
Abstract
Background
Understanding the molecular mechanisms underlying papillary thyroid cancer (PTC) proved to be vital not only for diagnostic purposes but also for tailored treatments. Despite the strong evidence of heritability, only a small subset of alterations has been implicated in PTC pathogenesis. To this reason, we used targeted next-generation sequencing (NGS) to identify candidate variants implicated in PTC pathogenesis, progression, and invasiveness.
Methods
A total of 42 primary PTC tissues were investigated using a targeted next-generation sequencing (NGS) panel enlisting 47 genes involved in DNA repair and tumor progression.
Results
We identified 57 point mutations in 78.5% of samples (n = 32). Thirty-two somatic mutations were identified exclusively in known thyroid cancer genes (BRAF, KRAS, NRAS, and TERT). Unpredictably, 45% of the all identified mutations (n = 25) resulted to be germline, most affecting DNA repair genes. Interestingly, none of the latter variants was in the main population databases. Following ACMG classification, 20% of pathogenic/likely pathogenic and 68% of variant of unknown significance were identified.
Conclusions
Overall, our results support the hypothesis that rare germline variants in DNA repair genes are accountable for PTC susceptibility. More data, including the segregation analysis in affected families, should be collected before definitely annotate these alterations and to establish their potential prognostic and treatment implications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Understanding the molecular mechanisms underlying tumorigenesis is vital for the accurate diagnosis and use of personalized treatments. Previously, monogenic assays were commonly used to find molecular alterations in tumor mass populations [1]. Nowadays, the use of next-generation sequencing (NGS) technology ensures the simultaneous analysis of hundreds of genes of interest, using targeted sequencing panels.
Thyroid cancers (TC) are the most widespread malignancies of the endocrine system and represent ~1–1.5% of all tumor-related diseases [2]. TC typically progresses from thyroid nodules, which are prevalent in the general population and mainly related to old age. Although surgery solve most cases, in some cases the tumors exhibit an aggressive behavior. TC can be categorized by histology. Medullary TC accounts for about 5% of all cases and it rises from parafollicular C cells [3]. The remaining 95% of all TC cases are follicular cell-derived TC and they are classified in papillary (PTC), follicular, Hürthle, poorly differentiated, and anaplastic TC [4]. PTC alone accounts for 80% of all TCs.
Over the past decades, the application of molecular technologies has shed considerably light on the genetic abnormalities associated with TC, elucidating novel approaches to tumor diagnosis, risk assessment of tumor progression, and potential novel therapeutic strategies [5]. Well-known somatic alterations include single-nucleotide variants (SNVs) affecting proto-oncogenes (BRAF, NRAS, HRAS, KRAS, EIF1AX) and chromosomal rearrangements (RET/PTC1, RET/PTC3, PAX8/PPARG), which vary with the histologic subtype [6]. Notwithstanding the tremendous effort to better characterize PTC etiology, the molecular bases of tumor development are still elusive.
DNA repair is a gatekeeper set of pathways meant to preserve genomic integrity upon exposure to genotoxic agents. Impairments in these fine-tuned systems are correlated to cancer susceptibility. DNA repair proteins functionally interact with each other, within the same DNA repair pathway and across different pathways, establishing ground for additive or even multiplicative effects on DNA repair activity and, hence, cancer risk [7]. Moreover, the identification of novel prognostic markers is critical for the improvement of the risk stratification for cancer death and recurrence. We created a targeted NGS panel enlisting genes directly involved in DNA repair and some of the most important genes involved in tumor progression. The final aim was to identify SNVs that could play a role in cancer susceptibility and to discover new biomarkers of PTC progression and invasiveness to be used in the future, allowing the development of tailored PTC prevention policies, treatment possibilities and perhaps implementation of guidelines.
