
Abstract
Psoriasis is one of the most common inflammatory skin diseases with a chronic, relapsing-remitting course. The last decades of intense research uncovered a pathological network of interactions between immune cells and other types of cells in the pathogenesis of psoriasis. Emerging evidence indicates that dendritic cells, TH17 cells, and keratinocytes constitute a pathogenic triad in psoriasis. Dendritic cells produce TNF-α and IL-23 to promote T cell differentiation toward TH17 cells that produce key psoriatic cytokines IL-17, IFN-γ, and IL-22. Their activity results in skin inflammation and activation and hyperproliferation of keratinocytes. In addition, other cells and signaling pathways are implicated in the pathogenesis of psoriasis, including TH9 cells, TH22 cells, CD8+ cytotoxic cells, neutrophils, γδ T cells, and cytokines and chemokines secreted by them. New insights from high-throughput analysis of lesional skin identified novel signaling pathways and cell populations involved in the pathogenesis. These studies not only expanded our knowledge about the mechanisms of immune response and the pathogenesis of psoriasis but also resulted in a revolution in the clinical management of patients with psoriasis. Thus, understanding the mechanisms of immune response in psoriatic inflammation is crucial for further studies, the development of novel therapeutic strategies, and the clinical management of psoriasis patients. The aim of the review was to comprehensively present the dysregulation of immune response in psoriasis with an emphasis on recent findings. Here, we described the role of immune cells, including T cells, B cells, dendritic cells, neutrophils, monocytes, mast cells, and innate lymphoid cells (ILCs), as well as non-immune cells, including keratinocytes, fibroblasts, endothelial cells, and platelets in the initiation, development, and progression of psoriasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Psoriasis is a chronic inflammatory skin disease whose prevalence varies from 0.1 to 8% depending on the geographical region and affects more than 125 million people worldwide [1,2,3]. The clinical features of psoriasis are heterogeneous and include cutaneous as well as systemic manifestations [3]. The most common variant of psoriasis is plaque psoriasis (psoriasis vulgaris) which accounts for more than 80% of cases [1]. Common symptoms include skin itching, burning, and soreness [3]. Moreover, several comorbidities have been associated with psoriasis including psoriatic arthritis, cardiovascular disease, metabolic syndrome, obesity, inflammatory bowel disease, and psychiatric disorders that are associated with systemic inflammation [3, 4].
The etiology of psoriasis is very complex and involves multiple intrinsic and extrinsic factors. Several genetic risk factors, including genes involved in antigen presentation (HLA-Cw6), cytokine signaling (IL12B, IL23R), interferon signaling, and NF-κB signaling (TNFAIP3, NFKBIA, NFKBIZ, TNIP1, and RELA) have been identified [3, 5,6,7]. Psoriasis is characterized by the activation of inflammatory pathways in both the innate and adaptive immune cells leading to the uncontrolled proliferation of keratinocytes (KCs), acanthosis, neovascularization, and potent skin infiltration by immune cells [8]. Over 80% of upregulated genes in psoriatic lesions are associated with activation of KCs and skin infiltration by T cells and macrophages [9]. Moreover, most significantly enriched transcripts are associated with immune response, defense response, and response to wounding [10].
Immune Response in Psoriasis
Skin is a complex immune organ that protects the organism from infection, enables wound healing, and interacts with the skin microbiome [11,12,13,14]. Moreover, it constitutes an important reservoir of immune cells and contains nearly twice the T cells present in the circulation [15]. Thus, disruption of the immune homeostasis may have detrimental effects on the human body and result in a variety of inflammatory diseases.
Psoriasis is characterized by the dysregulation of the cytokine network with multiple self-amplifying feeds accelerating pathogenic circuits. Psoriatic inflammation can be triggered in predisposed individuals by mechanical stress (Koebner phenomenon), air pollutants, sun exposure, drugs, infections, or vaccination [16]. It seems that pattern recognition receptors (PRRs), especially Toll-like receptors (TLRs), are crucial mediators of the response to these triggering factors [17]. Skin injury triggers the release of damage-associated molecular patterns (DAMPs) including dsRNA, ssRNA, and DNA from damaged cells which activate TLRs signaling in different types of cells, including KCs and dendritic cells (DCs) [18, 19]. Activation of TLRs by DAMPs or pathogen-associated molecular patterns (PAMPs) triggers the production of multiple cytokines and initiation of the psoriatic inflammation [19,20,21,22].
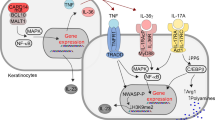
The first studies suggested that psoriasis is a classical TH1 inflammatory disease with IFN-γ as a key mediator of psoriatic inflammation [23]. Further intense studies revealed that it is associated with not only overactivated TH1 response but also TH17 and TH22 responses [24]. In general, proinflammatory cytokines and factors stimulating proliferation in psoriasis are produced mainly by T cells (IL-17, IL-21, IL-22, IFN-γ), DCs (TNF-α, IL-6, IL-20, IL-23, NO), and KCs (antimicrobial peptides (AMPs), IL-20, chemokines). These cells form a key pathogenic loop in psoriasis that involves a triad of IL-23-producing DCs, IL-17-producing TH17, and activated KCs (Fig. 1).

Dysregulation of immune response in psoriasis. Psoriasis may be triggered by different factors, including skin injury (Koebner phenomenon), infection, drugs, and autoantigens in predisposed individuals. In the early phase of psoriasis, neutrophils infiltrate the skin and release neutrophils extracellular traps (NETs), exosomes, metalloproteinase 9 (MMP9), and IL-17. Activated keratinocytes (KCs) have increased proliferative capacity and produce a variety of pro-inflammatory factors, including chemokines, antimicrobial peptides (AMPs), and alarmins. Dendritic cells (DCs) are activated by toll-like receptors (TLRs) ligands and AMPs which initiate T cell immune response. DCs activate a variety of T cells, including IL-17-producing TH17 and Tc17 cells, IL-9-producing TH9 cells, IFN-γ-producing TH1 cells, and IL-22-producing TH22 cells. Moreover, DCs and Langerhans cells (LCs) can present autoantigens stimulating autoreactive T cells. The impaired skin microbiome activates γδ T cells that produce IL-17 and IL-1β. In the late phase of inflammation, psoriatic lesions are characterized by the profound infiltration of immune cells, increased concentrations of multiple cytokines and chemokines, and hyperproliferation of KCs. Moreover, increased angiogenesis and endothelial cells activated by psoriatic cytokines facilitate the infiltration of immune cells into inflamed skin. Created with Biorender.com
Dysregulation of Cytokine Network Underlies Psoriasis Pathogenesis
Psoriasis has been recognized as a disease of dysregulated cytokine profile for over three decades [26]. Novel methods that enable the assessment of multiple cytokines [27] and transcriptomic genome-wide expression analysis [9, 28] uncovered a profoundly disrupted cytokine expression profile in psoriatic skin as well as a complex network between cells in psoriatic skin (Table 1).
Dysregulation of the cytokine profile is associated with overactivation of type 1, type 17, and type 22 pathways as well as innate inflammatory pathways (Fig. 2). TNF-α is the founder cytokine that initiates downstream inflammatory signaling in psoriasis. There are multiple triggers of TNF-α secretion, including skin injury, environmental stimuli, autoantigens, and TLRs agonists [105, 106]. In psoriatic skin, it is produced predominantly by activated T cells and antigen-presenting cells (APCs), including dermal DCs [102,103,104]. TNF-α synergizes with IFN-γ to induce the expression of chemokines and inflammatory adhesion molecules by endothelial cells which promote the infiltration of immune cells, especially T cells, into the skin [95]. Moreover, TNF-α triggers KCs and DCs to secrete IL-23 [53, 107].

Cytokine network and therapeutic targets in psoriasis. Immune response in psoriasis is associated with the activation of type 1, type 17, and type 22 pathways and innate inflammatory pathways. These signaling pathways induce the expression of different genes involved in the regulation of psoriatic inflammation (orange boxes). Several biological drugs targeting key psoriatic cytokines (indicated in red) or small molecule inhibitors of downstream cytokine signaling were developed and approved by the FDA. PsA, psoriasis arthritis; PP, pustular psoriasis. Created with Biorender.com
IL-23 is a heterodimer of p19 and p40 subunits. It is a key regulator of the type 17 pathway activating a variety of cells, including TH17, Tc17, γδ T cells, and innate lymphoid cells type 3 (ILC3) to produce IL-17 [108]. Moreover, IL-23 is a key cytokine that regulates the survival and pathogenic potential of TH17 cells [109,110,111,112].
TH17 cells orchestrate inflammation via multiple cytokines, especially IL-17. This cytokine regulates immune response to different pathogens and tissue repair processes. However, IL-17 is also implicated in a variety of TH17-mediated inflammatory autoimmune diseases [113, 114]. In lesional skin, the level of IL-17 family members, especially IL-17A, IL-17C, and IL-17F, is potently upregulated [25, 40, 80, 115,116,117,118]. It is caused by the potent infiltration, activation, and expansion of TH17 cells and positive feedback loops amplifying the production of IL-17 [119]. IL-17 exerts a variety of effects in inflamed psoriatic skin, including activation of KCs to produce AMPs, upregulation of ICAM-1 in endothelial cells to promote tissue inflammation and promotion of the infiltration of immune cells. Moreover, IL-17 potentiates inflammation by the induction of multiple pro-inflammatory cytokines and chemokines (Fig. 2) [40, 80, 115,116,117,118]. IL-17 synergizes with TNF-α to induce hyperproliferation of KCs, TH17-polarized inflammation, and upregulate psoriasis-related genes [61, 118, 120,121,122]. IL-17 also upregulates the level of psoriasis autoantigens which is potentiated by the vitamin D3 and IL-22 [123].
