, a novel biomarker for metastatic risk in pancreatic neuroendocrine tumors, in endoscopic ultrasound fine-needle aspiration. Diagn Cytopathol:Accepted for publication. https://doi.org/10.1002/dc.24368
Brosnan-Cashman JA, Graham MK, Heaphy CM (2018) Genetic alterations associated with ALTered telomeres. Oncotarget 9 (73):33739-33740. https://doi.org/10.18632/oncotarget.26111
Jonkers YM, Claessen SM, Perren A, Schmid S, Komminoth P, Verhofstad AA, Hofland LJ, de Krijger RR, Slootweg PJ, Ramaekers FC, Speel EJ (2005) Chromosomal instability predicts metastatic disease in patients with insulinomas. Endocr Relat Cancer 12 (2):435–447. https://doi.org/10.1677/erc.1.00960
Pea A, Yu J, Marchionni L, Noe M, Luchini C, Pulvirenti A, de Wilde RF, Brosens LA, Rezaee N, Javed A, Gobbo S, Regi P, Salvia R, Bassi C, He J, Weiss MJ, Cameron JL, Offerhaus GJA, Hruban RH, Lawlor RT, Scarpa A, Heaphy CM, Wood LD, Wolfgang CL (2018) Genetic Analysis of Small Well-differentiated Pancreatic Neuroendocrine Tumors Identifies Subgroups With Differing Risks of Liver Metastases. Ann Surg. https://doi.org/10.1097/SLA.0000000000003022
Bartsch DK, Kersting M, Wild A, Ramaswamy A, Gerdes B, Schuermann M, Simon B, Rothmund M (2000) Low frequency of p16(INK4a) alterations in insulinomas. Digestion 62 (2–3):171–177. https://doi.org/10.1159/000007810
Ueda K, Taira T, Hakoda H, Nakata S, Okata S, Nagai T, Aoki S, Mishima H, Sako A, Maruyama T, Okumura M (2016) Giant insulinoma: report of a case and review of published reports. Surg Case Rep 2 (1):136. https://doi.org/10.1186/s40792-016-0265-z
Crippa S, Zerbi A, Boninsegna L, Capitanio V, Partelli S, Balzano G, Pederzoli P, Di Carlo V, Falconi M (2012) Surgical management of insulinomas: short- and long-term outcomes after enucleations and pancreatic resections. Arch Surg 147 (3):261–266. https://doi.org/10.1001/archsurg.2011.1843
Wang Z, You J, Xu S, Hua Z, Zhang W, Deng T, Fang N, Fang Q, Liu H, Peng L, Wang P, Lou J (2015) Colocalization of insulin and glucagon in insulinoma cells and developing pancreatic endocrine cells. Biochem Biophys Res Commun 461 (4):598–604. https://doi.org/10.1016/j.bbrc.2015.04.072
Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, Melton DA, Yanai I (2016) A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst 3 (4):346–360 e344. https://doi.org/10.1016/j.cels.2016.08.011
Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ, van Oudenaarden A (2016) A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst 3 (4):385–394 e383. https://doi.org/10.1016/j.cels.2016.09.002
Bhatti TR, Ganapathy K, Huppmann AR, Conlin L, Boodhansingh KE, MacMullen C, Becker S, Ernst LM, Adzick NS, Ruchelli ED, Ganguly A, Stanley CA (2016) Histologic and Molecular Profile of Pediatric Insulinomas: Evidence of a Paternal Parent-of-Origin Effect. J Clin Endocrinol Metab 101 (3):914–922. https://doi.org/10.1210/jc.2015-2914
Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ (2003) Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Res 63 (16):4836–4841
Lu J, Herrera PL, Carreira C, Bonnavion R, Seigne C, Calender A, Bertolino P, Zhang CX (2010) Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 138 (5):1954–1965. https://doi.org/10.1053/j.gastro.2010.01.046
Arslan MS, Ozbek M, Karakose M, Tutal E, Ucan B, Yilmazer D, Dilli A, Gultekin SS, Cakal E, Delibasi T (2015) Transformation of nonfunctioning pancreatic tumor into malignant insulinoma after 3 years: an uncommon clinical course of insulinoma. Arch Endocrinol Metab 59 (3):270–272. https://doi.org/10.1590/2359-3997000000049
Alkatout I, Friemel J, Sitek B, Anlauf M, Eisenach PA, Stuhler K, Scarpa A, Perren A, Meyer HE, Knoefel WT, Kloppel G, Sipos B (2015) Novel prognostic markers revealed by a proteomic approach separating benign from malignant insulinomas. Mod Pathol 28 (1):69–79. https://doi.org/10.1038/modpathol.2014.82
Murat Cde B, da Rosa PW, Fortes MA, Correa L, Machado MC, Novak EM, Siqueira SA, Pereira MA, Correa-Giannella ML, Giannella-Neto D, Giorgi RR (2015) Differential expression of genes encoding proteins of the HGF/MET system in insulinomas. Diabetol Metab Syndr 7:84. https://doi.org/10.1186/s13098-015-0079-3
Acknowledgments
We thank T. Peeters and A. Meeker for their help with setting up the fluorescence in situ hybridization assay and use of Telometer and P. Cejas for confirming the IHC scoring protocol.
Funding
This work was supported by the Dutch Digestive Foundation/Maag Lever Darm Stichting (grant number CDG 14-020).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the University Medical Center (UMC) Utrecht Biobank Research Ethics Committee.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hackeng, W.M., Schelhaas, W., Morsink, F.H.M. et al. Alternative Lengthening of Telomeres and Differential Expression of Endocrine Transcription Factors Distinguish Metastatic and Non-metastatic Insulinomas. Endocr Pathol 31, 108–118 (2020). https://doi.org/10.1007/s12022-020-09611-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12022-020-09611-8