Materials and methods
Samples collection and DNA extraction
Informed consent was obtained from all individuals included in the study. Papillary thyroid tumors were obtained upon surgical resection at the Sapienza University of Rome Hospital. Immediately after surgery, tumor tissues were snap-frozen and stored in liquid nitrogen. Fresh-frozen tissue samples were reviewed by two different pathologists, who confirmed the diagnosis of PTC and excluded specimens in which tumor cells accounted for <60% of the total. Clinical data were collected by retrospective review of hospital charts, and tumors were staged according to the criteria of the AJCC/UICC TNM classification, 8th edition [8]. Tumors were risk-stratified based on clinical and histological data in accordance with the 2015 American Thyroid Association (ATA) risk of recurrence staging system [9]. Genomic DNA was isolated from surgical samples and peripheral blood as previously described [10].
Library preparation and next-generation sequencing (NGS)
Genomic DNA from 42 tumor tissues was quantified using the Qubit dsDNA HS Assay Kit (Life Technologies). Barcoded libraries were generated from 0.5 to 10 ng of DNA per sample using the Ion AmpliSeq HiFi mix (Ion AmpliSeq Library Kit Plus, Thermo Fisher Scientific) and two premixed pools of 952 primer pairs (Thermo Fisher Scientific), according to manufacturer’s instructions. Gene panel list is shown in Supplementary Table 1. Clonal amplification of libraries was performed by emulsion PCR on an Ion Chef Instrument, as previously described [11]. Sequencing was performed with the Ion S5 GeneStudio Sequencer using the Ion 540 Chip kit and the Ion 540™ Kit-Chef (all Thermo Fisher Scientific).
Data analysis and variant prioritization
The Variant Caller v5.12 was used to process data (Thermo Fisher Scientific). Annotation was performed with both Ion Reporter 5.12 (Thermo Fisher Scientific) and wANNOVAR, as previously described [12]. Briefly, somatic variants were called when a position was covered at least 500 times. High-quality variants were those with a depth of coverage (FDP) of ≥500, genotype quality scores of ≥30, a minimum alternate allele frequency of 5% (AF ≥ 5%), and absence of homopolymer regions (HRUN < 6). Finally, variants were prioritized based on their genomic location, with exclusion of intronic, intergenic, ncRNA-intronic, and UTR variants. Variant prioritization was based on population frequency, quality values, and functional consequences. Synonymous variants not affecting splice regions were excluded a priori. Variants were filtered based on their frequency among the European-descendent population (1000 Human Genomes Project, ESP6500SI, gnomAD, ExAC) and on clinical associations (NCBI dbSNP, ClinVar). In silico functional consequences were evaluated in seven databases (SIFT, Polyphen2_HVAR, Polyphen2_HDIV, LRT, MutationTaster, MutationAssessor, FATHMM, PROVEAN). After filtering and prioritization, missense, splice-site, stop-gain, stop-loss, and frameshift variants were retained for further evaluation. ACMG classification was used to annotate variants by the Varsome (https://varsome.com/) and wIntervar (http://wintervar.wglab.org/) online tools. The Catalogue Of Somatic Mutations In Cancer database was used to assess pathogenicity of somatic mutations (https://cancer.sanger.ac.uk/cosmic).
Sanger sequencing
Candidate variants and TERT promoter mutations were assessed by Sanger sequencing as previously described [13]. PCR conditions and sequencing primers are available upon request. TERT mutational analysis was performed in samples with remaining DNA after NGS analysis.
Statistical analysis
Data are reported as medians and range of values. Between-groups differences in categorical variables were analyzed using the Chi-square test or the Fisher exact test. Between-groups differences in continuous variables were assessed with the Mann–Whitney U test. p values lower than 0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism version 8.0 software (GraphPad Software Inc., San Diego, CA, USA).