Chemokines and Homing Receptors Regulate Immune Cell Trafficking in Psoriasis
Profound infiltration of immune cells into psoriatic skin is caused by, among others, dysregulated chemokine signaling (Table 2). The expression of chemokines and their receptors are regulated by psoriasis-associated cytokines, including IL-17 which upregulates CCL2, CCL7, CCL20, and CXCL1, or IFN-γ which upregulates CXCL9 and CXCL10 [113, 124]. Notably, some of them, including CXCL9 and CXCL10, are upregulated even in non-lesional skin of psoriasis patients which may contribute to the initiation of novel skin lesions [125].
In psoriasis, T cells infiltrating lesional skin have substantially upregulated expression of chemokine receptors, including a skin-homing receptor, CCR4, and CCR6 [127]. Moreover, there is an increased fraction of CCR4+ and CCR6+ T cells in peripheral blood [70]. Due to increased expression of chemokine receptors, skin-homing T cells of psoriasis patients respond to lower concentrations of chemokines, including CCL20, and exhibit stronger chemotactic responses compared to T cells of healthy individuals [132].
Immune Cells in Psoriasis
Multiple types of immune cells regulate the initiation, maintenance, and progression of psoriatic inflammation. T cells, especially TH17 cells, together with DCs are the main players in the pathogenesis of psoriasis. Nonetheless, a variety of other types of immune cells, including neutrophils, monocytes, macrophages, mast cells, and ILCs participate in the pathogenesis of psoriasis (Fig. 3). In general, innate immune cells, especially neutrophils, are key cells in the early phases of psoriasis development while T cell-dominated adaptive inflammation is a feature of stable plaques in the later phases [144].

The role of immune cells in psoriasis. The key populations of immune cells and their functions in the regulation of psoriatic inflammation. Created with Biorender.com
The Role of Neutrophils
Neutrophils are a heterogeneous innate immune cell population that can either suppress or enhance immune response [145]. In psoriasis, neutrophils participate in the initiation of the disease and early phases of progression of psoriatic inflammation [146, 147]. Early lesions and prepsoriatic skin adjacent to active lesions are characterized by the potent infiltration of CD15+ neutrophils [148]. Chronic lesions are also infiltrated by neutrophils, especially CD15posCD10pos and CD15posCD10neg neutrophils [146] Neutrophils are also enriched in the peripheral blood of psoriatic patients and are in a pre-activated state [149]. They are chemoattracted to the inflamed psoriatic skin mainly by IL-17E and CXCL8 [149]. Additionally, neutrophils release IL-17A and extracellular vesicles that induce the expression of proinflammatory cytokines and chemokines in KCs which enhance their migratory phenotype and promote infiltration into the skin [150, 151]. Notably, infiltrating neutrophils overexpress matrix metallopeptidase (MMP)-9 that activates vascular endothelial cells, enhances vascular permeability, and promotes CD4+ T cells infiltration [152].
In psoriatic lesions, IL-17+ neutrophils together with IL-17+ mast cells (MCs) are found in higher densities than IL-17+ T cells [152]. It seems that neutrophils do not express IL-17A, but can accumulate it [61] and release it during the formation of neutrophil extracellular traps (NETs) [152]. Indeed, neutrophils in psoriasis frequently undergo NETosis, even without any stimulation [152]. Moreover, they can be triggered and activated in psoriatic skin by different types of cells, especially by KCs [151, 153]. DNA- and RNA-containing NETs are highly abundant in the psoriatic skin as well as peripheral blood of psoriasis patients [152, 154]. Notably, NETosis in peripheral blood correlates with the clinical severity of psoriasis patients [154]. Moreover, low density granulocytes circulating in psoriasis patients blood seems to be more sensitive to cutaneus stimuli cousing their release of NETs then conventional neutrophils, leading to skin pathology [155]. Released nucleic acids form complexes with LL37 which triggers NETs and cytokine release by neutrophils in TLR8/TLR13- or TLR4/IL-36R-mediated mechanisms [156, 157]. Moreover, the release of NETs and complexes of LL37 with DNA/RNA activates other types of immune cells, promotes an inflammatory response in KCs, and induces IL-17 secretion by T cells [146, 154, 157].
The Role of Monocytes and Macrophages
In murine models, psoriatic inflammation is associated with the expansion of activated monocytes and macrophages in lesional skin and draining lymph nodes [158, 159]. These activated macrophages are a key source of TNF-α [76, 158]. Likewise, the lesional skin of psoriasis patients is characterized by the accumulation of activated macrophages in both the epidermis and dermis, especially in the late phase of psoriatic inflammation [76, 160].
Macrophages in psoriasis are “classically activated” by IFN-γ and produce IL-23p19, IL-12/23p40, iNOS, and TNF-α [161]. Moreover, IL-17 activates the pro-inflammatory phenotype of monocytes/macrophages and sensitizes them toward pathogen-derived TLR4 ligands [162]. Monocytes are attracted to inflamed psoriatic skin by different chemokines and cytokines [76]. Moreover, there is an expansion of immunomodulatory immature myeloid cells called myeloid-derived suppressor cells (MDSCs), especially monocytic MDSCs (M-MDSCs), in the peripheral blood and skin lesions of psoriatic patients that correlate with the clinical severity of psoriasis [165]. However, the role of MDSCs in the pathogenesis of psoriasis is unknown.
The Role of Mast Cells
Mast cells are key effector cells in acute allergic reactions and inflammatory diseases [166, 167]. Moreover, their expansion and activation contribute to psoriatic inflammation [168]. In psoriasis, mast cells express and release IL-17 and IL-22 [152]. Notably, analysis of psoriatic skin biopsies revealed that mast cells constitute most of the IL-17-containing and IL-22-containing cells in psoriatic lesions [152]. Psoriatic mast cells degranulate and form mast cell-extracellular traps (MCETosis) after stimulation with IL-1β and IL-23 which trigger the release of IL-17 [72, 152]. Notably, mechanical injury induces a pro-inflammatory response of mast cells which secrete tryptase, IL-6, IL-17, IL-18, and IL-36γ and activate KCs initiating psoriatic inflammation in the Koebner phenomenon [169].
The Role of Innate Lymphoid Cells
NK cells and NK-T cells are rare in lesional psoriasis skin [170]. However, their numbers significantly increase in the late-phase lesions [171]. In peripheral blood of psoriasis patients, the frequency of NK cells and NK-T cells is decreased, however, with a relative increase in NK-T17 cells [173, 174]. Despite their low numbers in psoriatic skin, NK cells produce high amounts of IFN-γ, TNF-α, IL-17A, and IL-22 [170, 172, 175]. Moreover, NK cells upregulate the expression of MHC class II proteins, ICAM-1, CXCL10, and CCL5 in psoriatic KCs [172]. It was suggested that also other innate lymphoid cells (ILC), including ILC2 and ILC3 participate in the pathogenesis of psoriasis [164, 176, 177]. However, the exact role of ILC in psoriasis patients remains to be determined.
The Role of Dendritic Cells
DCs are a major type of leukocytes in psoriatic lesions with numbers exceeding the number of T cells [104]. The main role of DCs in psoriasis is to orchestrate the immune response of T cells. Psoriatic DCs strongly induce both TH17 and TH1 T cells [178]. Notably, some of the T cells activated by psoriatic DCs simultaneously produce both IFN-γ and IL-17, which is not the case for healthy skin dermal DCs [178].
DCs in psoriatic lesions are divided into two main groups—CD11c+ BDCA-1+ resident DCs that are found also in healthy skin, and CD11c+ BDCA-1− inflammatory DCs that are more common in psoriasis lesional skin and are immature DCs that produce inflammatory cytokines [178]. These inflammatory DCs also include TNF-inducible NO synthase (iNOS)-producing DCs (TipDCs) [53, 179]. TipDCs are greatly increased in the lesional psoriatic skin [104, 180]. A similar population of DCs, that express IL-23p19, TNF-a, and iNOS, is called slan (6-sulfo LacNAc) DCs (slanDCs) and contributes to psoriasis by producing IL-17 and IL-22 [180, 181]. SlanDCs are enriched in lesional skin and are highly responsive to the TLR7 and TLR8 agonists as well as to complexes of self-RNA and LL37 [180, 181].
While in healthy skin DCs are restricted to the dermis, nearly half of DCs in psoriatic skin are located in the epidermis [104, 160, 182]. The population of epidermal DCs (eDCs) differs from a dermal counterpart and includes previously defined TipDCs, slan-DCs, and other types of DCs [180]. In general, eDCs express multiple genes regulating the activation of KCs and T cells as well as the recruitment of neutrophils, including IL-1β, IL-6, IL-8, CXCL1, and CCL17 [180].
Plasmacytoid dendritic cells (pDCs) are a rare population of DCs responsible for innate antiviral immunity [183]. These DCs are absent in healthy skin but accumulate in the skin of psoriasis patients [91]. Notably, they were identified to initiate psoriatic inflammation [91]. pDCs are activated by self-RNA-LL37 and self-DNA-LL37 complexes as well as other ligands via TLR signaling, especially TLR9 and TLR7. Complexes consisting of DNA or RNA and LL37 are abundant in lesional skin [19, 106]. In response to stimulation through TLR7 and TLR9, pDCs produce IFN-α that activates and induces expansion of T cells [91]. In the murine xenograft model of human psoriasis, inhibition of IFN-α completely prevented the development of T cell-dependent psoriasis [91].
pDCs are attracted to the early psoriatic lesions by chemerin, a chemotactic factor secreted by fibroblasts, mast cells, and endothelial cells [148]. Moreover, pDCs chemotaxis is promoted by VEGF-A-producing dermal IL-17A+ γδ T cells from the psoriatic skin [184]. Besides pDCs, conventional DCs (cDCs) participate in the induction and maintenance of psoriatic inflammation by the production of IL-23 [185, 186].
