
Abstract
Purpose
Adipokines produced by white adipose tissue are central in the development of lifestyle diseases. Individuals in industrialized countries spend a substantial part of life in the non-fasting, postprandial state, which is associated with increased oxidation and inflammation. The aim was to study postprandial adiponectin and leptin levels after an oral fat tolerance test (OFTT) and an oral glucose tolerance test (OGTT) in obese (OB) and healthy, normal weight individuals (NW).
Methods
Fifty adults with obesity (BMI ≥ 30) and 17 healthy, NW were included. Postprandial triglyceride (TG), adiponectin, and leptin levels were measured every second hour during an 8 h OFTT, and every half hour during a 2 h OGTT.
Results
Compared with the basal level, postprandial levels of adiponectin following OFTT showed a slight initial peak, followed by a significant decrease at 8 h, in the NW. In the OB these changes were abolished. Postprandial levels of leptin decreased significantly from basal levels in the OFTT, in the NW, whereas in the OB, leptin was unchanged except for a slight increase from 2 to 8 h. During the OGTT both adiponectin and leptin levels remained unchanged in the NW, but decreased significantly in the OB. In addition, the OB had delayed TG clearance at 6 h.
Conclusions
A fatty meal gives postprandial changes in the secretion of adiponectin and leptin in NW, but not in OB. Our observations indicate that a potential postprandial regulatory role of adiponectin and leptin is impaired in OB, and of importance in a more comprehensive understanding of the delayed postprandial TG clearance in obese individuals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Overweight and obesity are raising global health problems with several metabolic disturbances and comorbidities, such as type 2 diabetes and cardiovascular disease (CVD). CVD by itself is the leading cause of morbidity and mortality in industrialized countries, with obesity as an independent risk factor [1], and it was the main cause of death worldwide in 2012 [2].
In obesity the amount of white adipose tissue (WAT) is increased. WAT is a highly metabolically active endocrine organ (for review, see Ahima [3]). More than 600 adipokines have been described thus far [4], among which are leptin and adiponectin [3]. Increased levels of leptin, which is the case in obesity and leptin resistance (LR), are directly or indirectly associated to CVD [5]. In contrast, adiponectin, which is reduced in the obese and diabetic state, has shown to have protective and anti-atherogenic actions, opposing hyperglycemia, inflammation, lipotoxic damage, and insulin resistance (IR) [6, 7]. Furthermore, both leptin and adiponectin may facilitate responses of fibroblast growth factor 21 (FGF-21), which has effect on energy expenditure and whole-body glucose metabolism. FGF-21 is also a potent regulator of adiponectin secretion [8]. However, there are diverging reports about the role of adiponectin. In individuals with chronic illness and low body mass index (BMI), recent studies show that adiponectin might be associated with increased all-cause mortality, as well as increased cardiovascular mortality; the adiponectin-paradox [9,10,11]. Furthermore, adipokines play a pivotal role in the inflammation process and in the development of non-alcoholic-fatty liver disease (NAFLD) (for review, see Boutari [12]).
Individuals in industrialized countries spend a substantial part of life in the non-fasting, postprandial state, which is associated with increased oxidation and inflammation. Postprandial hyperlipidemia has been associated with overweight [13] and abdominal obesity [14,15,16,17] and is an independent risk factor for atherosclerosis. Furthermore, our knowledge of postprandial changes of leptin and adiponectin in normal weight and obese individuals is limited. The results are diverging, both for postprandial leptin and adiponectin secretion, with reports of no postprandial changes in leptin [18,19,20], as well as increased postprandial leptin levels in normal weight controls and decreased in obese individuals [21, 22]. For adiponectin, reports have found both increased [23,24,25] and unchanged [25,26,27,28,29] for both normal weight and obese individuals.
Due to the diverging results of the postprandial profile of adiponectin and leptin secretions both in normal weight and obese individuals, the aim of this study was to explore leptin and adiponectin in the postprandial state, in response to a fat and a carbohydrate load, separately, in obese and healthy, normal weight individuals.
Methods
Participants
Volunteers were recruited from the Centre of Obesity, Department of Gastroenterology, at the University Hospital of North Norway (UNN). The inclusion criteria for the obese individuals were BMI ≥ 30 kg/m2 and age 18–70 years. Exclusion criteria were pregnancy, current smoking, serious mental illness, and the use of medications to induce weight loss. The inclusion and exclusion criteria for the normal weight (BMI < 25 kg/m2) were the same, in addition to being normotensive, normoglycemic and to have triglycerides and cholesterol levels within the normal range. All of the study individuals had thyroxin (T4) and thyroid-stimulation hormone (TSH) levels within the normal range. All of the participants in the study were Caucasian.
Height, body weight, and waist circumference were measured. Blood pressure was measured three times on the right arm, after a 15-min rest. Appropriate cuff size was used. The mean of the two last measurements was used. All blood samples were collected at the laboratory, and at the same day, for the analysis of fasting glucose, total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, and fasting TG. The samples were taken from the antecubital vein, with the patient in a seated position. Serum lipids and apolipoprotein were measured according to a previous report from our group [30].