Results
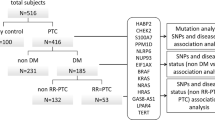
The aim of this study was to investigate the presence of novel point mutations and small insertions or deletions (indels) in genes related to DNA repair and to tumor progression in PTC, using a NGS approach. A total of 42 primary PTC tissues from 42 patients were investigated. Table 1 summarizes the characteristics of the patients enrolled in our cohort. Most of the cases were classic PTC (PTC-CT) (N = 31; 74%). The second largest histology subgroup was follicular variant PTC (PTC-FV) (N = 7; 17%), followed by: mixed PTC (N = 2; 5%), oxyphilic variant (N = 1; 2%), and trabecular variant (N = 1; 2%). The female:male ratio was 1.5:1 and 7% of cases were familial, defined as cases with two first- or second-degree relatives also affected with PTC. The 42 tissues were analyzed for mutations using small amount (0.5–10 ng) of DNA. Sequence coverage was assessed from the number and distribution of reads across the target DNA regions. Approximately 4.16 million mapped reads with the mean read length of 200 bp were generated. The mean depth of coverage was 4100X, with 96.7% reads on target. Overall, mutations were identified in 32 PTC specimens (78.5%). Fifty-seven single-nucleotide variants were detected in 18 coding genes. After filtering benign and likely benign SNVs, the median number of genetic alterations per ATA risk category was 2 (ranged from 0 to 4), which is consistent with most endocrine-related tumors having low mutational burden (Fig. 1). Overall, the totality of somatic mutations (N = 32) found in PTC specimens have been detected in BRAF, KRAS, and NRAS and possessed VAFs that ranged from 12 to 46%. The BRAF p.V600E mutation was confirmed as the most common hot-spot mutation in PTC (57%). Well-established RAS gene mutations (KRAS p.G12V, NRAS p.Q61K, and p.Q61R) were found in 5 PTCs (12%). TERT promoter mutations (TERT c. −124C > T (C228T) and c.-146C > T (C250T)) were identified in 12% of analyzed PTCs. In two patients TERT mutation was found in co-presence with BRAF p.V600E.

Association between the number of genetic alteration and clinical ATA category. No significant difference was assessed between the three ATA risk score-based categories
The remaining alterations identified, included SNVs with an allelic frequency ranging between 42 and 58%, suggesting the possibility that these variants might be germline. To this reason, Sanger sequencing was performed on DNA extracted from patient’s blood samples, confirming the germline origin of these variants. All the aforementioned variants and the clinical features of the patients are reported in Table 2.
Following ACMG classification, the identified germline SNVs included 2 pathogenic (8%), 3 likely pathogenic (12%), 17 variants of unknown significances (68%), and 3 (12%) variants having conflicting interpretation of pathogenicity, being simultaneously annotated as likely benign, VUS and likely pathogenic. The two pathogenic variants were detected in two patients both burdened by a classic PTC, with a tumor size falling into the upper 95% CI of the median. ERCC Excision Repair 5 (ERCC5, NM_000123) c.3004C > T is a missense variant introducing a stop codon (p. Q1002X). The second pathogenic variants is located in the splice region of the Transcription factor 7-like 2 (TCF7L2) exon 4 (NM_001146274: c.450 + 1G > T). Three likely pathogenic variants have been identified in four different patients. APC (NM_000038) c.5365G > T is a missense variant causing a valine to leucine change in position 1789 of APC protein sequence (p.V1789L). Alteration in the bone morphogenetic protein receptor type 1A (BMPR1A, NM_004329) c.1416G > T is a missense variant which causes a glutamic acid to be replaced by an aspartic acid in the amino acid chain (p.E472D) in the protein kinase domain. Transforming growth factor beta receptor 2 (TGFBR2, NM_001024847) c.1710C > G is a missense variant which causes an aspartic acid to be replaced by a glutamic acid in the amino acid chain (p.D570E). Three SNVs annotated with conflicting interpretation of pathogenicity were detected in MutL Homolog 1 (MLH1), MutL Homolog 3 (MLH3), and PMS1 homolog 2, mismatch repair system component (PMS2), all involved in the DNA mismatch repair (MMR) pathway: MLH1 (NM_000249) c.1013A > G (p.N388S); MLH3 (NM_014381) c.1343C > T (p. P448L); PMS2 (c.1004A > G; p.N335S).
We did not find any statistically significant association between the presence of a germline variant and any clinical characteristics, treatment outcome or somatic mutational status of the analyzed patients, except for more frequent lymph-node metastases observed in absence of germline alterations (p = 0.0236) (Table 3).
Discussion
The molecular basis of PTC development and progression is not completely understood. Despite the strong evidence of heritability, only a small subset of alterations has been convincingly implicated in PTC pathogenesis so far [14]. The best characterized markers of PTC aggressiveness are the somatic BRAF p.V600E mutation, which occurs in ~40% of all PTCs [15] and the coexisting BRAF p.V600E and TERT promoter mutations associated with PTC-specific mortality [16].