Another subset of DCs, Langerhans cells (LCs), which is a skin-resident population, contributes to the initial phase of psoriasis [186]. Activated psoriatic KCs produce bone morphogenetic protein 7 (BMP7) that promotes the differentiation of progenitor cells into inflammatory LCs [187]. LCs produce IL-6 and IL-23 leading to the infiltration of γδ T cells and CD4+ T cells into the skin [188, 189]. Notably, LCs migrate to the draining lymph nodes to induce expansion of IL-17A-producing γδ T cells [188]. In psoriasis, LCs have upregulated expression of S100A8 and S100A9 alarmins, increased production of IL-15, and produce IL-23 and IL-1β in response to TLR4 and TLR7/8 stimulation [180].
The Role of T cells
T cells are a crucial element of skin immunity [12]. There are approximately 20 billion antigen-experienced memory T cells in the skin of healthy adults [190]. Notably, the number of T cells is even up to 100 times higher in active psoriatic lesions than in healthy skin [191]. In psoriasis, activated T cells preferentially accumulate and expand in the epidermis [191, 193]. Accordingly, the substantial upregulation of IL-17A, IL-22, and IFN-γ is observed in epidermal T cells and to a lesser extent in their dermal counterparts [191]. Notably, while in healthy skin IL-17A is produced mainly by CD4+ T cells and IFN-γ is produced predominantly by CD8+ T cells, in psoriasis, both CD4+ T cells and CD8+ T cells co-produce both IL-17A and IFN-γ [164, 194].
TH Cells
CD4+ T helper (TH) cells orchestrate skin inflammation in psoriasis [195]. Both skin-infiltrating and circulating TH cells in psoriasis are predominantly TH1, TH9, TH17, and TH22 cells [70, 197].
TH1 Cells
Psoriasis was at first considered a typical TH1 inflammatory disease [23]. It was supported by the fact that epidermal T cells in psoriasis strongly produce type 1 cytokines, including IFN-γ, IL-2, and TNF-α [36]. TH1 cells and their main cytokine IFN-γ are increased in psoriatic lesions and peripheral blood of psoriasis patients [70]. Moreover, circulating CD4+ T cells in psoriasis are enriched in TNF-α+ cells compared to healthy controls [70].
TH17 Cells
The main factors regulating differentiation from naïve T cells toward TH17 lineage are IL-6 and TGF-β which activate the orphan nuclear receptor RORγt inducing the TH17 differentiation program [198, 199]. Both cytokines are upregulated in psoriasis (Table 1). Indeed, TH17 cells are substantially enriched in the circulation of psoriasis patients [70, 200]. Moreover, TH17 cells infiltrate and accumulate in psoriasis skin lesions and are predominantly localized in the dermis [201, 202]. It was found that 3–15% of T cells infiltrating psoriatic skin lesions produce IL-17A [25]. Notably, infiltrating TH17 cells accelerate further TH17 cells infiltration and expansion via the production of IL-6 in the dermis of psoriatic skin [39] as well as induction of CCL20 production by KCs [134]. Secreted IL-6 induces RORc, IL-17, and IL-23R expression supporting TH17 differentiation [68, 74]. The polarization of psoriatic inflammation toward the TH17 response is potentiated by IFN-γ produced by TH1 which reprograms APCs to produce IL-1, IL-23, and CCL20 [201, 203]. IL-23 produced by activated DCs supports T cell differentiation to TH17 cells and their expansion and survival [68, 74]. Thus, TH1 and TH17 colocalize in the pathological inflammatory environment of psoriasis and create a self-amplifying pathogenic loop. Moreover, cutaneous Staphylococcus aureus which is enriched in psoriatic and Streptococcal pyogenes that is associated with the onset of some types of psoriasis was identified to trigger a TH17 polarization of CD4+ T cells [204, 205].
TH17 cells are activated by factors that are upregulated in the psoriatic milieu, including IL-23, as well as IL-21 in an autocrine manner [68, 74]. TH17 cells express and secrete a variety of pro-inflammatory cytokines and chemokines that support psoriatic inflammation (Fig. 3) [198]. In lesional skin, IL-17F+ IFN-γ+ cells are the largest subset of T17 T cells and have a high expression of IL-1β, CSF-2, LTA, IL-24, and IL-34 [206].
TH22 Cells
It was demonstrated that IL-17 and IL-22 can be co-expressed by TH17 cells and synergize in the induction of the expression of AMPs by KCs in vitro [208]. However, a meta-analysis of healthy and psoriatic lesional skin transcriptome did not support the existence of dual-secreting IL-17A/IL-22 TH17 cells [209]. Furthermore, analysis of circulating immune cells in psoriasis confirmed that most of the TH17 cells do not co-express IL-17 and IL-22 [70]. Indeed, further studies revealed that a subpopulation of T cells producing IL-22 is distinct from TH17 cells and was named TH22 [71, 210]. These cells are crucial in the maintenance of skin homeostasis and contribute to different pathologies, including psoriasis [71, 196, 211]. The frequency of circulating TH22 cells is increased in psoriasis and correlates with the clinical severity [70, 73]. Moreover, TH22 cells infiltrate and accumulate in lesional psoriatic skin [202]. IL-22 produced by TH22 cells mediates acanthosis and immune cell infiltration induced by IL-23 [69]. Notably, TH22 cells are detected in the epidermis of resolved lesions after several years of treatment and form a disease memory in clinically healed psoriasis [191]. Thus, they are a promising therapeutic target.
TH9 Cells
IL-9-secreting TH9 cells are skin-tropic or skin-resident and produce TNF-α and granzyme B [212]. Moreover, TH9 cells induce the expression of multiple pro-inflammatory cytokines, including IFN-γ, IL-9, IL-13, and IL-17 [212]. Notably, TH9 cells are enriched in psoriatic skin lesions over three times compared to healthy skin [212]. Their main role in psoriasis is associated with the stimulation of angiogenesis via IL-9 secretion and potentiation of TH17 inflammation [48].
TH2 Cells
Unlike other populations of TH cells, the frequency of TH2 cells and levels of type 2 cytokines (IL-4 and IL-10) are decreased in psoriasis [36]. IL-4 suppresses the production of IL-23 by DCs while promoting the expression of IL-12p70 [213]. Thus, it impairs the induction and maintenance of pathogenic TH17 cell-mediated inflammation. Moreover, it suppresses the secretion of IL-1β and IL-6 and the expression of β-defensin 2 by psoriatic epidermal cells [214]. Due to its protective role in psoriatic inflammation, IL-4 therapy was proposed as a promising therapeutic strategy [35, 213].
TFH Cells
T follicular help (TFH) cells regulate humoral response by enabling the formation of the germinal center with B cells and regulating their maturation [215]. Despite psoriasis is not directly associated with humoral autoimmunity, the frequency of circulating TFH cells is increased in psoriatic patients [216]. Moreover, TFH cells infiltrate and accumulate in psoriasis lesions [216]. Notably, TFH cells in psoriasis are activated and have upregulated IL-21, IL-17, and IFN-γ production [216, 217]. Nonetheless, the mechanistic role of TFH cells in the pathogenesis of psoriasis is unclear.
CD8+Tc Cells
CD8+ cytotoxic T (Tc) cells are key effectors of the immune response against infection or cancer. Moreover, they are implicated in autoimmunity [219]. Tc cells, including Tc17 cells, are enriched in the circulation of psoriasis patients and potently infiltrate psoriatic lesions, especially the epidermis of lesional skin [194, 220]. CD8+ Tc cells are also a potent source of pro-inflammatory cytokines, including IL-17, IL-22, and IFN-γ in psoriatic lesions [194, 220]. Moreover, epidermal psoriatic CD8+ Tc cells have increased expression of granzyme A, granzyme B, and perforin 1, key proteins responsible for the cytotoxic activity of CD8+ T cells [194, 220]. The IL-17A-producing population of CD8+ T cells contains not only conventional T cells but also innate CD8+ mucosa-associated invariant T cell (MAIT cell) that contributes to psoriatic inflammation [221]. In mice, depletion of CD8+ Tc cells completely prevented the development of psoriasis [222]. Therefore, Tc cells together with TH cells are critically involved in the induction and execution of psoriatic inflammation.
γδ T Cells
Γδ T cells are selectively enriched in peripheral tissues, including skin, and can produce large amounts of cytokines in a short time [223]. Dermal γδ T cells express IL-23R, IL-17R, RORγt, and a variety of chemokine receptors, including CCR1, CCR2, CCR4, CCR5, CCR6, CXCR3, and CXCR4 [75, 224].
Emerging evidence suggests the pathogenic role of γδ T cells in psoriasis [225]. The frequency of γδ T cells in the dermis is substantially increased in patients with psoriasis [75]. Expansion and activation of γδ T cells in psoriatic lesions are stimulated by IL-1β via IL-1R [31].
In psoriatic skin, γδ T cells robustly produce proinflammatory cytokines, including IL-17, as well as proinflammatory chemokines CCL3, CCL4, CCL5, and CXCL8. Albeit, γδ T cells constitute about 1% of T cells in active psoriatic skin and are not observed in all resolved psoriatic lesions [226], their production of IL-17 is essential for psoriasis inflammation [31, 75, 119, 224]. Production of IL-17 by γδ T cells is induced by IL-1β and IL-23 and by IL-17-activated dermal fibroblast [31, 119]. Moreover, mediators secreted by γδ T cells activate KCs to produce multiple psoriasis-associated mediators, including NF-α, IL-6, CXCL9, CXCL10, and AMPs [227].
T Regulatory Cells (Tregs)
The frequency of Tregs in peripheral blood or lesional psoriatic skin compared to healthy individuals varies depending on the study [228]. Nonetheless, it is well-established that the immunoregulatory functions of Tregs are severely impaired in psoriasis [229]. Tregs isolated from lesional skin or peripheral blood of psoriasis patients fail to suppress effector T cell proliferation, in contrast to Tregs isolated from healthy individuals [229, 230]. Notably, in psoriasis Tregs not only fail to suppress the immune response in psoriasis but also actively contribute to psoriatic inflammation by the production of IL-17 [200].
Interestingly, obesity, especially long-chain FFAs, causes Treg loss in the skin and simultaneously increases IL-17A+ γδ T cells by reducing PPARγ+ skin Treg cells and a corresponding loss of control over IL-17A+ γδ T cell-mediated inflammation. Therefore, obesity plays a key role in the development of psoriasis [231].