Dual X-ray absorptiometry (DEXA, Lunar Prodigy Advance, GE healthcare, USA) measurements were collected of all the study participants. The DEXA measured total fat percent, abdominal fat percent, total fat mass (kg), and total muscle mass (kg).
Oral fat tolerance test
The oral fat tolerance test (OFTT) has proven to be a good, indirect and qualitative measure of postprandial TG clearance [31]. OFTT was performed as previously described, whereas most of the data from normal weight individuals have been published previously [30]. In short, blood samples for serum TG were collected at baseline before the high-fat test meal (1 g fat per kg body weight) and thereafter every second hour over the next 8 h. The TG clearance at 6 h was calculated by the following formula: \(Clearance\,\left( {6h} \right) = 100 \ast \left( {1 - \frac{{TG\left( {6h} \right) \,-\, TG\left( {0h} \right)}}{{TG\left( {max} \right) \,-\, TG\left( {0h} \right)}}} \right)\). We have previously demonstrated that the postprandial TG clearance at 6 h was the most suitable measure [30].
Oral glucose tolerance test
A standard oral glucose tolerance test (OGTT) was conducted after 12 h fasting, using an oral intake of 75 g glucose as previously described [30]. Glucose and insulin were measured every half hour for 2 h, and the individuals were at rest during the whole test. Serum insulin was analyzed directly by ELISA (DRG Insulin Elisa kit, DRG Instruments GmbH, Germany). IR determination was done by the homeostasis model assessment for IR (HOMA-IR) [32,33,34], and was calculated as followed: HOMA-IR = Fasting insulin (μmol /L) × Fasting glucose (mmol/L)/22.5 [35].
Indirect leptin resistance
We wanted to measure indirect LR, for comparison between the normal weight and the obese individuals. Resting energy expenditure (REE) measurements were performed by a canopy test with an indirect calorimetry device from Medical Graphics CPX metabolic cart (St Paul, MN, USA). The indirect calorimetry was performed in a supine position. Before start, the O2 and CO2 analyzers were calibrated (a combined internal and manual adjustment system), based on the ambient temperature and barometric pressure. In addition, the breathing capacity analyzer was calibrated with a three-calibration syringe using multiple measures. Measurements were taken in a resting and fasting state for 30 min. REE was derived from the respiratory exchange ratio and the respiratory quotient. At the completion of the REE, blood samples for measurements of serum leptin and adiponectin were obtained. Indirect LR was measured as; REE to serum leptin ratio [36].
Measurements for adipokines
ELISA kits (DRG Diagnostics, Marburg, Germany) were used to analyze the adipokines leptin (sandwich ref. EIA-2395) and adiponectin (human, ref. EIA-4574) at baseline and postprandial during OFTT and OGTT.
Statistics
Statistics were calculated on IBM SPSS 25 for Windows (SPSS Inc., IBM Corporation, Armonk, New York, USA). Parametric statistics were performed when the raw data (fasting glucose, fasting leptin and L:A ratio) or transformed data (Indirect LR, HOMA-IR and WBISI), using log transformation, followed a normal distribution using; otherwise, non-parametric tests were used. Tests for independent or paired samples were used as appropriate. Two-sided p-values < 0.05 were considered statistically significant. Repeated measures analysis of variance (RM-ANOVA) was used to analyze the postprandial timeline for normal weight individuals and obese individuals. We also did a subgroup analysis of only females, 15 normal weight and 40 obese females. Corrections of violations of sphericity were used as appropriate, according to the epsilon value.
Results
Anthropometric, metabolic- and clinical characteristics
Fifty obese and 17 normal weight individuals was included in the study, of these 40 obese and 15 normal weight females. Of the obese individuals; five had elevated fasting TG (≥1.7 mmol/L), three patients had untreated hypertension (≥130/ ≥ 85 mmHg), six patients had reduced HDL cholesterol (Women < 1.29 mmol/l, men < 1.03 mmol/L), and six patients had elevated fasting glucose (≥5.6 mmol/L). Furthermore, ten individuals had hypertension regulated within the normal range with antihypertensive medication (Ibersartan, Hydrochlortiazide, Losartan, Amlodipin, Enalapril, Selo-Zok), five individuals had type 2 diabetes (regulated with lifestyle, no anti-diabetes medication), eight patients used lipid lowering drugs (Simvastatin, Atorvastatin), and four patients were treated for hypothyreosis. The obese patients that were treated for hypothyreosis had started their medication years ago, were well substituted with stable T4 and TSH, within the normal range, and their bodyweight was stable before the study. Anthropometric, metabolic- and clinical characteristics of obese individuals and normal weight individuals are shown in Table 1. As expected, there were several significant differences; especially a higher baseline adiponectin and lower baseline leptin in the normal weight individuals compared with the obese individuals. Furthermore, delayed postprandial TG clearance at 6 h, lower insulin sensitivity, and higher indirect LR, were found in obese individuals compared with normal weight individuals (Table 1). Fasting leptin (OFTT) had a significant, positive correlation to fasting insulin (rs = 0.446, p = 0.000) and fasting glucose (rs = 0.264, p = 0.048), but not to fasting TG (rs= 0.204, p = 0.112) or TG clearance 6 h (rs = −0.257, p = 0.053). Fasting adiponectin (OFTT) had a significant, negative correlation to fasting TG (rs = −0.333, p = 0.008), fasting insulin (rs = −0.542, p = 0.000), and significant positive correlation to TG clearance 6 h (rs = 0.314, p = 0.012), but not to fasting glucose (rs = −0.203, p = 0.127).
