
Abstract
The recently described therapeutic benefits of the hemodialysate actovegin on neuropathic symptoms in diabetic patients with symptomatic polyneuropathy suggest a neuroprotective activity of the drug. To elucidate the possible cellular mechanism of the pharmacological effects of actovegin, we investigated its effects on cultured primary rat neurons in vitro. Primary neurons were cultured for up to 10 days in the presence of increasing doses of actovegin (0.3–1,000 mg/l). Total cell number, dendrite length and the number of excitatory synapses, i.e., the amount of the synaptic V-Glut1 protein, were measured by immunocytochemistry followed by fluorescence microscopy. The apoptotic level in neurons after induction of apoptosis by amyloid peptide Aβ25–35 was assessed by the level of activated caspase-3. In addition, the capability of the neurons to diminish oxidative stress was assessed by measuring the cellular level of reactive oxygen species ROS in the presence of actovegin. Actovegin treatment yielded an increased maintenance of neuronal cells and total number of synapses and could lower the level of activated caspase-3 in a dose-dependent manner. Dendrite lengths were not significantly affected. In addition, actovegin reduced the cellular level of ROS in cultured neurons. The cellular effects observed suggest neuroprotective and anti-oxidative effects of the drug Actovegin®, which could at least partially explain its therapeutic benefits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic distal symmetric polyneuropathy (DPN) affects approximately one-third of diabetic patients and is associated with substantial morbidity including excruciating neuropathic pain and foot ulcers leading to amputation (Ziegler et al. 2008; Boulton et al. 2005). Analgesics are effective in the treatment of neuropathic pain (Dworkin et al. 2007), but do not slow down the progression of the underlying neuropathy (Boulton et al. 2005). Various therapeutic approaches to treat DPN have been developed (Cameron et al. 2001), which address the pathology of the disorder, rather than just relieve pain (Ziegler et al. 1995; Boulton et al. 2005; Chalk et al. 2007). However, despite apparent recent progress, a potent sustainable therapy of DPN still remains an unsolved medical need.
Actovegin, a deproteinized hemodialysate produced from calf blood, containing low molecular weight compounds of up to 5,000 Da, has been shown to have substantial therapeutic benefits in DPN. Recently, a randomized, double-blind, placebo-controlled clinical trial with sequential intravenous and oral actovegin treatment of 567 patients with DPN was conducted over a period of 160 days (Ziegler et al. 2009). Actovegin treatment significantly improved neuropathic symptoms like vibration perception threshold, sensory function and quality of life of the DPN patients. The hemodialysate is approved as a drug (Actovegin®) in a number of countries and is applied to treat diabetic polyneuropathy and other diseases (for review see: Buchmayer et al. 2011).
The goal of the present investigations was to elucidate the cellular effects, by which actovegin may exert its beneficial therapeutic effects in DPN. Well-known pharmacological actions of actovegin are the stimulation of oxygen absorption, oxygen utilization, and cellular energy metabolism (Obermaier-Kusser et al. 1989; Buchmayer et al. 2011). Furthermore, it exerts insulin-like activity, such as stimulation of glucose transport, pyruvate dehydrogenase and glucose oxidation (Jacob et al. 1996). Because of these properties, actovegin has previously been used for treatment of cerebral vascular and degenerative disorders (Kanowski et al. 1995; Herrmann et al. 1992). In addition, hemodialysates administered as infusions have shown beneficial effects on the clinical signs of dementia (Schlaffer et al. 1991; Beiswenger et al. 2008), which may indicate regenerative effects on neuronal tissue.
The present experimental approach was based on earlier studies with a rat model of diabetic polyneuropathy, which showed a reduction in intraepidermal nerve fiber density in skin biopsies, as well as a decrease in the loss of intraepidermal innervation upon therapeutic interventions (Bianchi et al. 2004). Here, the cellular effects of increasing doses of the hemodialysate actovegin were assessed in a neuronal in vitro model, namely in cultures of dissociated hippocampal neurons from embryonic rat brains. Cultured embryonic rat neurons constitute an accepted and relevant model system in the scientific community and have been widely used to evaluate the effects of pharmacologic and genetic interventions. Moreover, well-described culturing protocols and procedures for phenotypic characterization of cultures of embryonal rat neurons exist (Araujo et al. 2004; Burkarth et al. 2007; Busciglio et al. 1995; Cambon et al. 2004; Maar et al. 1997; Neiiendam et al. 2004; Skibo et al. 2005; Walicke et al. 1986). The following morphometric parameters were measured: the number of viable neurons, neurite outgrowth and synaptic connectivity. In parallel experiments, we assessed the anti-apoptotic effect of actovegin by measuring the levels of caspase-3 in response to amyloid peptide (Aβ25–35). In addition, we assessed the anti-oxidative effect of actovegin by measuring the level of reactive oxygen species (ROS).