Several factors impair the immunoregulatory functions of Tregs, including IL-6, IL-21, and IL-23 which are potently enriched in the psoriatic milieu [39, 59, 230]. Inhibition of STAT3, a downstream protein of these cytokines, partially restores suppressive properties of Tregs resulting in the inhibited production of pro-inflammatory cytokines, including IFN-γ, TNF-α, and IL-17, by psoriatic T cells [230]. The mechanisms of impaired immunoregulatory Tregs functions in psoriasis are associated with downregulated CD73/AMPK signaling pathway [232] and an increased level of miR-210 that downregulates FOXP3, a key regulator of the Tregs transcriptional program [233]. However, this phenomenon is most probably much more complex and requires further studies to identify pathways that may be modulated to restore Tregs functions in psoriasis.
Memory T Cells
Human skin contains four main populations of memory T cells, CD69+CD103− and CD69+CD103+ non-recirculating resident memory T cells (TRM), CCR7+/L-selectin+ central memory T cells (TCM), and CCR7+/L-selectin− migratory memory T cells (TMM) [234]. TRM cells have increased expression of adhesion molecules and produce a variety of pro-inflammatory and regulatory cytokines [235]. Notably, skin TRM cells are long-lived and, in mice, persist for over a year even in the absence of local antigen presentation [236].
In psoriasis, approximately half of epidermal CD8+ Tc cells in the psoriatic skin co-express TRM cell markers CD103 and CD49a and are profoundly enriched compared to healthy skin [191, 237, 238]. The density of infiltrating CD8+ TRM cells in the dermis correlates with the thickness of the psoriatic epidermis [239]. CD8+ TRM cells in psoriatic skin are an important source of IFN-γ, IL-17A, and IL-22 [191, 237, 238]. CD8+ TRM cells can be divided based on the expression of PD-1 into two groups. In psoriasis, PD-1+ CD8+ TRM cells preferentially expressed IL-23R and produce IL-17A, while PD-1− CD8+ TRM cells are a source of IFN-γ [239]. Unlike CD8+ Tc cells, only a small fraction of CD4+ TH cells express CD103, a marker of TRM cells [237].
TRM cells are a crucial population of immune cells from the clinical point of view since they are key cells that evoke the recurrence of psoriasis after therapy. Psoriatic lesions are often recurrent in the same sites [240], which is caused by the “lesional memory” phenomenon in which TRM cells play a pivotal role [241]. Both IL-17A-producing CD8+ T cells and IL-22-producing CD4+ T cells form a psoriasis disease–localized memory in the skin even 6 years after the beginning of the successful anti-TNF-α therapy [191, 226]. The latest clinical trial with antibodies against IL-17A and IL-17F (bimekizumab) showed cessation of symptoms in patients with psoriatic arthritis and an inadequate response or intolerance to TNF-α inhibitors [242].
Similarly, a small number of T cells, including TRM cells is also detected after 24 weeks of treatment with anti-IL-17A therapeutics [243]. Further analysis revealed that the IL-17A-producing CD8+ TRM cells are associated with early relapse of psoriasis after therapy [239]. In addition to memory αβ T cells, studies in mice identified a subset of IL-17-producing γδ T cells that are long-lived, persist in the skin after psoriasis resolution, and are capable of establishing memory population in the psoriatic skin [244]. Therefore, there is a great clinical interest in the development of therapies targeting T cells in psoriasis skin that constitute immunological memory to prevent disease relapse.
T Cells Autoantigens
Besides the initial activation of innate immunity, psoriasis may be also triggered by the initiation of the adaptive immune response. ADAMTS-like protein 5 (ADAMTSL5), a melanocyte protein, was identified as an autoantigen in psoriasis presented by HLA-C*06:02, the main psoriasis risk gene [105, 245]. Activation of autoreactive T cells by ADAMTSL5 triggers a TH17 phenotype with strong IL-17A and IFN-γ production [105]. These autoreactive T cells are present in the peripheral blood of two-thirds of psoriasis patients [105].
The antimicrobial peptide cathelicidin (LL37) has been recognized as another autoantigen by T cells in peripheral blood in about half of psoriasis patients [246]. Circulating T cells in psoriasis patients triggered by LL37 produce IFN-γ, IL-17, IL-22, and CXCL8 as well as upregulate perforins and granzyme B [246]. These autoreactive T cells express skin-homing receptors and are detected in lesional psoriatic skin [246]. Notably, LL37 also regulates the fate of non-autoreactive T cells. It suppresses TH1 differentiation and promotes TH17 differentiation and survival of non-LL37-specific T cells [247]. To exert its effects, LL37 needs to be present during antigen presentation and the T cell activation process, which is indeed a case in vivo since LL37 can be released in the lymph nodes by neutrophils [247]. Both ADAMTSL5 and LL37 are enriched in psoriatic lesional skin, and their levels are upregulated by IL-17 and TNF-α [246, 248].
Another identified autoantigen in psoriasis is the neolipid antigen produced by phospholipase A2 (PLA2) [249]. PLA2G4D, a cytosolic PLA2 group IVD, is highly expressed in psoriatic lesions, especially by the mast cells in the dermis and KCs, while it is not detected in healthy skin [249]. PLA2G4D generates ligands for CD1a from phospholipids in plasma membranes [249, 250]. Notably, mast cells not only produce CD1a ligands but also secrete them in the extracellular vesicles delivering them to the APCs [249]. Several types of cells can present lipid antigens with CD1a; however, Langerhans cells in the skin are crucial in this process [251]. A subset of T cells recognizes lipid antigens presented by CD1a [252]. These CD1a autoreactive T cells are enriched in the peripheral blood and accumulate in the lesional skin of psoriasis patients [249]. In response to CD1a ligands, they produce high amounts of IFN-γ, IL-17A, and IL-22, contributing to psoriatic inflammation [249].
Additionally, T cell receptor (TCR) sequencing in psoriasis revealed that T cell clones that are expanded in psoriasis are detectable also in the non-skin-homing (CLA−) population [253]. Thus, they may recognize autoantigens that are not exclusively expressed or located in the skin [253]. However, sequencing of TCR revealed that T cell response in the psoriatic skin is highly polyclonal [254], which excludes one potent autoantigen in psoriasis and suggests a more systemic character of inflammation.
The Role of B Cells
Despite the recent progress in the studies on the role of skin-associated B cells in the maintenance of skin homeostasis and regulation of inflammatory response [255], the role of B cells in psoriasis has been neglected so far. Nonetheless, psoriasis patients have an increased fraction of B cells, including activated B cells in peripheral blood which correlates with the clinical severity of psoriasis [217, 256]. Moreover, the frequency of B cells is also increased in psoriatic lesional skin [257]. Besides autoreactive T cells, several autoantigens for antibodies were proposed, including heterogeneous nuclear ribonucleoprotein A1 (hnRNP-A1), keratin 13, and Rab coupling protein isoform 3 (FLJ00294) (RAB11FIP1) [258, 259]. Moreover, a group of psoriasis patients has IgG autoantibodies against LL-37 and ADAMTS-L5 [260]. However, the mechanistic role of B cells and autoantibodies in the regulation of psoriatic inflammation remains unknown.
Some evidence of the role of B cells in psoriasis was derived from murine models of psoriasis. Regulatory B cells (Bregs) that suppress the immune response by secretion of IL-10 were found to potently suppress psoriatic inflammation by promoting Tregs expansion while inhibiting TH17 cells differentiation [261]. In mice, splenic Bregs enter circulation and migrate to lymph nodes suppressing the production of IFN-γ and IL-17 by lymphocytes in draining lymph nodes and inflamed skin [262]. Nonetheless, studies in psoriasis patients are required to determine the role of Bregs in the pathogenesis of psoriasis.
Non-Immune Cells Regulating Immunity in Psoriasis
Activation of immune cells in the psoriatic milieu affects all cells present in the skin. Importantly, these cells are not only victims but also actively participate in the amplification of psoriatic inflammation. Here, we described the role of KCs, key non-immune cells, as well as fibroblast, endothelial cells, and platelets in psoriasis.
The Role of Keratinocytes
KCs constitute about 90% of epidermal cells and are crucial non-immune cells that orchestrate psoriasis initiation and progression [263,264,265]. Activated KCs contribute to the pathogenesis of psoriasis via two main mechanisms. Firstly, psoriatic KCs have increased proliferative activity which results in epidermal hyperplasia, which is a hallmark of psoriatic plaques. Psoriatic skin is characterized by the dysregulated differentiation of KCs and enrichment of differentiated KCs [164]. Secondly, KCs are a crucial source of multiple immune mediators, including AMPs, chemokines, cytokines, and S100 alarmins (S100A7, S100A8, S100A9) that potentiate inflammation [164, 182, 206]. One of the crucial chemokines produced in a large amount by psoriatic KCs is CCL20 which chemoattracts IL-17-producing CCR6+ TH17 cells and γδ T cells [132, 266,267,268,269].
KCs are crucial responders to psoriatic cytokines, including IL-17, IL-22, IL-36, and IFN-γ [28, 265, 270,271,272]. In response to activation with these cytokines, KCs upregulate the expression of alarmins and AMPs [208]. Additionally, IL-17A induces the expression and release of IL-25 from KCs [80]. IL-25 in an autocrine manner induces a pro-inflammatory phenotype and hyperproliferation of KCs which is mediated by the activation of STAT3 [80]. Moreover, IFN-γ produced by activated immune cells primes KCs by rapid and long-lasting suppression of miRNA-149, a suppressor of the inflammatory response [273, 274]. In addition to cytokines, KCs are activated in psoriasis by DAMPs [18, 275]. dsRNA-LL37 complexes trigger IFN-β production in KCs which promotes activation and maturation of DCs that in turn drive T cell response [18]. Moreover, enhanced production of highly charged polyamines by psoriatic KCs promotes self-RNA sensing by DCs via TLR signaling [276].