Oral fat tolerance test
Postprandial triglycerides after oral fat tolerance test
Normal weight individuals
When comparing fasting TG with postprandial TG levels, in normal weight individuals, there was a significant increase in TG at 2 h (p = 0.000), and close to significant increase at 4 h (p = 0.062) and 8 h (p = 0.084) during the OFTT (Fig. 1c and Supplementary Table 2).

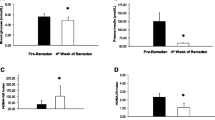
Oral fat tolerance test. Eight hours oral fat tolerance test in normal weight (circle) and obese (square) individuals measuring adiponectin (panel a), leptin (panel b), and triglycerides (panel c). Significant differences (p < 0.05) from baseline values are marked * in the separate groups
Obese individuals
When comparing fasting TG with postprandial TG levels, in obese individuals, there was a significant increase in TG at 2 h (p = 0.000), 4 h (p = 0.000) and 6 h (p = 0.000) (Fig. 1c and Supplementary Table 2).
Postprandial adiponectin after oral fat tolerance test
Normal weight individuals
For the healthy, normal weight individuals, when compared with baseline values there was a slight, non-significant, increase of adiponectin at 2 h (p = 0.052), and a subsequent significant decrease towards 8 h postprandial (p = 0.046) (Fig. 1a and Supplementary Table 2).
Normal weight individuals (females only)
For the healthy, normal weight individuals, when compared with baseline values there was a significant increase of adiponectin at 2 h (p = 0.026), and a subsequent non-significant decrease towards 8 h postprandial (p = 0.068).
Obese individuals
In the obese individuals, when compared with baseline values there were no significant differences in adiponectin levels postprandial during the OFTT (Fig. 1a and Supplementary Table 2).
Obese individuals (females only)
For the obese individuals, when compared with baseline values there were no significant differences in adiponectin levels postprandial during the OFTT.
Postprandial leptin after oral fat tolerance test
Normal weight individuals
When compared with baseline values the postprandial leptin levels were significantly decreased at 2 h (p = 0.001), 4 h (p = 0.001), 6 h (p = 0.001) and 8 h (p = 0.004). Compared with adiponectin an initial increase of leptin was not observed (Fig. 1b and Supplementary Table 2).
Normal weight individuals (females only)
When compared with baseline values the postprandial leptin levels were significantly decreased at 2 h (p = 0.001), 4 h (p = 0.001), 6 h (p = 0.001) and 8 h (p = 0.003). Compared with adiponectin an initial increase of leptin was not observed.
Obese individuals
When compared with baseline values the postprandial leptin levels were unchanged at all time points (Fig. 1b and Supplementary Table 2), but they had a slight, non-significant, increase at 8 h (p = 0.052).
Obese individuals (females only)
When compared with baseline values the postprandial leptin levels were unchanged at all time points, but they had a slight, non-significant, increase at 8 h (p = 0.054).
Oral glucose tolerance test
Postprandial adiponectin after oral glucose tolerance test
Normal weight individuals
When compared with baseline levels there was a non-significant trend towards increased adiponectin at 30 min (p = 0.064) (Fig. 2a and Supplementary Table 3).

Oral glucose tolerance test. Two hours oral glucose tolerance test in normal weight (circle) and obese (square) individuals measuring adiponectin (panel a), leptin (panel b), insulin (panel c), and glucose (panel d). Significant differences (p < 0.05) from baseline values are marked * in the separate groups
Normal weight individuals (females only)
When compared with baseline levels there were no significant changes in adiponectin during OGTT in females.
Obese individuals
Compared with baseline values of adiponectin there was a significant decrease of adiponectin at 90 min (p = 0.009) (Fig. 2a and Supplementary Table 3).
Obese individuals (females only)
When compared with baseline levels, adiponectin had a gradual and significant decrease at 90 min (p = 0.013), and also at 120 min (p = 0.009) during OGTT in females.
Postprandial leptin in oral glucose tolerance test
Normal weight individuals
When compared with the baseline values leptin decreased slightly (Fig. 2b and Supplementary Table 3), but not significant (n.s.).