In this study, we used a targeted NGS panel to investigate the molecular profiles of a cohort of PTC specimens, identifying point mutations in 78.5% of samples. We once again confirmed the high prevalence of the BRAF p.V600E somatic mutation in classic PTC and of KRAS p.G12V, NRAS p.Q61R, and p.Q61K somatic mutations in the follicular variants of PTC. The prevalence of TERT promoter somatic mutations in PTC from this study was consistent with that reported by TCGA (9%) [16].
We did not find any somatic mutations in the other genes associated with cancer aggressiveness analyzed. Unpredictably, 45% of the identified mutations resulted to be germline. Following ACMG classification, 20% of pathogenic/likely pathogenic and 68% of variant of unknown significances were identified.
We highlighted a nonsense mutation in ERCC5 gene in a 70-years-old female patient who also harbors the BRAF p.V600E somatic mutation (VAF = 30%). ERCC5, also known as XPG, encodes a structure-specific endonuclease that has multiple functions during nucleotide excision repair, a DNA repair pathway triggered by ultraviolet (UV)-induced damage [7]. The amino acid change falls into the C-terminal PCNA-binding domain, whose function is crucial for repair efficiency [14]. Alterations affecting its coding sequence or its mRNA levels can impair DNA repair resulting in genomic instability and carcinogenesis. Homozygous or compound heterozygous mutation in ERCC5 are associated to the xeroderma pigmentosum complementation group G (XPG), a rare disease characterized by photosensitive erythema, keratoses and skin and eye disorders due to a high sensitivity to UV radiations. Indeed, ERCC5 has been associated with an increased risk of developing skin tumors (basalomas, squamous cell carcinomas, melanomas). Interestingly, a patient’s relative was affected by a uterine carcinoma, that is also reported to be also be associated to the ERCC5 p.Q1002X. Indeed, previous studies have linked ERCC5 SNVs to soft tissues cancer susceptibility, including endometrial and thyroid [7, 17]. The tumor was positive for the BRAF p.V600E mutation.
In a 37-years-old female patient, a splice-site mutation has been identified in TCF7L2. TCF7L2 is an important component of the Wnt signaling pathway. TCF7L2 (previously reported as TCF-4) is a transcription factor that interacts with β-catenin in the nucleus inducing the expression of target genes, including CCND1 and c-MYC, involved in cellular proliferation, evasion of apoptosis, and tissue invasion and metastasis [18]. Loss of function mutations in TCF7L2 have been associated to both prostate [18] and colon cancer [19] susceptibility and to an increased risk of Type II diabetes [20]. We identified a variant in the splice region of TCF7L2, which alters RNA splicing reducing the amount of the functional codified protein. Accordingly, the genome aggregation database (gnomAD) identifies TCF7L2 as a strong loss-of-function intolerant gene. The pLI (probability of being loss-of-function intolerant), representing the intolerance toward protein-truncating variation, is 1, strengthening the concept of being extremely intolerant toward loss-of-function alterations. Thus, in silico data corroborate the possibility that this alteration to be strongly deleterious.
Mutations in APC are known to be causative of familial adenomatous polyposis, colon hepatocellular, and gastric carcinoma together with desmoid tumors. We found an APC p.V1789L substitution in a 65-years-old female patient in whose medical history, in addition to the PTC, a previous duodenal ulcer and breast fibroadenoma were also reported. Moreover, her family history turned to be positive for TC, liver, and genitourinary cancers. This variant was not previously reported and is not enlisted in any population databases (i.e., GnomAD). In contrast, this amino acid appears to be only partially phylogenetically conserved, hindering the possibility to clearly annotate it as a deleterious variant. It should be noted that the patient turned to be also positive for the BRAF p.V600E and the TERT c.‐124C > T (C228T).