Psoriatic KCs have impaired several self-regulatory pathways. For instance, cholecystokinin (CCK), a peptide hormone that suppresses KC inflammatory response, is potently decreased in lesional psoriatic skin [277]. Moreover, KCs in psoriasis exhibit increased reactivity to various cytokines. KCs from the psoriatic lesions more robustly respond to the TNF-α and IL-17 by the IL-23 production that KCs from healthy patients [120]. Moreover, some cytokines in the psoriatic milieu, including IL-25 and IL-29, increase the sensitivity of KCs towards their action by upregulating STAT proteins [80, 207].
KCs in psoriasis have potently upregulated inflammatory NF-κB signaling [278]. This pathway is activated by multiple stimuli, including TNF-α, plasmin, and TLRs ligands that are increased in the psoriatic microenvironment [279, 280]. Moreover, several KCs-associated mutations activating the NF-κB pathway were identified in psoriasis patients, including gain-of-function mutations in the CARD14 gene that encodes an activator of NF-κB signaling or loss-of-function in the TNFAIP3 gene that encodes an inhibitor of NF-κB pathway [265].
In inflamed skin, KCs act as nonprofessional antigen-presenting cells (APCs). KCs activated by IFN-γ co-stimulate naïve T cells via CD58/CD2 and to a lesser extent CD54/LFA-1 [192]. Notably, epidermal T cells in psoriatic lesions express CD2 but not CD28, suggesting that interaction of CD58/CD2 may be crucial for T cell activation in the skin [192]. Naïve T cells activated by psoriatic KCs selectively differentiate into TH1 and TH17 cells [192]. T cells activated by KCs selectively express skin-homing factors, including CCR4 and CCR8 [192]. Moreover, KCs via TGF-β induce CD103 (integrin αE) expression by T cells which is required for immune cell entry into the epidermis [281]. This, together with the fact that activated KCs secrete chemokines that attract IL-17-producing cells, makes KCs key regulators of the maintenance and amplification of immune cells infiltration in psoriatic skin.
The Role of Fibroblasts
Fibroblasts in the skin create a microenvironment regulating KCs activity, proliferation, and differentiation. In psoriasis, fibroblasts contribute to the pathogenesis of the disease mainly by inducing hyperproliferation of KCs [282]. Insulin-like growth factor-I (IGF-I) is one of the growth factors secreted by fibroblasts promoting KCs proliferation [283]. Moreover, dermal fibroblast of psoriasis patients has increased expression of α5 integrin, fibronectin, keratinocyte growth factor, and fibroblast growth factor receptor 2 that regulate proliferation and differentiation of KCs [284].
Besides the regulation of KCs, fibroblasts also contribute to the regulation of psoriatic inflammation. Fibroblasts in psoriasis have increased expression of IL-6, CXCL2, CXCL12, and CCL19 [164, 285]. Moreover, they produce CXCL8 which is potentiated by TNF-α [286]. Notably, the expression of IL-6 and CXCL8 in activated psoriatic fibroblasts is much higher than in psoriatic KCs [285]. Moreover, fibroblasts triggered by IL-17 promote the production of IL-17 by skin-infiltrating γδ T cells amplifying psoriatic inflammation [119]. Thus, fibroblasts can modulate chemotaxis and activity of neutrophils, T cells, and other types of immune cells in psoriatic skin.
The Role of Endothelial Cells
One of the histological hallmarks of psoriatic skin is increased dermal vascularity [287]. It is caused by increased concentrations of VEGF, angiopoietin, and TNF-α and is regulated predominantly by psoriatic γδ T cells and TH9 cells [48, 184]. Moreover, dermal endothelial cells are activated by psoriasis cytokines, including IL-36γ and IL-17A, which induce their proliferation, secretion of proinflammatory cytokines and chemokines, and upregulate ICAM-1 expression [288]. Transcriptional studies of psoriatic endothelial cells identified enrichment clusters relating to leukocyte adhesion, T cell activation, and IL-8 response in psoriatic lesional skin [164]. Increased angiogenesis and activation of endothelial cells facilitate diapedesis and infiltration of immune cells into the skin amplifying psoriatic inflammation.
Insights Into the Immune Network in Psoriasis from “Omics” Studies
Advances in research techniques enable more comprehensive studies on multiple types of cells and their interactions contributing to the initiation and progression of psoriasis (Table 3). These global analyses not only confirmed observations described in murine psoriasis models [289] but also revealed a complex network of interaction between immune cells and between immune and non-immune cells and uncovered novel mechanisms regulating psoriatic inflammation.
High-dimensional flow cytometry enables the identification of major immune cell types contributing to the pathogenesis of psoriasis as well as their dynamics during therapy. It identified specific myeloid cell populations that are the main producers of IL-23 while T cells are predominantly responsible for IL-17 production [163]. Moreover, it identified a novel population of CD3– CD4+ lymphoid tissue inducer cells abundant in psoriatic lesions [290]. Mass cytometry also uncovered the general sequence of the events during psoriasis initiation, progression, and maintenance and the composition of the epidermal immune microenvironment [186].
High-throughput analysis of psoriatic transcriptome confirmed a global character of inflammatory response in KCs, melanocytes, and immune cells [182] and the key role of the IL-17 signaling in inflamed skin [28]. Single-cell RNA sequencing identified subsets of T17 cells [206] and CD8+ T cells [218], dysregulation of skin-resident T cells [301], the key population of DCs in psoriatic lesions producing IL-1β and IL-23A [297], and IL-10-expressing regulatory DCs [206]. Moreover, TCR sequencing of T cells of psoriasis patients revealed polyclonal character of immune response and suggested the existence of autoantigens outside the skin [253, 254]. Nonetheless, further mechanistic studies are required to confirm these observations and to dissect the role of newly identified cell populations in psoriasis.
Conclusions
Exact understanding the mechanisms of psoriasis development and thus the use of effective treatment is important because psoriasis can be associated with distant complications and an even more frequent incidence of lymphohematologic malignancies [302]. The last decades clarified the view of psoriasis as a TH17 disease with the important components of TH1 and TH22 response and revealed crucial dysregulated components leading to the development of the disease. Emerging evidence demonstrated a key role of IL-17, IL-23, and TNF-α and resulted in the development and approval of biological therapies that revolutionized psoriasis management. It is well established that T cells orchestrate psoriatic inflammation with the help of multiple cells, including DCs, neutrophils, and KCs. Moreover, the role of several types of cells, including regulatory B and T cells, or autoantibodies in psoriasis remains largely unknown. Moreover, the role of the recently identified immunoregulatory population of cells named CD71+ erythroid cells (CECs) in psoriasis remains unknown [303, 304]. Recently, there has been a growing awareness of the role of the microbiota in the regulation of immunity. Indeed, growing evidence suggests the role of impaired skin and gut microbiome composition in the pathogenesis of psoriasis [292, 305]. Our understanding of the dysregulation of immune response in psoriasis has improved significantly over the past decade which offers the potential for the further development of even more effective and durable therapeutic strategies.
Data Availability
No datasets were generated or analysed during the current study.