Normal weight individuals (females only)
When compared with baseline levels there were no significant changes in adiponectin during OGTT in females.
Obese individuals
When compared with baseline levels, leptin had a gradual and significant decrease at all time points 30 min, 60 min, 90 min, and 120 min (all: p = 0.000) during OGTT (Fig. 2b and Supplementary Table 3).
Obese individuals (females only)
When compared with baseline levels, leptin had a gradual and significant decrease at all timepoints 30 min (p = 0.000), 60 min (p = 0.011), 90 min (p = 0.000), and 120 min (p = 0.001) during OGTT in obese females.
As expected, both insulin and glucose increased significantly at all time points in both groups during the OGTT (Fig. 2c, d). The obese individuals had significant higher basal (Table 1) and postprandial levels of both insulin and glucose, as expected. No measurements of glucose and insulin were done in the OFTT.
Discussion
In this study we report postprandial adiponectin, leptin, and TG responses after an 8 h fat load and a 2 h carbohydrate load in normal weight and obese individuals. In addition to insulin resistance, indirect leptin resistance was observed in the obese individuals, in addition to delayed TG clearance. In general, an apparent time effect of a postprandial suppression of leptin and adiponectin was observed in normal weight individuals in response to a fat load, whereas these regulations were more or less abolished in the obese individuals. These results were also supported by doing analysis only in normal weight and obese females. Our data indicate that adiponectin and leptin might have a postprandial regulatory role that can be overruled in obese individuals. These interactions are tightly associated to postprandial TG clearance in an apparent complex and not well understood regulatory mechanisms in the white adipose tissue.
The postprandial adiponectin levels observed in response to a fat load in our study is in conflict with other studies. In normal weight individuals, other studies have found postprandial adiponectin to be both increased [23, 24], or unchanged [25,26,27,28], whereas in obese individuals both increased [25] and unchanged levels [26, 29] have been observed. The early, slight increase of adiponectin seen in our study, after a fat load, indicates that there might be a triggered exocytic pathway in the adipocyte. This is supported from studies in mice, where a response time of 10–45 min for translocation of adiponectin to the plasma membranesome [8]. In mice exogenous adiponectin enhanced free fatty acid (FFA) oxidation by activating the adenosine monophosphate-activated protein kinase, to reduce the postprandial FFA increase [37, 38]. Based on these reports, and our observations, the exact physiological role of adiponectin is still hard to understand. Most likely, adiponectin appears to play a tuning role in FFA oxidation especially in fat tissue to accommodate storage of postprandial excess of TG, to enhance FFA oxidation in skeletal muscle, to improve insulin sensitivity and to suppress glucose production in liver (for review, see Wang [39]). This is mainly achieved by a large number of hormones released from each organ.
Recently, much attention has been on the FGF-21-adiponectin axis [8], which has been proposed to protect against a various cardio-metabolic disorders via mediating multi-organ communications (for review, see Hui [40]). It has been proposed that FGF-21 regulates postprandial lipid metabolism and permits better clearance of triglyceride-rich lipoprotein fractions [41], especially in healthy individuals, and that adiponectin might mediate this response. In our study there was a trend towards a slight, non-significant, increase of adiponectin 2 h in the OFTT, followed by a significant decrease, in normal weight individuals, whereas no changes in the obese individuals. Therefore, it is tempting to speculate that the fat induced response of adiponectin observed in the normal weight individuals in our study, could be explained by an FGF-21 mechanism, but this FGF-21-adiponectin axis is overruled in obesity, perhaps due to FGF-21 resistance. Furthermore, a report has also shown that impaired leptin signaling, in relation to increased caveolin-1-expression, in obesity, may prevent a concordant increase in adiponectin despite high levels of leptin [42]. This might indicate that leptin resistance and adiponectin resistance are connected in complex mechanisms. One might also speculate that leptin resistant individuals, has a different postprandial profile than leptin sensitive individuals. However, this awaits future studies that have the correct study design to explore this further. None of the individuals included in this study had heart failure, CVD, low BMI, or renal disease. In individuals with these conditions, adiponectin has shown to be high, and related to mortality; the adiponectin-paradox [9,10,11]. This is most likely because of higher levels of brain naturetic peptide, and is also connected to individuals with cachexia (for review, see Woodward [43]). It is unknown whether adiponectin resistance also has a role in this process. Further studies are needed on this patient group to explore postprandial adiponectin levels, but also postprandial leptin levels.