Alteration in BMPR1A have been strongly associated to an increased susceptibility to the juvenile polyposis syndrome and to colorectal cancer [21]. We identified a p.E472D substitution in BMPR1A in a 45-years-old male patient. Although this amino acid change does not suggest the abolition of the protein kinase domain functionality, this variant was not found in the gnomAD database, suggesting a possible impact in protein activity. Literature regarding the role of BMPR1A in thyroid carcinogenesis are still scanty. It should be noted that the tumor was positive for BRAF p.V600E (VAF = 15%) and TERT C228T mutations and was reclassified in the ATA high risk category at the last follow-up.
Interestingly the two PTCs harboring both the TERT and BRAF mutations have a very different prognosis (Table 2) suggesting that the different germline mutational status may play a role in the modulation of cancer aggressiveness.
A missense mutation was identified in a 27-years-old male patient who also carries the NRAS p.Q61R mutation (VAF = 46.5%). Alteration in TGFBR2 have been associated to the Lynch syndrome, squamous cell carcinoma of the esophagus and the connectivopathies Loeys-Dietz and type-2 Marfan syndromes. TGFBR2 p.D570E falls in a hot-spot region where most missense variants are annotated as pathogenic. Notwithstanding the aminoacidic change occurs between two negative-charged residues, the position is quite phylogenetically conserved.
Moreover, three SNVs were classified having conflicting interpretation of pathogenicity, being simultaneously annotated as likely benign, VUS and likely pathogenic. Indeed, inactivating mutations in both MLH1 and PMS2 are associated to the Lynch syndrome, a heritable condition associated with a greatly increased risk of colorectal, endometrial, stomach, and ovarian cancers together with tumors of the small intestine, the biliary tract, brain, ureters, and renal pelvis [22]. MLH1 p.N388S is located in a hot-spot region where 24 variants, all pathogenic, have been identified. Nevertheless, this SNV has been previously assessed, suggesting a potential lack of impact in the protein function [23, 24]. MLH3 p.P448L is simultaneously annotated as benign and VUS and is not present in gnomAD database. Besides that, this residue is not strongly conserved and the Varsome database annotated this variant as a polymorphism. Since this SNP was never been published before nor in vitro experiments have been performed to assess its impact on MLH3 functions, we are unable to strongly associate this alteration to papillary thyroid carcinogenesis. Finally, the PMS2 p.N335S was detected in the same PTC sample that also harbored the APC p.V1789L. This residue is highly conserved during phylogenesis and in silico predictors strongly suggest its pathogenic effect. Therefore, the co-presence of the two alterations, the complexity of the familial history and the fact that PMS2 has a much lower penetrance for Lynch Syndrome than the other MMR genes [25], prevents us from assessing whether it is deleterious or not. More data, including the segregation analysis in affected families, should be collected before definitely annotate these alterations.
The only statistically significant association we found with the patient’s clinical features is between the presence of a germline variation and a less frequent lymph-node involvement. However, patients with a germline mutation tended to be male and to co-occur with a somatic mutation. Unfortunately, the patient’s cohort analyzed is admittedly too small to reach reliable conclusions.
The main finding of our study is the frequent involvement of alterations in DNA repair genes in PTC predisposition. This is in accordance with recent findings that reported the presence of germline mutations in DNA repair genes in a concurrent composite mucoepidermoid carcinoma and papillary thyroid carcinoma suggesting their putative role in the predisposition to TC development [26]. Interestingly, all the pathogenic and putatively pathogenic mutations found in our PTC cohort are not present in the major population databases (i.e., gnomAD). In our opinion this result is extremely relevant. Since typical PTC-related pedigrees do not often follow Mendelian inheritance but, instead, are small with irregular transmission of the cancer phenotype, the most widely used approach for the investigation on PTC genetics relied on genome-wide association studies. Despite the efforts, this kind of studies has failed to explain the “missing” heritability of PTC [7]. Indeed, our results tell a different story: the predisposition to PTC may be due to very rare germinal variants, not investigated in polygenic and gene-environment interactions-based studies.
Regardless, the results presented here should be observed only as proof of concept and must therefore be validated through replication in larger populations and implemented with familial segregation analysis. Moreover, our data are derived from a targeted panel while the use of massive technologies such as the whole exome sequencing will surely help to shed better light on familial predisposition to PTC.