References
Armstrong AW, Read C (2020) Pathophysiology, Clinical presentation, and treatment of psoriasis: a review. JAMA 323:1945–1960
Parisi R, Iskandar IYK, Kontopantelis E, Augustin M, Griffiths CEM, Ashcroft DM, Global Psoriasis A (2020) National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ 369:m1590-m
Greb JE, Goldminz AM, Elder JT, Lebwohl MG, Gladman DD, Wu JJ et al (2016) Psoriasis Nature Reviews Disease Primers 2:16082
Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, Gelfand JM (2017) Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol 76:377–390
Harden JL, Krueger JG, Bowcock AM (2015) The immunogenetics of psoriasis: a comprehensive review. J Autoimmun 64:66–73
Ran D, Cai M, Zhang X (2019) Genetics of psoriasis: a basis for precision medicine. Precision Clinical Medicine 2:120–130
Tsoi LC, Stuart PE, Tian C, Gudjonsson JE, Das S, Zawistowski M et al (2017) Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat Commun 8:15382
Rendon A, Schäkel K (2019) Psoriasis pathogenesis and treatment. Int J Mol Sci 20:1475
Swindell WR, Johnston A, Voorhees JJ, Elder JT, Gudjonsson JE (2013) Dissecting the psoriasis transcriptome: inflammatory- and cytokine-driven gene expression in lesions from 163 patients. BMC Genomics 14:527
Gudjonsson JE, Ding J, Johnston A, Tejasvi T, Guzman AM, Nair RP et al (2010) Assessment of the psoriatic transcriptome in a large sample: additional regulated genes and comparisons with in vitro models. J Invest Dermatol 130:1829–1840
Kabashima K, Honda T, Ginhoux F, Egawa G (2019) The immunological anatomy of the skin. Nat Rev Immunol 19:19–30
Ho AW, Kupper TS (2019) T cells and the skin: from protective immunity to inflammatory skin disorders. Nat Rev Immunol 19:490–502
Belkaid Y, Harrison OJ (2017) Homeostatic immunity and the microbiota. Immunity 46:562–576
Quaresma JAS (2019) Organization of the skin immune system and compartmentalized immune responses in infectious diseases. Clin Microbiol Rev 32:e00034-e118
Clark RA (2010) Skin-resident T cells: the ups and downs of on site immunity. J Invest Dermatol 130:362–370
Kamiya K, Kishimoto M, Sugai J, Komine M, Ohtsuki M (2019) Risk factors for the development of psoriasis. Int J Mol Sci 20:4347
Sun L, Liu W, Zhang L-J (2019) The role of toll-like receptors in skin host defense, psoriasis, and atopic dermatitis. J Immunol Res 2019:1824624
Zhang LJ, Sen GL, Ward NL, Johnston A, Chun K, Chen Y et al (2016) Antimicrobial peptide LL37 and MAVS signaling drive interferon-β production by epidermal keratinocytes during skin injury. Immunity 45:119–130
Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B et al (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449:564–569
Liu S, Wu F, Wu Z, Li Y, Zhang S, Yu N (2019) IL-17A synergistically enhances TLR3-mediated IL-36γ production by keratinocytes: a potential role in injury-amplified psoriatic inflammation. Exp Dermatol 28:233–239
Re F, Strominger JL (2001) Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem 276:37692–37699
Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J et al (2011) IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 12:1159–1166
Krueger JG (2002) The immunologic basis for the treatment of psoriasis with new biologic agents. J Am Acad Dermatol 46:1–23
Krueger JG (2012) Hiding under the skin: a welcome surprise in psoriasis. Nat Med 18:1750–1751
Wang A, Fogel AL, Murphy MJ, Panse G, McGeary MK, McNiff JM et al (2021) Cytokine RNA in situ hybridization permits individualized molecular phenotyping in biopsies of psoriasis and atopic dermatitis. JID Innovations 1:100021
Nickoloff BJ (1991) The cytokine network in psoriasis. Arch Dermatol 127:871–884
Johnston A, Guzman AM, Swindell WR, Wang F, Kang S, Gudjonsson JE (2014) Early tissue responses in psoriasis to the antitumour necrosis factor-α biologic etanercept suggest reduced interleukin-17 receptor expression and signalling. Br J Dermatol 171:97–107
Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A et al (2014) Transcriptome analysis of psoriasis in a large case–control sample: RNA-seq provides insights into disease mechanisms. J Investig Dermatol 134:1828–1838
Natsuaki Y, Egawa G, Nakamizo S, Ono S, Hanakawa S, Okada T et al (2014) Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin. Nat Immunol 15:1064–1069
Mee JB, Johnson CM, Morar N, Burslem F, Groves RW (2007) The psoriatic transcriptome closely resembles that induced by interleukin-1 in cultured keratinocytes: dominance of innate immune responses in psoriasis. Am J Pathol 171:32–42
Cai Y, Xue F, Quan C, Qu M, Liu N, Zhang Y et al (2019) A critical role of the IL-1β-IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J Invest Dermatol 139:146–156
Iznardo H, Puig L (2021) The interleukin-1 family cytokines in psoriasis: pathogenetic role and therapeutic perspectives. Expert Rev Clin Immunol 17:187–199
Witte E, Kokolakis G, Witte K, Philipp S, Doecke WD, Babel N et al (2014) IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J Invest Dermatol 134:2757–2767
Uyemura K, Yamamura M, Fivenson DF, Modlin RL, Nickoloff BJ (1993) The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol 101:701–705
Hahn M, Ghoreschi K (2017) The role of IL-4 in psoriasis. Expert Rev Clin Immunol 13:171–173
Austin LM, Ozawa M, Kikuchi T, Walters IB, Krueger JG (1999) The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol 113:752–759
Hochrein H, O’Keeffe M, Luft T, Vandenabeele S, Grumont RJ, Maraskovsky E, Shortman K (2000) Interleukin (IL)-4 is a major regulatory cytokine governing bioactive IL-12 production by mouse and human dendritic cells. J Exp Med 192:823–833
Grossman RM, Krueger J, Yourish D, Granelli-Piperno A, Murphy DP, May LT et al (1989) Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci USA 86:6367–6371
Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD (2009) IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol 183:3170–3176
Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M et al (2008) Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 29:628–636
Arican O, Aral M, Sasmaz S, Ciragil P (2005) Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm 2005:201561
Szepietowski JC, Bielicka E, Nockowski P, Noworolska A, Wasik F (2000) Increased interleukin-7 levels in the sera of psoriatic patients: lack of correlations with interleukin-6 levels and disease intensity. Clin Exp Dermatol 25:643–647
Bonifati C, Trento E, Cordiali-Fei P, Carducci M, Mussi A, D’Auria L et al (1997) Increased interleukin-7 concentrations in lesional skin and in the sera of patients with plaque-type psoriasis. Clin Immunol Immunopathol 83:41–44
Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S et al (2015) Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med 21:1272–1279
Duan H, Koga T, Kohda F, Hara H, Urabe K, Furue M (2001) Interleukin-8-positive neutrophils in psoriasis. J Dermatol Sci 26:119–124
Schulz BS, Michel G, Wagner S, Süss R, Beetz A, Peter RU et al (1993) Increased expression of epidermal IL-8 receptor in psoriasis. Down-regulation by FK-506 in vitro. J Immunol 151:4399–406
Giustizieri ML, Mascia F, Frezzolini A, De Pità O, Chinni LM, Giannetti A et al (2001) Keratinocytes from patients with atopic dermatitis and psoriasis show a distinct chemokine production profile in response to T cell-derived cytokines. J Allergy Clin Immunol 107:871–877
Singh TP, Schön MP, Wallbrecht K, Gruber-Wackernagel A, Wang X-J, Wolf P (2013) Involvement of IL-9 in Th17-associated inflammation and angiogenesis of psoriasis. PLoS ONE 8:e51752
Midde HS, Priyadarssini M, Rajappa M, Munisamy M, Mohan Raj PS, Singh S, Priyadarshini G (2021) Interleukin-9 serves as a key link between systemic inflammation and angiogenesis in psoriasis. Clin Exp Dermatol 46:50–7
Szodoray P, Alex P, Chappell-Woodward CM, Madland TM, Knowlton N, Dozmorov I et al (2006) Circulating cytokines in Norwegian patients with psoriatic arthritis determined by a multiplex cytokine array system. Rheumatology 46:417–425
Trepicchio WL, Ozawa M, Walters IB, Kikuchi T, Gilleaudeau P, Bliss JL et al (1999) Interleukin-11 therapy selectively downregulates type I cytokine proinflammatory pathways in psoriasis lesions. J Clin Investig 104:1527–1537
Yawalkar N, Karlen S, Hunger R, Brand CU, Braathen LR (1998) Expression of interleukin-12 is increased in psoriatic skin. J Invest Dermatol 111:1053–1057
Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F et al (2004) Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 199:125–130
Swindell WR, Xing X, Stuart PE, Chen CS, Aphale A, Nair RP et al (2012) Heterogeneity of inflammatory and cytokine networks in chronic plaque psoriasis. PLoS ONE 7:e34594
de Jesús-Gil C, Ruiz-Romeu E, Ferran M, Sagristà M, Chiriac A, García P et al (2020) IL-15 and IL-23 synergize to trigger Th17 response by CLA+ T cells in psoriasis. Exp Dermatol 29:630–638
Purzycka-Bohdan D, Szczerkowska-Dobosz A, Zablotna M, Wierzbicka J, Piotrowska A, Zmijewski MA et al (2016) Assessment of interleukin 16 serum levels and skin expression in psoriasis patients in correlation with clinical severity of the disease. PloS One 11:e0165577-e
Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K (2009) Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol 160:319–324
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD et al (2007) Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 8:950–957
Liu Y, Zhang C, Li B, Yu C, Bai X, Xiao C et al (2021) A novel role of IL-17A in contributing to the impaired suppressive function of Tregs in psoriasis. J Dermatol Sci 101:84–92
Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N et al (2019) Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol 139:1480–1489
Tollenaere MAX, Hebsgaard J, Ewald DA, Lovato P, Garcet S, Li X et al (2021) Signalling of multiple interleukin (IL)-17 family cytokines via IL-17 receptor A drives psoriasis-related inflammatory pathways. Br J Dermatol 185:585–594
Ohta Y, Hamada Y, Katsuoka K (2001) Expression of IL-18 in psoriasis. Arch Dermatol Res 293:334–342
Gangemi S, Merendino RA, Guarneri F, Minciullo PL, DiLorenzo G, Pacor M, Cannavò SP (2003) Serum levels of interleukin-18 and s-ICAM-1 in patients affected by psoriasis: preliminary considerations. J Eur Acad Dermatol Venereol 17:42–46