The postprandial leptin response to a fat load was significantly decreased at 2–8 h in normal weight individuals; on the contrary, the obese individuals had a slightly increased leptin at 8 h, however non-significant. These results contradict for the most other reports. For normal weight individuals, one study showed that leptin decreased at 6 h [44] such as in our study, unchanged in other studies [18, 19], whereas an increase was observed in other reports [20,21,22]. In obese individuals two reports showed a significant postprandial decrease in leptin [21, 22], as well as no postprandial leptin changes were observed in other studies [18, 19]. Finally, opposite to our observation of the secretion of adiponectin, no initial close to significant, increase of leptin was observed. This is in agreement with a report that human leptin secretion has a constitutively slow profile [45]. The discrepancies between these reports are hard to explain but can to some extent be explained by differences in the postprandial observation time, that for most studies were less than 3 h. In addition, the studies often have a small study group. Altogether, the exact postprandial physiological role of leptin, if any, is still unsettled. However, it is tempting to speculate that leptin increases the postprandial FFA oxidation (expenditure) in healthy, normal weight individuals, at least in muscle tissue [46], and that this is abolished in obese individuals with established leptin resistance. This may explain the delayed TG clearance; however, this awaits further studies.
There is increasing knowledge of adipocyte physiology that act to nutritional changes, that by systemic effect either can be beneficial, or harmful with various metabolic disturbances, such as in obesity. This is most likely a fine tuning of interactions in the adipokine and myokine secretome. Moreover, the cell biology of the fat expansion is complex, but the increased understanding of the pathophysiological changes in the fat tissue explaining the detrimental systemic effects (for review, see Rutkowski [47]). Our observations suggest that a postprandial increase of TG trigger a fine tuning adipokine response in normal weight individuals, but is overruled in obese patients with leptin resistance, and most likely also with adiponectin resistance [48].
The strengths of this study are, first, individuals were included from the everyday practice at the obesity out-patient clinic, which underlines the clinical utility and transferability of our observations. Second, the postprandial measurements of the adipokines were done over a long observation time of 8 h for the fat-load and 2 h for the carbohydrate load, and documented the TG clearance. Third, the number of study participants was higher than previous studies that have investigated postprandial adipokines. The most prominent weaknesses are, first, a lack of match between the groups studied according to number of individuals, sex and age. Moreover, we did not measure or record any type of exercise, nor did we monitor the diet, or intake of different types of fat, as for example N-3-PUFA in the period before the postprandial studies. Finally, a model of adipokine measurements directly in interstitial fat tissue is highly preferable to get a more precise postprandial response profile of adipokines.
Conclusion
In conclusion, postprandial changes were observed in both adiponectin and leptin suggesting a physiological role after a fatty meal in normal weight individuals. In obese individuals with leptin resistance and delayed TG clearance, these regulatory mechanisms seem to be overruled, but of importance in a more comprehensive understanding of the delayed postprandial TG clearance in obese individuals.
Abbreviations
- CVD:
-
Cardiovascular disease
- WAT:
-
White adipose tissue
- FGF-21:
-
Fibroblast growth factor 21
- L:A ratio:
-
Leptin to Adiponectin ratio
- HOMA-IR:
-
Homeostasis model assessment of insulin resistance
- NAFLD:
-
Non-alcoholic fatty liver disease
- TG:
-
Triglycerides
- BMI:
-
Body mass index
- PUFA:
-
Poly-unsaturated fatty acids
- UNN:
-
University Hospital of North Norway
- kg:
-
Kilograms
- T4:
-
Thyroxin
- TSH:
-
Thyroid-stimulation hormone
- NCEP/ATPIII:
-
National Cholesterol Education Panel/Adult Treatment panel
- LDL:
-
Low-density lipoprotein