In conclusion, notwithstanding the limitations in our approach, our data support the hypothesis that rare germline variants in genes involved in DNA repair are accountable for PTC susceptibility, rather than genome-wide-based risk scoring.
References
Y.J. Cha, J.S. Koo, Next-generation sequencing in thyroid cancer. J. Transl. Med. 14 (2016). https://doi.org/10.1186/s12967-016-1074-7
C. Mio, E. Lavarone, K. Conzatti, F. Baldan, B. Toffoletto, C. Puppin, S. Filetti, C. Durante, D. Russo, A. Orlacchio, A. Di Cristofano, C. Di Loreto, G. Damante, MCM5 as a target of BET inhibitors in thyroid cancer cells. Endocr. Relat. Cancer 23, 335–347 (2016)
S. Liyanarachchi, J. Gudmundsson, E. Ferkingstad, H. He, J.G. Jonasson, V. Tragante, F.W. Asselbergs, L. Xu, L.A. Kiemeney, R.T. Netea-Maier, J.I. Mayordomo, T.S. Plantinga, H. Hjartarson, J. Hrafnkelsson, E.M. Sturgis, P. Brock, F. Nabhan, G. Thorleifsson, M.D. Ringel, K. Stefansson, A. de la Chapelle, Assessing thyroid cancer risk using polygenic risk scores. PNAS 117, 5997–6002 (2020). https://doi.org/10.1073/pnas.1919976117
F. Baldan, C. Mio, L. Allegri, K. Conzatti, B. Toffoletto, C. Puppin, S. Radovic, C. Vascotto, D. Russo, C.D. Loreto, G. Damante, F. Baldan, C. Mio, L. Allegri, K. Conzatti, B. Toffoletto, C. Puppin, S. Radovic, C. Vascotto, D. Russo, C. Di Loreto, G. Damante, Identification of tumorigenesis-related mRNAs associated with RNA-binding protein HuR in thyroid cancer cells. Oncotarget 7, 63388–63407 (2016). https://doi.org/10.18632/oncotarget.11255
R.A. DeLellis, Pathology and genetics of thyroid carcinoma. J. Surgical Oncol. 94, 662–669 (2006). https://doi.org/10.1002/jso.20700
A. Prete, P. Borges de Souza, S. Censi, M. Muzza, N. Nucci, M. Sponziello, Update on fundamental mechanisms of thyroid cancer. Front. Endocrinol. 11 (2020). https://doi.org/10.3389/fendo.2020.00102
L.S. Santos, B.C. Gomes, H.N. Bastos, O.M. Gil, A.P. Azevedo, T.C. Ferreira, E. Limbert, S.N. Silva, J. Rueff, Thyroid cancer: the quest for genetic susceptibility involving DNA repair genes. Genes (Basel) 10 (2019). https://doi.org/10.3390/genes10080586
M. Tuttle, L.F. Morris, B. Haugen, J. Shah, J.A. Sosa, E. Rohren, R.M. Subramaniam, J.L. Hunt, N.D. Perrier, M.B. Amin, S.B. Edge, F. Greene, D. Byrd, R.K. Brookland, M.K. Washington, C.C. Compton, K.R. Hess, D.C. Sullivan, J.M. Jessup, Thyroid-Differentiated and Anaplastic Carcinoma (Chapter 73) In: Amin M.B., Edge S.B., Greene F., et al. (eds). AJCC Cancer Staging Manual. 8th (Springer International Publishing, New York City, 2017).