Konrad RJ, Higgs RE, Rodgers GH, Ming W, Qian Y-W, Bivi N et al (2019) Assessment and clinical relevance of serum IL-19 levels in psoriasis and atopic dermatitis using a sensitive and specific novel immunoassay. Sci Rep 9:5211
He Z, Jin L, Liu ZF, Hu L, Dang EL, Feng ZZ et al (2012) Elevated serum levels of interleukin 21 are associated with disease severity in patients with psoriasis. Br J Dermatol 167:191–193
Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M et al (2008) IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature 454:350–352
Wang Y, Wang LL, Yang HY, Wang FF, Zhang XX, Bai YP (2016) Interleukin-21 is associated with the severity of psoriasis vulgaris through promoting CD4+ T cells to differentiate into Th17 cells. Am J Transl Res 8:3188–3196
Wei L, Laurence A, Elias KM, O’Shea JJ (2007) IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem 282:34605–34610
Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W (2007) Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445:648–651
Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A (2010) Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol 130:1373–1383
Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H (2009) Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol 10:864–871
Mashiko S, Bouguermouh S, Rubio M, Baba N, Bissonnette R, Sarfati M (2015) Human mast cells are major IL-22 producers in patients with psoriasis and atopic dermatitis. J Allergy Clin Immunol 136:351–9.e1
Luan L, Ding Y, Han S, Zhang Z, Liu X (2014) An increased proportion of circulating Th22 and Tc22 cells in psoriasis. Cell Immunol 290:196–200
Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL (2003) Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem 278:1910–1914
Cai Y, Shen X, Ding C, Qi C, Li K, Li X et al (2011) Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity 35:596–610
Wang Y, Edelmayer R, Wetter J, Salte K, Gauvin D, Leys L et al (2019) Monocytes/macrophages play a pathogenic role in IL-23 mediated psoriasis-like skin inflammation. Sci Rep 9:5310
Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S et al (2006) IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med 203:2577–2587
Kumari S, Bonnet MC, Ulvmar MH, Wolk K, Karagianni N, Witte E et al (2013) Tumor necrosis factor receptor signaling in keratinocytes triggers interleukin-24-dependent psoriasis-like skin inflammation in mice. Immunity 39:899–911
Kunz S, Wolk K, Witte E, Witte K, Doecke W-D, Volk H-D et al (2006) Interleukin (IL)-19, IL-20 and IL-24 are produced by and act on keratinocytes and are distinct from classical ILs. Exp Dermatol 15:991–1004
Xu M, Lu H, Lee YH, Wu Y, Liu K, Shi Y et al (2018) An interleukin-25-mediated autoregulatory circuit in keratinocytes plays a pivotal role in psoriatic skin inflammation. Immunity 48:787–98.e4
Itoh T, Hatano R, Komiya E, Otsuka H, Narita Y, Aune TM et al (2019) Biological effects of IL-26 on T cell–mediated skin inflammation, including psoriasis. J Investig Dermatol 139:878–889
Hatano R, Itoh T, Otsuka H, Okamoto S, Komiya E, Iwata S et al (2019) Characterization of novel anti-IL-26 neutralizing monoclonal antibodies for the treatment of inflammatory diseases including psoriasis. MAbs 11:1428–1442
Balato A, Lembo S, Mattii M, Schiattarella M, Marino R, De Paulis A et al (2012) IL-33 is secreted by psoriatic keratinocytes and induces pro-inflammatory cytokines via keratinocyte and mast cell activation. Exp Dermatol 21:892–894
Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL et al (2011) IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol 186:2613–2622
D’Erme AM, Wilsmann-Theis D, Wagenpfeil J, Hölzel M, Ferring-Schmitt S, Sternberg S et al (2015) IL-36γ (IL-1F9) is a biomarker for psoriasis skin lesions. J Invest Dermatol 135:1025–1032
Tortola L, Rosenwald E, Abel B, Blumberg H, Schäfer M, Coyle AJ et al (2012) Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest 122:3965–3976
Teng X, Hu Z, Wei X, Wang Z, Guan T, Liu N et al (2014) IL-37 ameliorates the inflammatory process in psoriasis by suppressing proinflammatory cytokine production. J Immunol 192:1815–1823
Rønholt K, Nielsen AL, Johansen C, Vestergaard C, Fauerbye A, López-Vales R et al (2020) IL-37 expression is downregulated in lesional psoriasis skin. Immunohorizons 4:754–761
Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G et al (2018) IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis 9:1104
Han Y, Mora J, Huard A, da Silva P, Wiechmann S, Putyrski M et al (2019) IL-38 ameliorates skin inflammation and limits IL-17 production from γδ T cells. Cell Rep 27:835–46.e5
Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O et al (2005) Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med 202:135–143
Eriksen KW, Lovato P, Skov L, Krejsgaard T, Kaltoft K, Geisler C, Ødum N (2005) Increased sensitivity to interferon-alpha in psoriatic T cells. J Invest Dermatol 125:936–944
Tohyama M, Yang L, Hanakawa Y, Dai X, Shirakata Y, Sayama K (2012) IFN-α enhances IL-22 receptor expression in keratinocytes: a possible role in the development of psoriasis. J Invest Dermatol 132:1933–1935
Bielenberg DR, McCarty MF, Bucana CD, Yuspa SH, Morgan D, Arbeit JM et al (1999) Expression of interferon-beta is associated with growth arrest of murine and human epidermal cells. J Invest Dermatol 112:802–809
Mehta NN, Teague HL, Swindell WR, Baumer Y, Ward NL, Xing X et al (2017) IFN-γ and TNF-α synergism may provide a link between psoriasis and inflammatory atherogenesis. Sci Rep 7:13831
Li AG, Wang D, Feng XH, Wang XJ (2004) Latent TGFbeta1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J 23:1770–1781
Di Fusco D, Laudisi F, Dinallo V, Monteleone I, Di Grazia A, Marafini I et al (2017) Smad7 positively regulates keratinocyte proliferation in psoriasis. Br J Dermatol 177:1633–1643
Flisiak I, Porębski P, Flisiak R, Chodynicka B (2003) Plasma transforming growth factor β1 as a biomarker of psoriasis activity and treatment efficacy. Biomarkers 8:437–443
Flisiak I, Chodynicka B, Porebski P, Flisiak R (2002) Association between psoriasis severity and transforming growth factor beta(1) and beta (2) in plasma and scales from psoriatic lesions. Cytokine 19:121–125
Mussi A, Bonifati C, Carducci M, D’Agosto G, Pimpinelli F, D’Urso D et al (1997) Serum TNF-alpha levels correlate with disease severity and are reduced by effective therapy in plaque-type psoriasis. J Biol Regul Homeost Agents 11:115–118
Patel AB, Tsilioni I, Weng Z, Theoharides TC (2018) TNF stimulates IL-6, CXCL8 and VEGF secretion from human keratinocytes via activation of mTOR, inhibited by tetramethoxyluteolin. Exp Dermatol 27:135–143
Haider AS, Cohen J, Fei J, Zaba LC, Cardinale I, Toyoko K et al (2008) Insights into gene modulation by therapeutic TNF and IFNγ antibodies: TNF regulates IFNγ production by T cells and TNF-regulated genes linked to psoriasis transcriptome. J Investig Dermatol 128:655–666
Johansen C, Funding AT, Otkjaer K, Kragballe K, Jensen UB, Madsen M et al (2006) Protein expression of TNF-α in psoriatic skin is regulated at a posttranscriptional level by MAPK-activated protein kinase 2. J Immunol 176:1431–1438
Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, Nussbaum R et al (2005) Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci USA 102:19057–19062
Arakawa A, Siewert K, Stöhr J, Besgen P, Kim SM, Rühl G et al (2015) Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med 212:2203–2212
Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V et al (2009) Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 206:1983–1994
Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M, Nograles KE, Tian S, Cardinale I et al (2011) Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 131:677–687
Liu T, Li S, Ying S, Tang S, Ding Y, Li Y et al (2020) The IL-23/IL-17 pathway in inflammatory skin diseases: from bench to bedside. Front Immunol 11:594735
McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM et al (2009) The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol 10:314–324
Haines CJ, Chen Y, Blumenschein WM, Jain R, Chang C, Joyce-Shaikh B et al (2013) Autoimmune memory T helper 17 cell function and expansion are dependent on interleukin-23. Cell Rep 3:1378–1388
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD et al (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240
Whitley SK, Li M, Kashem SW, Hirai T, Igyártó BZ, Knizner K et al (2022) Local IL-23 is required for proliferation and retention of skin-resident memory T(H)17 cells. Sci Immunol 7:eabq3254
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH et al (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141
Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL (2019) IL-17 receptor–based signaling and implications for disease. Nat Immunol 20:1594–1602
Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M et al (1998) IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol 160:3513–3521
Kawaguchi M, Kokubu F, Odaka M, Watanabe S, Suzuki S, Ieki K et al (2004) Induction of granulocyte-macrophage colony-stimulating factor by a new cytokine, ML-1 (IL-17F), via Raf I-MEK-ERK pathway. J Allergy Clin Immunol 114:444–450
Albanesi C, Cavani A, Girolomoni G (1999) IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-gamma and TNF-alpha. J Immunol 162:494–502
Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM et al (2013) Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol 190:2252–2262
Ha H-L, Wang H, Pisitkun P, Kim J-C, Tassi I, Tang W et al (2014) IL-17 drives psoriatic inflammation via distinct, target cell-specific mechanisms. Proc Natl Acad Sci 111:E3422–E3431
Ehst B, Wang Z, Leitenberger J, McClanahan D, De La Torre R, Sawka E et al (2021) Synergistic induction of IL-23 by TNFα, IL-17A, and EGF in keratinocytes. Cytokine 138:155357
Chiricozzi A, Nograles KE, Johnson-Huang LM, Fuentes-Duculan J, Cardinale I, Bonifacio KM et al (2014) IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS ONE 9:e90284
Chiricozzi A, Suárez-Fariñas M, Fuentes-Duculan J, Cueto I, Li K, Tian S et al (2016) Increased expression of interleukin-17 pathway genes in nonlesional skin of moderate-to-severe psoriasis vulgaris. Br J Dermatol 174:136–145