- LR:
-
Leptin resistance
- HDL:
-
High-density lipoprotein
- DEXA:
-
Dual X-ray absorptiometry
- OGTT:
-
Oral glucose tolerance test
- OFTT:
-
Oral fat tolerance test
- REE:
-
Resting energy expenditure
- IR:
-
Insulin resistance
- RM-ANOVA:
-
Repeated measures analysis of variance
- n.s.:
-
Not significant
- FFA:
-
Free fatty acid
References
H.B. Hubert, M. Feinleib, P.M. McNamara, W.P. Castelli, Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham heart study. Circulation 67(5), 968–977 (1983)
S. Abu-Abid, A. Szold, J. Klausner, Obesity and cancer. J. Med. 33(1–4), 73–86 (2002)
R.S. Ahima, Adipose tissue as an endocrine organ. Obes. (Silver Spring) 14(Suppl 5), 242S–249S (2006). https://doi.org/10.1038/oby.2006.317
S. Lehr, S. Hartwig, H. Sell, Adipokines: a treasure trove for the discovery of biomarkers for metabolic disorders. Proteom. Clin. Appl. 6(1–2), 91–101 (2012). https://doi.org/10.1002/prca.201100052
J. Beltowski, Leptin and atherosclerosis. Atherosclerosis 189(1), 47–60 (2006). https://doi.org/10.1016/j.atherosclerosis.2006.03.003
T. Kadowaki, T. Yamauchi, N. Kubota, K. Hara, K. Ueki, K. Tobe, Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116(7), 1784–1792 (2006). https://doi.org/10.1172/JCI29126
A.T. Turer, P.E. Scherer, Adiponectin: mechanistic insights and clinical implications. Diabetologia 55(9), 2319–2326 (2012). https://doi.org/10.1007/s00125-012-2598-x
W.L. Holland, A.C. Adams, J.T. Brozinick, H.H. Bui, Y. Miyauchi, C.M. Kusminski, S.M. Bauer, M. Wade, E. Singhal, C.C. Cheng, K. Volk, M.S. Kuo, R. Gordillo, A. Kharitonenkov, P.E. Scherer, An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 17(5), 790–797 (2013). https://doi.org/10.1016/j.cmet.2013.03.019
C. Menzaghi, V. Trischitta, The adiponectin paradox for all-cause and cardiovascular mortality. Diabetes 67(1), 12–22 (2018). https://doi.org/10.2337/dbi17-0016
S.H. Choi, E.J. Ku, E.S. Hong, S. Lim, K.W. Kim, J.H. Moon, K.M. Kim, Y.J. Park, K.S. Park, H.C. Jang, High serum adiponectin concentration and low body mass index are significantly associated with increased all-cause and cardiovascular mortality in an elderly cohort, “adiponectin paradox”: the Korean longitudinal study on health and aging (KLoSHA). Int. J. Cardiol. 183, 91–97 (2015). https://doi.org/10.1016/j.ijcard.2015.01.057
J.R. Kizer, Adiponectin, cardiovascular disease, and mortality: parsing the dual prognostic implications of a complex adipokine. Metab.: Clin. Exp. 63(9), 1079–1083 (2014). https://doi.org/10.1016/j.metabol.2014.06.011
C. Boutari, N. Perakakis, C.S. Mantzoros, Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol. Metab. (Seoul.) 33(1), 33–43 (2018). https://doi.org/10.3803/EnM.2018.33.1.33
S. Tonstad, J.P. Despres, Treatment of lipid disorders in obesity. Expert Rev. Cardiovasc. Ther. 9(8), 1069–1080 (2011). https://doi.org/10.1586/erc.11.83
P. Blackburn, B. Lamarche, C. Couillard, A. Pascot, A. Tremblay, J. Bergeron, I. Lemieux, J.P. Despres, Contribution of visceral adiposity to the exaggerated postprandial lipemia of men with impaired glucose tolerance. Diabetes Care 26(12), 3303–3309 (2003)
R.R. Emmons, C.E. Garber, C.M. Cirnigliaro, J.M. Moyer, S.C. Kirshblum, M.D. Galea, A.M. Spungen, W.A. Bauman, The influence of visceral fat on the postprandial lipemic response in men with paraplegia. J. Am. Coll. Nutr. 29(5), 476–481 (2010)
N. Mekki, M.A. Christofilis, M. Charbonnier, C. Atlan-Gepner, C. Defoort, C. Juhel, P. Borel, H. Portugal, A.M. Pauli, B. Vialettes, D. Lairon, Influence of obesity and body fat distribution on postprandial lipemia and triglyceride-rich lipoproteins in adult women. J. Clin. Endocrinol. Metab. 84(1), 184–191 (1999). https://doi.org/10.1210/jcem.84.1.5397
G. Vansant, A. Mertens, E. Muls, Determinants of postprandial lipemia in obese women. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 23(Suppl 1), 14–21 (1999)
S. Dagogo-Jack, C. Fanelli, D. Paramore, J. Brothers, M. Landt, Plasma leptin and insulin relationships in obese and nonobese humans. Diabetes 45(5), 695–698 (1996)
M. Bueno, S. Esteba-Castillo, R. Novell, O. Gimenez-Palop, R. Coronas, E. Gabau, R. Corripio, N. Baena, M. Vinas-Jornet, M. Guitart, D. Torrents-Rodas, J. Deus, J. Pujol, M. Rigla, A. Caixas, Lack of postprandial peak in brain-derived neurotrophic factor in adults with Prader–Willi syndrome. PLoS ONE 11(9), e0163468 (2016). https://doi.org/10.1371/journal.pone.0163468