Materials and Methods
Preparation of Neuronal Cultures
Dissociated cultures of rat hippocampal neurons were prepared according to a standard protocol (Maar et al. 1997). Hippocampal tissue from embryonic day 18 rat fetuses was collected in sterile Hanks’ balanced salt solution (HBSS buffer [Invitrogen, Karlsruhe, Germany] containing 7 mM HEPES, pH 7.3) and trypsinized using 1× trypsin–EDTA (PAA, Pasching, Austria) containing 10 mM HEPES, pH 7.3. The serum-free medium and culture conditions were tailored for the growth of neuronal cells in order to minimize the growth of contaminating other, e.g., glial cell types. Combination of nuclear staining and phase contrast microscopy ensured that the numbers of cells counted reflect changes in neuronal cell numbers. Subsequently, tissue was triturated in HBSS–HEPES buffer until no tissue clumps were visible. Cell number was determined and 2 × 104 cells/well were seeded in 100 μl NMEM-B27 medium (Burkarth et al. 2007; Goetze et al. 2003) into poly-l-lysine-coated 96-well plates (Greiner Bio-One, Frickenhausen, Germany) and cultured at 37°C and 5% CO2.
Actovegin Treatment of Rat Hippocampal Neurons
Rat hippocampal neurons were treated with increasing doses (0.3, 1, 3, 10, 30, 100, 300, 1,000 μg/ml) of actovegin (Actovegin®, Nycomed, Linz, Austria) by the addition of 50 μl of prediluted stock solution to the cultures, 5 h after plating. A broad dose range was tested, as it was not possible to define a dose that relates to the dose applied in the therapy of diabetic patients. Cells were cultivated at 37°C and 5% CO2 in a humidified atmosphere. Actovegin treatment effects were explored with respect to culture maintenance and synaptic connectivity, neurite outgrowth and neuroprotection (i.e., protection against Aβ25–35-induced apoptosis). Furthermore, the protection by actovegin from oxidative stress (ROS levels) was tested in the rat neurons.
Immunocytochemistry
For immunocytochemical labeling of dendrites with antibodies against microtubule-associated protein 2 (MAP2; at days 3 and 6 of in vitro culture) to be used in subsequent analysis of neurite outgrowth, and excitatory synapses (Vesicular Glutamate Transporter 1 (VGlut1); day 10) for the purpose of assessment of synaptic connectivity, cultivated hippocampal neurons were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature.
Cells were washed once with PBS and then blocked and soaked with blocking reagent (Roche Diagnostics GmbH, Mannheim, Germany) containing 0.2% Triton X-100 (Sigma-Aldrich, Steinheim, Germany) for 1 h at room temperature. Primary antibodies against microtubule-associated protein 2 (MAP2; Sigma-Aldrich) and vesicular glutamate transporter 1 (VGlut1; Synaptic Systems, Göttingen, Germany) were diluted [1:1,000] in blocking reagent containing 0.2% Triton X-100 and incubated with the cells at 4°C overnight. Cells were washed with PBS, and primary antibodies were detected with Cy3-conjugated goat anti-mouse IgG (diluted 1:600; Dianova, Hamburg, Germany) during a 2-h incubation at room temperature. Nuclei were stained with Hoechst 33258 (1 mg/ml; Invitrogen GmbH, Darmstadt, Germany). The working concentration of 1 μg/ml was achieved by a 1:1,000 dilution in PBS. Cells were washed and stored at 4°C until examination by fluorescence microscopy. Image acquisition was carried out on a Zeiss Axiovert 200.
Cell Number, Neurite Outgrowth and Synaptic Connectivity of Rat Hippocampal Neurons
Average neurite lengths of MAP2-labeled neuronal cultures were determined using the ImageJ-Plugin Neurite Average (Natural and Medical Sciences Institute, Reutlingen, Germany), which employs the principles of stereology (Ronn et al. 2000). Determination of the number of synaptic connections of VGlut1-labeled neuronal cultures and counting of cell numbers was carried out using ImageJ’s Analyze Particles function: http://rsb.info.nih.gov/ij/.
Briefly, Neurite Average projects a virtual grid onto micrographs of MAP2-labeled neuronal cultures and determines the number of intersections of this grid with MAP2-positive dendritic structures. The number of these intersections corresponds to the degree of dendritic arborization at a given time point. Micrographs of either Hoechst 33258- or VGlut1-stained neuronal cultures were thresholded and binearized and the resulting signals were counted.