B.R. Haugen, E.K. Alexander, K.C. Bible, G.M. Doherty, S.J. Mandel, Y.E. Nikiforov, F. Pacini, G.W. Randolph, A.M. Sawka, M. Schlumberger, K.G. Schuff, S.I. Sherman, J.A. Sosa, D.L. Steward, R.M. Tuttle, L. Wartofsky, 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 26, 1–133 (2016). https://doi.org/10.1089/thy.2015.0020
V. Pecce, A. Verrienti, L. Abballe, R. Carletti, G. Grani, R. Falcone, V. Ramundo, C. Durante, C. Di Gioia, D. Russo, S. Filetti, M. Sponziello, Loss of function SETD2 mutations in poorly differentiated metastases from two hürthle cell carcinomas of the thyroid. Cancers 12, 1892 (2020). https://doi.org/10.3390/cancers12071892
M. Codrich, M. Comelli, M.C. Malfatti, C. Mio, D. Ayyildiz, C. Zhang, M.R. Kelley, G. Terrosu, C.E.M. Pucillo, G. Tell, Inhibition of APE1-endonuclease activity affects cell metabolism in colon cancer cells via a p53-dependent pathway. DNA Repair 82, 102675 (2019). https://doi.org/10.1016/j.dnarep.2019.102675
V. Pecce, M. Sponziello, G. Damante, F. Rosignolo, C. Durante, L. Lamartina, G. Grani, D. Russo, C.R. di Gioia, S. Filetti, A. Verrienti, A synonymous RET substitution enhances the oncogenic effect of an in-cis missense mutation by increasing constitutive splicing efficiency. PLoS Genet. 14 (2018). https://doi.org/10.1371/journal.pgen.1007678
A. Verrienti, A. Carbone, P. Bellitti, M.C. Fabiano, R.F. De Rose, M. Maranghi, P. Lucia, C. Durante, F. Rosignolo, V. Pecce, M. Sponziello, C. Puppin, G. Costante, R. Bruno, A novel double mutation val648ile and val804leu of ret proto-oncogene in multiple endocrine neoplasia type 2. Endocr. Pract. 21, 1248–1254 (2015). https://doi.org/10.4158/EP15838.OR
J. Jendrzejewski, S. Liyanarachchi, R. Nagy, L. Senter, P.E. Wakely, A. Thomas, F. Nabhan, H. He, W. Li, K. Sworczak, M.D. Ringel, L.S. Kirschner, A. de la Chapelle, Papillary thyroid carcinoma: association between germline DNA variant markers and clinical parameters. Thyroid 26, 1276–1284 (2016). https://doi.org/10.1089/thy.2015.0665
R.P. Tufano, G.V. Teixeira, J. Bishop, K.A. Carson, M. Xing, BRAF mutation in papillary thyroid cancer and its value in tailoring initial treatment: a systematic review and meta-analysis. Medicine 91, 274–286 (2012). https://doi.org/10.1097/MD.0b013e31826a9c71
R. Liu, J. Bishop, G. Zhu, T. Zhang, P.W. Ladenson, M. Xing, Mortality risk stratification by combining BRAF V600E and TERT promoter mutations in papillary thyroid cancer: genetic duet of BRAF and TERT promoter mutations in thyroid cancer mortality. JAMA Oncol. 3, 202–208 (2017). https://doi.org/10.1001/jamaoncol.2016.3288
J. Joo, K.-A. Yoon, T. Hayashi, S.-Y. Kong, H.-J. Shin, B. Park, Y.M. Kim, S.-H. Hwang, J. Kim, A. Shin, J.-Y. Kim, Nucleotide excision repair gene ERCC2 and ERCC5 variants increase risk of uterine cervical cancer. Cancer Res. Treat. 48, 708–714 (2016). https://doi.org/10.4143/crt.2015.098
I. Agalliu, M. Suuriniemi, L. Prokunina-Olsson, B. Johanneson, F.S. Collins, J.L. Stanford, E.A. Ostrander, Evaluation of a variant in the transcription factor 7-like 2 (TCF7L2) gene and prostate cancer risk, progression, and mortality in a population-based study. Prostate 68, 740–747 (2008). https://doi.org/10.1002/pros.20732
A. Hazra, C.S. Fuchs, A.T. Chan, E.L. Giovannucci, D.J. Hunter, Association of the TCF7L2 polymorphism with colorectal cancer and adenoma risk. Cancer Causes Control 19, 975–980 (2008). https://doi.org/10.1007/s10552-008-9164-3