M.P. Maziarz, S. Preisendanz, S. Juma, V. Imrhan, C. Prasad, P. Vijayagopal, Resistant starch lowers postprandial glucose and leptin in overweight adults consuming a moderate-to-high-fat diet: a randomized-controlled trial. Nutr. J. 16(1), 14 (2017). https://doi.org/10.1186/s12937-017-0235-8
E. Korek, H. Krauss, M. Gibas-Dorna, J. Kupsz, M. Piatek, J. Piatek, Fasting and postprandial levels of ghrelin, leptin and insulin in lean, obese and anorexic subjects. Prz. Gastroenterol. 8(6), 383–389 (2013). https://doi.org/10.5114/pg.2013.39922
P. Imbeault, E. Doucet, P. Mauriege, S. St-Pierre, C. Couillard, N. Almeras, J.P. Despres, A. Tremblay, Difference in leptin response to a high-fat meal between lean and obese men. Clin. Sci. (Lond.) 101(4), 359–365 (2001)
G. Musso, R. Gambino, M. Durazzo, G. Biroli, M. Carello, E. Faga, G. Pacini, F. De Michieli, L. Rabbione, A. Premoli, M. Cassader, G. Pagano, Adipokines in NASH: postprandial lipid metabolism as a link between adiponectin and liver disease. Hepatology 42(5), 1175–1183 (2005). https://doi.org/10.1002/hep.20896
A. Kennedy, J.P. Spiers, V. Crowley, E. Williams, F.E. Lithander, Postprandial adiponectin and gelatinase response to a high-fat versus an isoenergetic low-fat meal in lean, healthy men. Nutrition 31(6), 863–870 (2015). https://doi.org/10.1016/j.nut.2015.01.009
P.J. English, S.R. Coughlin, K. Hayden, I.A. Malik, J.P. Wilding, Plasma adiponectin increases postprandially in obese, but not in lean, subjects. Obes. Res. 11(7), 839–844 (2003). https://doi.org/10.1038/oby.2003.115
L.K. Phillips, J.M. Peake, X. Zhang, I.J. Hickman, D.R. Briskey, B.E. Huang, P. Simpson, S.H. Li, J.P. Whitehead, J.H. Martin, J.B. Prins, Postprandial total and HMW adiponectin following a high-fat meal in lean, obese and diabetic men. Eur. J. Clin. Nutr. 67(4), 377–384 (2013). https://doi.org/10.1038/ejcn.2013.49
P.W. Peake, A.D. Kriketos, G.S. Denyer, L.V. Campbell, J.A. Charlesworth, The postprandial response of adiponectin to a high-fat meal in normal and insulin-resistant subjects. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 27(6), 657–662 (2003). https://doi.org/10.1038/sj.ijo.0802289
A. Lozano, P. Perez-Martinez, C. Marin, F.J. Tinahones, J. Delgado-Lista, C. Cruz-Teno, P. Gomez-Luna, F. Rodriguez-Cantalejo, F. Perez-Jimenez, J. Lopez-Miranda, An acute intake of a walnut-enriched meal improves postprandial adiponectin response in healthy young adults. Nutr. Res. 33(12), 1012–1018 (2013). https://doi.org/10.1016/j.nutres.2013.08.010
T.D.S. Ferreira, V.P. Antunes, P.M. Leal, A.F. Sanjuliani, M. Klein, The influence of dietary and supplemental calcium on postprandial effects of a high-fat meal on lipaemia, glycaemia, C-reactive protein and adiponectin in obese women. Br. J. Nutr. 118(8), 607–615 (2017). https://doi.org/10.1017/S0007114517002525
M.A. Larsen, R. Goll, S. Lekahl, O.S. Moen, J. Florholmen, Delayed clearance of triglyceride-rich lipoproteins in young, healthy obese subjects. Clin. Obes. 5(6), 349–357 (2015). https://doi.org/10.1111/cob.12118
J.C. Cohen, Chylomicron triglyceride clearance: comparison of three assessment methods. Am. J. Clin. Nutr. 49(2), 306–313 (1989)
A. Esteghamati, H. Ashraf, O. Khalilzadeh, A. Zandieh, M. Nakhjavani, A. Rashidi, M. Haghazali, F. Asgari, Optimal cut-off of homeostasis model assessment of insulin resistance (HOMA-IR) for the diagnosis of metabolic syndrome: third national surveillance of risk factors of non-communicable diseases in Iran (SuRFNCD-2007). Nutr. Metab. 7, 26 (2010). https://doi.org/10.1186/1743-7075-7-26
P. Gayoso-Diz, A. Otero-Gonzalez, M.X. Rodriguez-Alvarez, F. Gude, F. Garcia, A. De Francisco, A.G. Quintela, Insulin resistance (HOMA-IR) cut-off values and the metabolic syndrome in a general adult population: effect of gender and age: EPIRCE cross-sectional study. BMC Endocr. Disord. 13, 47 (2013). https://doi.org/10.1186/1472-6823-13-47
P. Gayoso-Diz, A. Otero-Gonzalez, M.X. Rodriguez-Alvarez, F. Gude, C. Cadarso-Suarez, F. Garcia, A. De Francisco, Insulin resistance index (HOMA-IR) levels in a general adult population: curves percentile by gender and age. The EPIRCE study. Diabetes Res. Clin. Pract. 94(1), 146–155 (2011). https://doi.org/10.1016/j.diabres.2011.07.015