Aβ25–35 Treatment and Apoptosis Induction of Rat Hippocampal Neurons
Rat hippocampal neurons were cultivated until day 8, and cell culture medium was aspirated; 100 μl of Aβ25–35 stock solution (Bachem Distribution Services GmbH, Weil am Rhein, Germany; 225 μM in cell culture medium) was added to each well, and desired actovegin concentrations were adjusted by the addition of 50 μl of a prediluted actovegin stock solution (final Aβ25–35 concentration, 150 μM). Plates were further cultivated until day 10, the time point for the assessment of apoptosis induction. Quantitation of apoptotic levels was carried out using the Caspase-Glo® 3/7 Assay (Promega GmbH, Mannheim, Germany) after induction of apoptosis with synthetic Aβ25–35. The cell culture medium was aspirated at day 10, and 50 μl PBS was added to each well. Cells were lysed by addition of 50 μl Caspase-Glo® 3/7 reagent and incubation for 30 min at room temperature. Luminescence signals (which were proportional to total caspase-3/7 activity) were recorded using a Pherastar microplate reader (BMG Labtech, Offenburg, Germany). For normalization purposes, cell numbers were quantitated in cultures set up and treated with Aβ25–35 peptides and actovegin in parallel using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). Briefly, medium was aspirated at day 10 and 50 μl PBS was added to each well. Cells were lysed by the addition of 50 μl CellTiter-Glo® Reagent and a 10-min incubation at room temperature. Luminescence signals were recorded using a Pherastar microplate reader. Activated caspase-3 levels were determined on a per-cell basis; data are reported as activated caspase-3 per cell. Each iteration of activated caspase-3 determination was normalized to three independent determinations of cell number.
Measurement of Oxidative Stress
Levels of oxidative stress, i.e., the level of ROS, in rat hippocampal neurons were determined at day 8 after tertiary-butyl hydroperoxide (TBHP) treatment to induce ROS production experimentally or day 10 after actovegin treatment, respectively. In the case of TBHP treatment, cell culture medium was replaced at day 8 by 100 μl of fresh NMEM-B27 and different concentrations of TBHP were added to the cell culture medium by addition of 50 μl of prediluted TBHP stock solution in NMEM-B27 to achieve final concentrations of 0.1, 0.2, 0.3, 0.5, 0.8, 1, 2 and 3 mM, respectively. Cells were incubated at 37°C and 5% CO2 for one hour before being subjected to ROS measurements.
For determination of cellular ROS levels, neuronal cultures, TBHP or actovegin treated, were washed twice with prewarmed HBSS buffer, and 50 μl of a 15-μM c-H2DCFDA stock solution was added after the last washing step (c-H2DCFDA concentration, 5 μM per well). Cells were incubated for 25 min at 37°C and 5% CO2, and excess c-H2DCFDA was washed off with prewarmed HBSS. Fluorescence intensity (proportional to total ROS content) was measured with a Pherastar microplate reader using a 485/520 nm filter set. In order to calculate ROS content on a per-cell basis, cell numbers were quantified in the same plate, directly following recording of the fluorescent signal using the CellTiter-Glo® Luminescent Cell Viability Assay described above.
Statistical Analysis
Experimental data sets were analyzed with the help of Statview 5.0 software (SAS-Institute Inc.) using one-way analysis of variance and Fisher’s PLSD (protected least significant difference) post hoc tests. At least three replicates were performed for all experiments. All results are presented as mean values ± SEM. Data were considered to be significantly different at P values < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Maintenance of Rat Primary Neurons
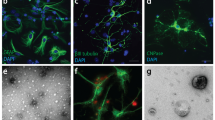
Following incubation of primary rat neurons for 10 days in the presence or absence of actovegin, cell numbers in actovegin-treated cultures (as quantified using nuclear staining) were significantly elevated up to 2.4-fold compared with untreated cultures (P < 0.001 at concentrations of actovegin greater than 10 μg/ml; Fig. 1a). The increase in the cell number observed was dose dependent. Micrographs of representative nuclear and synaptic staining of actovegin-treated and untreated cultures on day 10 are presented in Fig. 1c–f. Cultures that were inspected at day 7 and 9 in vitro using phase contrast microscopy revealed an improved maintenance of neuronal cell numbers and an increase in the density of the dendritic network in actovegin-treated compared with untreated cultures; this result was verified by manual counting of neuronal cells (Fig. 1g–n).