S.F.A. Grant, G. Thorleifsson, I. Reynisdottir, R. Benediktsson, A. Manolescu, J. Sainz, A. Helgason, H. Stefansson, V. Emilsson, A. Helgadottir, U. Styrkarsdottir, K.P. Magnusson, G.B. Walters, E. Palsdottir, T. Jonsdottir, T. Gudmundsdottir, A. Gylfason, J. Saemundsdottir, R.L. Wilensky, M.P. Reilly, D.J. Rader, Y. Bagger, C. Christiansen, V. Gudnason, G. Sigurdsson, U. Thorsteinsdottir, J.R. Gulcher, A. Kong, K. Stefansson, Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 38, 320–323 (2006). https://doi.org/10.1038/ng1732
A. Rohlin, E. Rambech, A. Kvist, T. Törngren, F. Eiengård, U. Lundstam, T. Zagoras, S. Gebre-Medhin, Å. Borg, J. Björk, M. Nilbert, M. Nordling, Expanding the genotype–phenotype spectrum in hereditary colorectal cancer by gene panel testing. Fam. Cancer 16, 195–203 (2017). https://doi.org/10.1007/s10689-016-9934-0
H. Kobayashi, S. Ohno, Y. Sasaki, M: Matsuura, Hereditary breast and ovarian cancer susceptibility genes (Review). Oncol. Rep. 30, 1019–1029 (2013). 10.3892/or.2013.2541
M. Morak, A. Ibisler, G. Keller, E. Jessen, A. Laner, D. Gonzales-Fassrainer, M. Locher, T. Massdorf, A.M. Nissen, A. Benet-Pagès, E. Holinski-Feder, Comprehensive analysis of the MLH1 promoter region in 480 patients with colorectal cancer and 1150 controls reveals new variants including one with a heritable constitutional MLH1 epimutation. J. Med. Genet. 55, 240–248 (2018). https://doi.org/10.1136/jmedgenet-2017-104744
N. Köger, L. Paulsen, F. López‐Kostner, A.D. Valle, C.A. Vaccaro, E.I. Palmero, K. Alvarez, C. Sarroca, F. Neffa, P.G. Kalfayan, M.L. Gonzalez, B.M. Rossi, R.M. Reis, A. Brieger, S. Zeuzem, I. Hinrichsen, M. Dominguez‐Valentin, G. Plotz, Evaluation of MLH1 variants of unclear significance. Genes Chromosomes Cancer 57, 350–358 (2018). https://doi.org/10.1002/gcc.22536
M.F. Hansen, J. Johansen, A.E. Sylvander, I. Bjørnevoll, B.A. Talseth‐Palmer, L.A.S. Lavik, A. Xavier, L.F. Engebretsen, R.J. Scott, F. Drabløs, W. Sjursen, Use of multigene-panel identifies pathogenic variants in several CRC-predisposing genes in patients previously tested for Lynch Syndrome. Clin. Genet. 92, 405–414 (2017). https://doi.org/10.1111/cge.12994
R. Falcone, M. Sponziello, R. Carletti, C. Di Gioia, F. Nardi, C. Mio, V. Pecce, L. Abballe, G. Grani, V. Ramundo, G. Damante, C. Durante, M. Filetti, M. Roberto, P. Marchetti, A. Verrienti, Exploring the molecular insights of concurrent composite mucoepidermoid carcinoma and papillary thyroid carcinoma. Endocrine 68, 230–232 (2020). https://doi.org/10.1007/s12020-020-02221-8
Acknowledgements
We thank the Genomic Platform at the University of Udine for the services to support our research.
Funding
Open access funding provided by Università degli Studi di Udine within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
M.S. and G.D. conceptualized the study. C.M. and M.S. developed the method. C.M. performed the next-generation sequencing and wrote the manuscript draft. A.V. and V.P. performed the statistics and aided in data interpretation. G.D. supervised the project. All authors were major contributors to revision, editing, and proof-reading the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
This study was approved by the Ethics Committee of Azienda Universitaria Policlinico Umberto I, Rome, Italy.
Informed consent
Informed consent was obtained prior to samples collection.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mio, C., Verrienti, A., Pecce, V. et al. Rare germline variants in DNA repair-related genes are accountable for papillary thyroid cancer susceptibility. Endocrine 73, 648–657 (2021). https://doi.org/10.1007/s12020-021-02705-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-021-02705-1