D.R. Matthews, J.P. Hosker, A.S. Rudenski, B.A. Naylor, D.F. Treacher, R.C. Turner, Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28(7), 412–419 (1985)
R.H. Lustig, S. Sen, J.E. Soberman, P.A. Velasquez-Mieyer, Obesity, leptin resistance, and the effects of insulin reduction. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 28(10), 1344–1348 (2004). https://doi.org/10.1038/sj.ijo.0802753
T. Yamauchi, J. Kamon, Y. Minokoshi, Y. Ito, H. Waki, S. Uchida, S. Yamashita, M. Noda, S. Kita, K. Ueki, K. Eto, Y. Akanuma, P. Froguel, F. Foufelle, P. Ferre, D. Carling, S. Kimura, R. Nagai, B.B. Kahn, T. Kadowaki, Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8(11), 1288–1295 (2002). https://doi.org/10.1038/nm788
J. Fruebis, T.S. Tsao, S. Javorschi, D. Ebbets-Reed, M.R. Erickson, F.T. Yen, B.E. Bihain, H.F. Lodish, Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. USA 98(4), 2005–2010 (2001). https://doi.org/10.1073/pnas.041591798
Z.V. Wang, P.E. Scherer, Adiponectin, the past two decades. J. Mol. Cell Biol. 8(2), 93–100 (2016). https://doi.org/10.1093/jmcb/mjw011
X. Hui, T. Feng, Q. Liu, Y. Gao, A. Xu, The FGF21-adiponectin axis in controlling energy and vascular homeostasis. J. Mol. Cell Biol. 8(2), 110–119 (2016). https://doi.org/10.1093/jmcb/mjw013
A. Iroz, A. Montagner, F. Benhamed, F. Levavasseur, A. Polizzi, E. Anthony, M. Regnier, E. Fouche, C. Lukowicz, M. Cauzac, E. Tournier, M. Do-Cruzeiro, M. Daujat-Chavanieu, S. Gerbal-Chalouin, V. Fauveau, S. Marmier, A.F. Burnol, S. Guilmeau, Y. Lippi, J. Girard, W. Wahli, R. Dentin, H. Guillou, C. Postic, A specific ChREBP and PPARalpha cross-talk is required for the glucose-mediated FGF21 response. Cell Rep. 21(2), 403–416 (2017). https://doi.org/10.1016/j.celrep.2017.09.065
P. Singh, P. Sharma, K.R. Sahakyan, D.E. Davison, F.H. Sert-Kuniyoshi, A. Romero-Corral, J.M. Swain, M.D. Jensen, F. Lopez-Jimenez, T. Kara, V.K. Somers, Differential effects of leptin on adiponectin expression with weight gain versus obesity. Int J. Obes. (Lond.) 40(2), 266–274 (2016). https://doi.org/10.1038/ijo.2015.181
L. Woodward, I. Akoumianakis, C. Antoniades, Unravelling the adiponectin paradox: novel roles of adiponectin in the regulation of cardiovascular disease. Br. J. Pharm. 174(22), 4007–4020 (2017). https://doi.org/10.1111/bph.13619
S.D. Poppitt, F.E. Leahy, G.F. Keogh, Y. Wang, T.B. Mulvey, M. Stojkovic, Y.K. Chan, Y.S. Choong, B.H. McArdle, G.J. Cooper, Effect of high-fat meals and fatty acid saturation on postprandial levels of the hormones ghrelin and leptin in healthy men. Eur. J. Clin. Nutr. 60(1), 77–84 (2006). https://doi.org/10.1038/sj.ejcn.1602270
P.G. Cammisotto, M. Bendayan, Leptin secretion by white adipose tissue and gastric mucosa. Histol. Histopathol. 22(2), 199–210 (2007). https://doi.org/10.14670/HH-22.199
Y. Minokoshi, C. Toda, S. Okamoto, Regulatory role of leptin in glucose and lipid metabolism in skeletal muscle. Indian J. Endocrinol. Metab. 16(Suppl 3), S562–S568 (2012). https://doi.org/10.4103/2230-8210.105573
J.M. Rutkowski, J.H. Stern, P.E. Scherer, The cell biology of fat expansion. J. Cell Biol. 208(5), 501–512 (2015). https://doi.org/10.1083/jcb.201409063
A. Engin, Adiponectin-resistance in obesity. Adv. Exp. Med Biol. 960, 415–441 (2017). https://doi.org/10.1007/978-3-319-48382-5_18
Acknowledgements
We are grateful to expert assistance performed by Marian Remijn, Line Wilsgaard and Odd-Sverre Moen at the laboratory, UNN.
Funding
This study was funded by the Fund of Gastroenterology, UNN and by the Northern Norway Regional Health Authority (Helse Nord RHF) (ID: SPF 1200-14), Norway.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study was approved by The Regional Committee for Medical and Health Research Ethics of Northern Norway (2007, ID: 200704595-10/MRO/400) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The data bank approved by Norwegian Social Science Data Services (ID: 2206), registered on the 15th of April 2008.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Larsen, M.A., Isaksen, V.T., Paulssen, E.J. et al. Postprandial leptin and adiponectin in response to sugar and fat in obese and normal weight individuals. Endocrine 66, 517–525 (2019). https://doi.org/10.1007/s12020-019-02102-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-019-02102-9