Evaluation of a cell number via nuclei staining and b total excitatory synapse number via vesicular glutamate transporter 1 (VGlut1) staining on day 10 in vitro following incubation of rat primary neurons with increasing concentrations of Actovegin® (Act.; 0.3–1,000 μg/ml) compared with untreated cells. Each plotted point represents the mean value obtained from six replicate wells; the error bars represent the SEM. ***P < 0.001, **P < 0.01; Fisher-PLSD test (comparison with untreated cells). c–f Show corresponding micrographs of representative cultures at day 10 in vitro treated with 1,000 μg/ml Actovegin® or left untreated (scale bar: 200 μm [C], 50 μm [E]). g–n: Representative phase contrast micrographs and manual determination of neurons in untreated (g–j) and Actovegin® 300 μg/ml-treated (k–n) primary neuronal cultures at day 7 and day 9 in vitro, respectively (scale bar in Fig. 1c: 200 μm; scale bars in Fig. 1e, g, k: 50 μm; DIV = days in vitro)
Synaptic Connectivity of Rat Primary Neurons
An antibody against VGlut1 (a marker protein for excitatory presynaptic terminals) was used in indirect immunocytochemistry to label excitatory synapses, the major synaptic type in the cultures analyzed. After 10 days of culture, the numbers of synaptic contacts in cultures of rat primary neurons were significantly elevated up to 3.6-fold in response to the administration of increasing doses of actovegin when compared to untreated cultures (P < 0.001 at actovegin doses greater than 300 μg/ml; Fig. 1b). Although the total number of synaptic contacts increased significantly as a result of an overall elevated neuronal cell number, the occurrence of synaptic contacts was not significantly increased by actovegin on a per-cell basis when compared to untreated cultures (Fig. 2). There was a decrease at a concentration of 1 μg/ml of actovegin.

Evaluation of vesicular glutamate transporter 1 (VGlut1)/cell ratio of rat primary neurons as calculated from VGlut1 and nuclear stainings on day 10 in vitro following incubation with increasing concentrations of Actovegin® (Act.; 0.3–1,000 μg/ml). Signals were compared with those of untreated cells. Each plotted point represents the mean value obtained from six replicate wells; the error bars represent the SEM. **P < 0.01; Fisher-PLSD test (comparison with untreated cells)
Neurite Outgrowth of Rat Primary Neurons
Neurite outgrowth of primary neurons treated with actovegin was assessed to test for a possible growth-promoting action. Addition of different doses of diluted actovegin to cultivated rat hippocampal neurons resulted in no significant change in neurite length compared with untreated cells, when measured after 3 and 6 days of culture (data not shown).
Neuroprotection of Rat Primary Neurons
We employed Aβ25–35 peptides for the induction of apoptosis in primary embryonic rat neurons to test for a potential anti-apoptotic effect of actovegin. Aβ25–35 represents the core fragment of Morbus Alzheimer-associated Aβ1–40 and Aβ1–42 amyloid peptides and is attributed to the latter’s neurotoxicity (Klementiev et al. 2007; Pike et al. 1995). Additionally, intra-cerebroventricular (i.c.v.) injection of Aβ25–35 into rodent brain results in neurotoxic effects that have been also observed after administration of full-length Aβ1-42 (Maurice et al. 1998; Stepanichev and Moiseeva et al. 2003a, b, Trubetskaya et al. 2003; Cheng et al. 2006). Most interestingly, an occurrence of Aβ25–35 as a cleavage product of Aβ1–40 in the brains of Alzheimer patients has been described recently (Kubo et al. 2002).
Aβ25–35 peptide-induced apoptosis in primary neurons cultured in the presence or absence of actovegin was determined by measuring the levels of activated caspase-3. Cell numbers were determined in cultures treated in parallel by using the CellTiter-Glo® assay. The aim was to normalize the levels of activated caspase-3, taking into account the increase in cell numbers observed with actovegin treatment. In the absence of Aβ25–35 peptides, levels of apoptosis induction in cells treated with increasing doses of actovegin were not consistently altered compared with untreated cells (Fig. 3). In the presence of Aβ25–35 peptides, rat primary neurons treated with increasing amounts of actovegin exhibited a significant decline in the induction of apoptosis (a decrease in the levels of activated caspase-3 per cell; P < 0.001 at concentrations of actovegin greater than 300 μg/ml). This was apparent at the mid- to high-dose range and followed a trend toward dose dependency (Fig. 3).

Evaluation of apoptosis induction on day 10 in vitro in rat neurons cultured in the presence of different concentrations of Actovegin® (Act.; 0.3–1,000 μg/ml) after treatment with 150 μM Aβ25–35 compared to controls. 150 μM Aβ25–35 was added on day 8; control/untreated cultures were included. Each plotted point represents the mean value obtained from three replicate wells; the error bars represent the SEM. ***P < 0.001, **P < 0.01; Fisher’s PLSD test
Oxidative Stress in Rat Primary Neurons
Preliminary experiments were carried out to test the suitability of a fluorescence-based assay system using c-H2DCFDA (5-[and-6]-carboxy-2′7′-dichlorodihydrofluorescein diacetate, Sigma-Aldrich) as a fluorogenic indicator to detect ROS in the cultures of primary rat neurons used. Treatment of neurons with increasing amounts of tert-butyl hydroperoxide (TBHP), a common inducer of ROS production, resulted in significantly elevated levels of intracellular ROS (P < 0.001 at concentrations of TBHP greater than 0.2 mM; Fig. 4a). The ability of TBHP to induce oxidative stress was dose dependent, with higher concentrations of TBHP resulting in higher levels of ROS generation. Thus, the cell-permeant fluorescent indicator of intracellular oxidative status c-H2DCFDA was confirmed as a suitable marker in the determination of levels of ROS in the neuronal culture system used.

Evaluation of levels of reactive oxygen species (ROS) in rat neurons after treatment with a increasing concentrations of tert-butyl hydroperoxide (TBHP) (0.1–3 mM) and b TBHP (3 mM) or increasing concentrations of Actovegin® (Act.; 0.3–1,000 μg/ml), compared with untreated cells. Each plotted point represents the mean value obtained from six replicate wells; the error bars represent the SEM. ***P < 0.001, *P < 0.05; Fisher-PLSD test
With respect to actovegin treatment, levels of ROS were determined in neuronal cultures grown for 10 days in vitro under the influence of actovegin to test for a possible effect on oxidative stress in neuronal cells. A significant reduction in ROS content (on a per-cell basis) was detected in cells cultivated in the presence of increasing amounts of actovegin (P < 0.001 at concentrations of actovegin greater than 0.3 μg/ml; Fig. 4b).
Discussion
In the present study, we used hippocampal neurons cultured from embryonic rat brains as an in vitro model to study drug effects on neuronal tissues. The neurons were treated with increasing actovegin concentrations in order to identify possible cellular effects, which may explain the beneficial therapeutic effects in patients with symptomatic diabetic polyneuropathy (DPN).
Briefly, we found that the hemodialysate actovegin has substantial dose-dependent protective effects on rat neurons over a wide range of concentrations. It significantly supported the maintenance of cultured neurons by increasing cell survival and by reducing apoptotic levels when challenged with the neurotoxic peptide Aβ25–35. In addition, it increased the total number of synaptic connections and decreased the burden of oxidative stress in the neurons. No significant effect of actovegin, however, was observed with respect to neurite outgrowth. The protective effects of actovegin observed in the in vitro model may in concert contribute to the therapeutic benefits of the drug actovegin, which was observed in a clinical trial (Ziegler et al. 2009). In the respective randomized, double-blind, placebo-controlled clinical trial, it was recently shown that sequential parenteral and oral treatment with actovegin was associated with a positive effect on neuropathic symptoms, vibration perception threshold, sensory nerve function and mental health-related quality of life in patients with type 2 diabetes and symptomatic polyneuropathy. Since nerve ischemia and hypoxia are believed to play a role in the pathogenesis of diabetic neuropathy, actovegin as a drug may protect against hypoxic cell injury, as previously observed in cultured hepatocytes (de Groot et al. 1990). The results of the clinical study (Ziegler et al. 2009) are in line with the data obtained in vitro presented herein. The increased survival and synaptic density of neurons cultured in vitro in the presence of Actovegin® suggest that protection and enhanced maintenance of neuronal networks may occur under conditions such as diabetic polyneuropathy or following trauma and stroke. The anti-apoptotic effect of Actovegin® observed in vitro suggests a neuroprotective effect in neurodegenerative disorders, such as Alzheimer’s disease (Schlaffer et al. 1991; Saletu et al. 1990; Semlitsch et al. 1990; Ziegler 2008).
As the neuronal cultures were derived from rat hippocampal tissue, it is possible that non-neuronal cell types such as astrocytes were present and may have contributed to the increase in cell numbers observed here. However, neuronal cells could be readily identified by phase contrast microscopy owing to their morphology, and an expansion of glial cells was not observed either in actovegin-treated or untreated cultures. Moreover, it is unlikely that the much higher cell numbers observed after actovegin treatment were due to an increase in numbers of non-neuronal cells, as there was no evidence for a change (i.e., reduction) in synaptic contacts on a per-cell basis as could have been caused by an increase in the number of non-neuronal cells.
In order to shed light on the nature of the active properties of Actovegin®, a number of experiments were conducted to assess the effects of treatment of rat primary neurons with increasing amounts of fetal calf serum (FCS), which is also derived from calf blood like actovegin. Here, a slight but statistically insignificant trend toward increased cell numbers and synaptic connections at submaximal doses of FCS was observed. This may argue for a stronger beneficial effect of Actovegin® due to an enrichment of the active physiological substance(s) during the production of Actovegin®.
Although cell numbers were found to be maintained in the presence of Actovegin®, experiments that explored the induction of apoptosis suggested that there was no effect of Actovegin® per se on reducing apoptosis when assessed on day 10 of culture in the absence of apoptosis-inducing agents like amyloid peptides. This apparent discrepancy suggests that loss of neuronal cells may occur through mechanisms other than apoptosis.
While analgesic drugs, such as antidepressants, anticonvulsants and opioids, are used for the symptomatic treatment of painful diabetic polyneuropathy, other compounds, such as antioxidants (alpha-lipoic acid) and aldose reductase inhibitors, have been developed to address the putative pathogenetic mechanisms of this condition (Boulton et al. 2005; Ziegler et al. 2004, 2006). Treatment with alpha-lipoic acid has been shown to reduce oxidative stress in vitro, to prevent nerve dysfunction in experimental diabetic neuropathy, and to reduce neuropathic symptoms and deficits in patients with diabetes (Cameron et al. 1998). Thus, the potential of Actovegin® to reduce oxidative stress levels in vitro as demonstrated here may at least partially explain its beneficial effect in patients with diabetes and symptomatic polyneuropathy (Ziegler et al. 2009). Results from previous clinical studies have demonstrated positive therapeutic effects of Actovegin® on age-associated memory impairment and dementia (Schlaffer et al. 1991; Saletu et al. 1990; Semlitsch et al. 1990; Ziegler et al. 2008). Compared with placebo, Actovegin® significantly provided better outcomes in assessments at the psychopathological, psychometric and behavioral levels. The experiments employing amyloid peptides described in the current study not only demonstrate the benefits of Actovegin® in protection against Aβ25–35-induced apoptosis, but also the relevance in the context of Alzheimer’s disease, in which the accumulation and neurotoxic action of amyloid peptides in the brain plays a role in the development of this neurodegenerative disorder. Interestingly, an increased level of reactive oxygen species (ROS) has also been observed after experimental addition of these peptides in earlier studies using Alzheimer’s models. Although not specifically tested in the present setting, it is likely that the reduction in apoptosis in the rat neurons is triggered by a reduction in ROS through Actovegin® (Dumont and Beal 2011; Nunomura et al. 2001).
It should, however, be kept in mind that, despite its clarity, the relevance of the current in vitro study for the in vivo beneficial effects of Actovegin® observed in patients with diabetic polyneuropathy is limited and needs to be further clarified by experimental in vivo approaches including animal and human studies. It has to be noted that the highest experimentally used concentrations of Actovegin, which are in the supra-physiological range, have shown a tendency to be less effective in respect to VGlut-1 expression and cell numbers.
In conclusion, the neuroprotective effects observed in vitro are in line with the results from previous clinical trials (Ziegler et al. 2009; Saletu et al. 1990; Semlitsch et al. 1990) and indicate that Actovegin® could offer a potential treatment option for neurodegenerative disorders.
References
Araujo, I. M., Ambrosio, A. F., et al. (2004). Neurotoxicity induced by antiepileptic drugs in cultured hippocampal neurons: A comparative study between carbamazepine, oxcarbazepine, and two new putative antiepileptic drugs, BIA 2–024 and BIA 2–093. Epilepsia, 45(12), 1498–1505.
Beiswenger, K. K., Calcutt, N. A., & Mizisin, A. P. (2008). Dissociation of thermal hypoalgesia and epidermal denervation in streptozotocin-diabetic mice. Neuroscience Letter, 442, 267–272.
Bianchi, R., Buyukakilli, B., Brines, M., Savino, C., Cavaletti, G., Oggioni, N., et al. (2004). Erythropoietin both protects from and reverses experimental diabetic neuropathy. Proceedings of the National Academy of Sciences of the USA, 101, 823–828.
Boulton, A. J. M., Vinik, A. I., Arezzo, J. C., Bril, V., Feldman, E. L., Freeman, R., et al. (2005). Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care, 28, 956–962.
Buchmayer, F., Pleiner, J., Elmlinger, M. W., Lauer, G., Neel, G., & Sitte, H. H. (2011). Actovegin®: A biological drug for more than 5 decades. Wiener Medizinische Wochenschrift, 161(3–4), 80–88.
Burkarth, N., Kriebel, M., Kranz, E. U., & Volkmer, H. (2007). Neurofascin regulates the formation of gephyrin clusters and their subsequent translocation to the axon hillock of hippocampal neurons. Molecular and Cellular Neuroscience, 36, 59–70.
Busciglio, J., Lorenzo, A., Yeh, J., & Yankner, B. A. (1995). Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron, 14, 879–888.
Cambon, K., Hansen, S. M., Venero, C., Herrero, A. I., Skibo, G., Berezin, V., et al. (2004). A synthetic neural cell adhesion molecule mimetic peptide promotes synaptogenesis, enhances presynaptic function, and facilitates memory consolidation. Journal of Neuroscience, 24, 4197–4204.
Cameron, N. E., Cotter, M. A., Horrobin, D. H., & Tritschler, H. J. (1998). Effects of alpha-lipoic acid on neurovascular function in diabetic rats: Interaction with essential fatty acids. Diabetologia, 41(4), 390–399.
Cameron, N. E., Eaton, S. E., Cotter, M. A., & Tesfaye, S. (2001). Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia, 44, 1973–1988.
Chalk, C., Benstead, T. J., & Moore, F. (2007). Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. The Cochrane Database of Systematic Reviews, 4, CD004572.
Cheng, G., Whitehead, S. N., et al. (2006). Effects of pyrrolidine dithiocarbamate on beta-amyloid (25–35)-induced inflammatory responses and memory deficits in the rat. Neurobiology of Diseases, 23(1), 140–151.
de Groot, H., Brecht, M., & Machicao, F. (1990). Evidence for a factor protective against hypoxic liver parenchymal cell injury in a protein-free blood extract. Esearch Communications in Chemical Pathology and Pharmacology, 68(1), 125–128.
Dumont, M., & Beal, M. F. (2011). Neuroprotective strategies involving ROS in Alzheimer disease. Free Radical Biology and Medicine, 51(5), 1014–1026.
Dworkin, R. H., O’Connor, A. B., Backonja, M., Farrar, J. T., Finnerup, N. B., Jensen, T. S., et al. (2007). Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain, 132, 237–251.
Goetze, B., Grunewald, B., Kiebler, M.A., & Macchi, P. (2003). Coupling the iron-responsive element to GFP–an inducible system to study translation in a single living cell. Sci STKE, (204), PL12.
Herrmann, W. M., Bohn-Olszewsky, W. J., & Kuntz, G. (1992). Infusionstherapie mit Actovegin bei Patienten mit primärer degenerativer Demenz vom Alzheimertyp und Multiinfarktdemenz. Ergebnisse einer prospektiven, placebo-kontrollierten Doppelblindstudie bei stationären Patienten. Z. Geriatrie, 5, 46–55. Article in German.
Jacob, S., Dietze, G. J., Machicao, F., Kuntz, G., & Augustin, H. J. (1996). Improvement of glucose metabolism in patients with type II diabetes after treatment with a hemodialysate. Arzneimittelforschung, 46, 269–272.
Kanowski, S., Kinzler, E., Lehmann, E., Schweizer, A., & Kuntz, G. (1995). Confirmed clinical efficacy of Actovegin in elderly patients with organic brain syndrome. Pharmacopsychiatry, 28(4), 125–133.
Klementiev, B., Novikova, T., Novitskaya, V., Walmod, P. S., Dmytriyeva, O., Pakkenberg, B., et al. (2007). A neural cell adhesion molecule-derived peptide reduces neuropathological signs and cognitive impairment induced by Abeta25–35. Neuroscience, 145(1), 209–224.
Kubo, T., Nishimura, S., et al. (2002). In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1–40) to truncated and toxic fragments ([D-Ser 26]A beta 25–35/40) and fragment presence in the brains of Alzheimer’s patients. Journal of Neuroscience Research, 70(3), 474–483.
Maar, T. E., Rønn, L. C., Bock, E., Berezin, V., Moran, J., Pasantes-Morales, H., et al. (1997). Characterization of microwell cultures of dissociated brain tissue for studies of cell–cell interactions. Journal of Neuroscience Research, 47, 163–172.
Maurice, T., Su, T. P., et al. (1998). Sigma1 (sigma 1) receptor agonists and neurosteroids attenuate B25–35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience, 83(2), 413–428.
Neiiendam, J. L., Køhler, L. B., Christensen, C., Li, S., Pedersen, M. V., Ditlevsen, D. K., et al. (2004). An NCAM-derived FGF-receptor agonist, the FGL-peptide, induces neurite outgrowth and neuronal survival in primary rat neurons. Journal of Neurochemistry, 91, 920–935.
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology and Experimental Neurology, 60(8), 759–767.
Obermaier-Kusser, B., Mühlbacher, C., Mushack, J., Seffer, E., Ermel, B., Machicao, F., et al. (1989). Further evidence for a two-step model of glucose-transport regulation Inositol phosphate-oligosaccharides regulate glucose-carrier activity. Biochemical Journal, 261, 699–705.
Pike, C. J., Walencewicz-Wasserman, A. J., et al. (1995). Structure-activity analyses of beta-amyloid peptides: Contributions of the beta 25–35 region to aggregation and neurotoxicity. Journal of Neurochemistry, 64(1), 253–265.
Ronn, L. C., Ralets, I., Hartz, B. P., Bech, M., Berezin, A., Berezin, V., et al. (2000). A simple procedure for quantification of neurite outgrowth based on stereological principles. Journal of Neuroscience Methods, 100(1–2), 25–32.
Saletu, B., Grünberger, J., Linzmayer, L., Anderer, P., & Semlitsch, H. V. (1990). EEG brain mapping and psychometry in age-associated memory impairment after acute and 2-week infusions with the hemoderivative Actovegin: Double-blind, placebo-controlled trials. Neuropsychobiology, 24(3), 135–148.
Schlaffer, K., Wauschkuhn, C. H., & Hauser, B. (1991). Study to evaluate the encephalotropic potency of a hemodialysate. Controlled study using electro-retinography and visual evoked potentials under hypoxic conditions in human volunteers (preliminary communication). Arzneimittelforschung, 41, 699–704.
Semlitsch, H. V., Anderer, P., Saletu, B., & Hochmayer, I. (1990). Topographic mapping of cognitive event-related potentials in a double-blind, placebo-controlled study with the hemoderivative Actovegin in age-associated memory impairment. Neuropsycho-Biology, 24(1), 49–56.
Skibo, G. G., Lushnikova, I. V., Voronin, K. Y., Dmitrieva, O., Novikova, T., Klementiev, B., et al. (2005). A synthetic NCAM-derived peptide, FGL, protects hippocampal neurons from ischemic insult both in vitro and in vivo. European Journal of Neuroscience, 22(7), 1589–1596.
Stepanichev, M. Y., Moiseeva, Y. V., et al. (2003a). Single intracerebroventricular administration of amyloid-beta (25–35) peptide induces impairment in short-term rather than long-term memory in rats. Brain Research Bulletin, 61(2), 197–205.
Stepanichev, M. Y., Zdobnova, I. M., et al. (2003b). Effects of tumor necrosis factor-alpha central administration on hippocampal damage in rat induced by amyloid beta-peptide (25–35). Journal of Neuroscience Research, 71(1), 110–120.
Trubetskaya, V. V., Stepanichev, M. Y., et al. (2003). Administration of aggregated beta-amyloid peptide (25–35) induces changes in long-term potentiation in the hippocampus in vivo. Neuroscience and Behavioral Physiology, 33(2), 95–98.
Walicke, P., Cowan, W. M., et al. (1986). Fibroblast growth factor promotes survival of dissociated hippocampal neurons and enhances neurite extension. Proceedings of the National Academy of Sciences of the USA, 83(9), 3012–3016.
Ziegler, D. (2008). Treatment of diabetic neuropathy and neuropathic pain: How far have we come? Diabetes Care, 31(2), S255–S261.
Ziegler, D., Hanefeld, M., Ruhnau, K. J., Meißner, H. P., Lobisch, M., Schütte, K., et al. (1995). The ALADIN Study Group: Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant-lipoic acid: A 3-week multicentre randomized controlled trial (ALADIN Study). Diabetologia, 38, 1425–1433.
Ziegler, D., Nowak, H., Kempler, P., Vargha, P., & Low, P. A. (2004). Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: A meta-analysis. Diabetic Medicine, 21(2), 114–121.
Ziegler, D., Ametov, A., Barinov, A., Dyck, P. J., Gurieva, I., Low, P. A., et al. (2006). Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: The SYDNEY 2 trial. Diabetes Care, 29(11), 2365–2370.
Ziegler, D., Rathmann, W., Dickhaus, T., Meisinger, C., & Mielck, A. (2008). The KORA Study Group. Prevalence of polyneuropathy in pre-diabetes and diabetes is associated with abdominal obesity and macroangiopathy: The MONICA/KORA Augsburg Surveys S2 and S3. Diabetes Care, 31, 464–469.
Ziegler, D., Movsesyan, L., Mankovsky, B., Gurieva, I., Abylaiuly, Z., & Strokov, I. (2009). Treatment of symptomatic polyneuropathy with actovegin in type 2 diabetic patients. Diabetes Care, 32, 1479–1484.
Acknowledgments
This study was sponsored by Nycomed GmbH, Konstanz, Germany. The authors thank Andreas Dieckmann, Tatiana Bobrova and Katrine Husum from Nycomed for their scientific input to the study and manuscript.
Conflict of interest
This study was sponsored by Nycomed GmbH, Konstanz, Germany. Martin Elmlinger is employee of Nycomed. Dan Ziegler has received honoraria for speaking and consulting activities from Nycomed GmbH. Martin Kriebel, i.e., the NMI Reutlingen, Germany, has no conflicts of interest to declare.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Elmlinger, M.W., Kriebel, M. & Ziegler, D. Neuroprotective and Anti-Oxidative Effects of the Hemodialysate Actovegin on Primary Rat Neurons in Vitro. Neuromol Med 13, 266–274 (2011). https://doi.org/10.1007/s12017-011-8157-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-011-8157-7