Peric M, Koglin S, Kim SM, Morizane S, Besch R, Prinz JC et al (2008) IL-17A enhances vitamin D3-induced expression of cathelicidin antimicrobial peptide in human keratinocytes. J Immunol 181:8504–8512
Rauschenberger T, Schmitt V, Azeem M, Klein-Hessling S, Murti K, Grän F et al (2019) T cells control chemokine secretion by keratinocytes. Front Immunol 10:1917
Chen SC, de Groot M, Kinsley D, Laverty M, McClanahan T, Arreaza M et al (2010) Expression of chemokine receptor CXCR3 by lymphocytes and plasmacytoid dendritic cells in human psoriatic lesions. Arch Dermatol Res 302:113–123
Vestergaard C, Just H, Baumgartner Nielsen J, Thestrup-Pedersen K, Deleuran M (2004) Expression of CCR2 on monocytes and macrophages in chronically inflamed skin in atopic dermatitis and psoriasis. Acta Derm Venereol 84:353–8
Teraki Y, Miyake A, Takebayashi R, Shiohara T (2004) Homing receptor and chemokine receptor on intraepidermal T cells in psoriasis vulgaris. Clin Exp Dermatol 29:658–663
de Groot M, Teunissen MB, Ortonne JP, Lambert JR, Naeyaert JM, Picavet DI et al (2007) Expression of the chemokine receptor CCR5 in psoriasis and results of a randomized placebo controlled trial with a CCR5 inhibitor. Arch Dermatol Res 299:305–313
Nomura I, Gao B, Boguniewicz M, Darst MA, Travers JB, Leung DYM (2003) Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J Allergy Clin Immunol 112:1195–1202
Johansen C, Rittig AH, Mose M, Bertelsen T, Weimar I, Nielsen J et al (2017) STAT2 is involved in the pathogenesis of psoriasis by promoting CXCL11 and CCL5 production by keratinocytes. PLoS ONE 12:e0176994
Bosè F, Petti L, Diani M, Moscheni C, Molteni S, Altomare A et al (2013) Inhibition of CCR7/CCL19 axis in lesional skin is a critical event for clinical remission induced by TNF blockade in patients with psoriasis. Am J Pathol 183:413–421
Homey B, Dieu-Nosjean MC, Wiesenborn A, Massacrier C, Pin JJ, Oldham E et al (2000) Up-regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J Immunol 164:6621–6632
Gombert M, Dieu-Nosjean M-C, Winterberg F, Bünemann E, Kubitza RC, Da Cunha L et al (2005) CCL1-CCR8 interactions: an axis mediating the recruitment of T cells and Langerhans-type dendritic cells to sites of atopic skin inflammation. J Immunol 174:5082–5091
Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I et al (2009) Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol 129:2175–2183
Weninger W, Carlsen HS, Goodarzi M, Moazed F, Crowley MA, Baekkevold ES et al (2003) Naive T cell recruitment to nonlymphoid tissues: a role for endothelium-expressed CC chemokine ligand 21 in autoimmune disease and lymphoid neogenesis. J Immunol 170:4638–4648
Kim HO, Cho SI, Chung BY, Ahn HK, Park CW, Lee CH (2012) Expression of CCL1 and CCL18 in atopic dermatitis and psoriasis. Clin Exp Dermatol 37:521–526
Homey B, Alenius H, Müller A, Soto H, Bowman EP, Yuan W et al (2002) CCL27-CCR10 interactions regulate T cell-mediated skin inflammation. Nat Med 8:157–165
Kakinuma T, Saeki H, Tsunemi Y, Fujita H, Asano N, Mitsui H et al (2003) Increased serum cutaneous T cell-attracting chemokine (CCL27) levels in patients with atopic dermatitis and psoriasis vulgaris. J Allergy Clin Immunol 111:592–597
Kulke R, Bornscheuer E, Schlüter C, Bartels J, Röwert J, Sticherling M, Christophers E (1998) The CXC receptor 2 is overexpressed in psoriatic epidermis. J Invest Dermatol 110:90–94
Suárez-Fariñas M, Li K, Fuentes-Duculan J, Hayden K, Brodmerkel C, Krueger JG (2012) Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate-to-severe psoriasis. J Invest Dermatol 132:2552–2564
Rottman JB, Smith TL, Ganley KG, Kikuchi T, Krueger JG (2001) Potential role of the chemokine receptors CXCR3, CCR4, and the integrin alphaEbeta7 in the pathogenesis of psoriasis vulgaris. Lab Invest 81:335–347
Zgraggen S, Huggenberger R, Kerl K, Detmar M (2014) An important role of the SDF-1/CXCR4 axis in chronic skin inflammation. PloS One 9:e93665-e
Fraticelli P, Sironi M, Bianchi G, D’Ambrosio D, Albanesi C, Stoppacciaro A et al (2001) Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J Clin Invest 107:1173–1181
Christophers E, Metzler G, Röcken M (2014) Bimodal immune activation in psoriasis. Br J Dermatol 170:59–65
Németh T, Sperandio M, Mócsai A (2020) Neutrophils as emerging therapeutic targets. Nat Rev Drug Discovery 19:253–275
Rodriguez-Rosales YA, Langereis JD, Gorris MAJ, van den Reek JMPA, Fasse E, Netea MG et al (2021) Immunomodulatory aged neutrophils are augmented in blood and skin of psoriasis patients. Journal of Allergy and Clinical Immunology 148:1030–1040
Katayama H (2018) Development of psoriasis by continuous neutrophil infiltration into the epidermis. Exp Dermatol 27:1084–1091
Albanesi C, Scarponi C, Pallotta S, Daniele R, Bosisio D, Madonna S et al (2009) Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J Exp Med 206:249–258
Senra L, Mylonas A, Kavanagh RD, Fallon PG, Conrad C, Borowczyk-Michalowska J et al (2019) IL-17E (IL-25) enhances innate immune responses during skin inflammation. J Invest Dermatol 139:1732–42.e17
Shao S, Fang H, Zhang J, Jiang M, Xue K, Ma J et al (2019) Neutrophil exosomes enhance the skin autoinflammation in generalized pustular psoriasis via activating keratinocytes. Faseb j 33:6813–6828
Liu XT, Shi ZR, Lu SY, Hong D, Qiu XN, Tan GZ et al (2022) Enhanced migratory ability of neutrophils toward epidermis contributes to the development of psoriasis via crosstalk with keratinocytes by releasing IL-17A. Front Immunol 13:817040
Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S et al (2011) Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol 187:490–500
Guérard S, Allaeys I, Martin G, Pouliot R, Poubelle PE (2013) Psoriatic keratinocytes prime neutrophils for an overproduction of superoxide anions. Arch Dermatol Res 305:879–889
Hu SC-S, Yu H-S, Yen F-L, Lin C-L, Chen G-S, Lan C-CE (2016) Neutrophil extracellular trap formation is increased in psoriasis and induces human β-defensin-2 production in epidermal keratinocytes. Sci Rep 6:31119
Skrzeczynska-Moncznik J, Zabieglo K, Osiecka O, Morytko A, Brzoza P, Drozdz L et al (2020) Differences in staining for neutrophil elastase and its controlling inhibitor SLPI reveal heterogeneity among neutrophils in psoriasis. J Invest Dermatol 140:1371–8.e3
Herster F, Bittner Z, Archer NK, Dickhöfer S, Eisel D, Eigenbrod T et al (2020) Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat Commun 11:105
Shao S, Fang H, Dang E, Xue K, Zhang J, Li B et al (2019) Neutrophil extracellular traps promote inflammatory responses in psoriasis via activating epidermal TLR4/IL-36R crosstalk. Front Immunol 10:746
Wang H, Peters T, Kess D, Sindrilaru A, Oreshkova T, Van Rooijen N et al (2006) Activated macrophages are essential in a murine model for T cell-mediated chronic psoriasiform skin inflammation. J Clin Invest 116:2105–2114
Stratis A, Pasparakis M, Rupec RA, Markur D, Hartmann K, Scharffetter-Kochanek K et al (2006) Pathogenic role for skin macrophages in a mouse model of keratinocyte-induced psoriasis-like skin inflammation. J Clin Invest 116:2094–2104
Terhorst D, Chelbi R, Wohn C, Malosse C, Tamoutounour S, Jorquera A et al (2015) Dynamics and transcriptomics of skin dendritic cells and macrophages in an imiquimod-induced, biphasic mouse model of psoriasis. J Immunol 195:4953–4961
Fuentes-Duculan J, Suárez-Fariñas M, Zaba LC, Nograles KE, Pierson KC, Mitsui H et al (2010) A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J Investig Dermatol 130:2412–2422
Erbel C, Akhavanpoor M, Okuyucu D, Wangler S, Dietz A, Zhao L et al (2014) IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J Immunol 193:4344–4355
Mehta H, Mashiko S, Angsana J, Rubio M, Hsieh YM, Maari C et al (2021) Differential changes in inflammatory mononuclear phagocyte and T-cell profiles within psoriatic skin during treatment with guselkumab vs. secukinumab. J Invest Dermatol 141:1707–18.e9
Reynolds G, Vegh P, Fletcher J, Poyner EFM, Stephenson E, Goh I, Botting RA, Huang N, Olabi B, Dubois A, Dixon D, Green K, Maunder D, Engelbert J, Efremova M, Polański K, Jardine L, Jones C, Ness T, Horsfall D, McGrath J, Carey C, Popescu DM, Webb S, Wang XN, Sayer B, Park JE, Negri VA, Belokhvostova D, Lynch MD, McDonald D, Filby A, Hagai T, Meyer KB, Husain A, Coxhead J, Vento-Tormo R, Behjati S, Lisgo S, Villani AC, Bacardit J, Jones PH, O’Toole EA, Ogg GS, Rajan N, Reynolds NJ, Teichmann SA, Watt FM, Haniffa M (2021) Developmental cell programs are co-opted in inflammatory skin disease. Science 371(6527):eaba6500. https://doi.org/10.1126/science.aba6500
Liu P, Peng C, Chen X, Wu L, Yin M, Li J, Qin Q, Kuang Y, Zhu W (2021) Acitretin promotes the differentiation of myeloid-derived suppressor cells in the treatment of psoriasis. Front Med 8:625130. https://doi.org/10.3389/fmed.2021.625130
Kolkhir P, Elieh-Ali-Komi D, Metz M, Siebenhaar F, Maurer M (2022) Understanding human mast cells: lesson from therapies for allergic and non-allergic diseases. Nat Rev Immunol 22(5):294–308. https://doi.org/10.1038/s41577-021-00622-y. Epub 2021 Oct 5
Galli SJ, Gaudenzio N, Tsai M (2020) Mast cells in inflammation and disease: recent progress and ongoing concerns. Annu Rev Immunol 38:49–77
Harvima IT, Nilsson G, Suttle MM, Naukkarinen A (2008) Is there a role for mast cells in psoriasis? Arch Dermatol Res 300:461–478
Ji YZ, Liu SR (2019) Koebner phenomenon leading to the formation of new psoriatic lesions: evidences and mechanisms. Biosci Rep 39(12):BSR20193266. https://doi.org/10.1042/BSR20193266
Polese B, Zhang H, Thurairajah B, King IL (2020) Innate lymphocytes in psoriasis. Front Immunol 11:242. https://doi.org/10.3389/fimmu.2020.00242
Vissers WH, Arndtz CH, Muys L, Van Erp PE, de Jong EM, van de Kerkhof PC (2004) Memory effector (CD45RO+) and cytotoxic (CD8+) T cells appear early in the margin zone of spreading psoriatic lesions in contrast to cells expressing natural killer receptors, which appear late. Br J Dermatol 150:852–859
Ottaviani C, Nasorri F, Bedini C, de Pità O, Girolomoni G, Cavani A (2006) CD56brightCD16(-) NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur J Immunol 36:118–128
Hu Y, Chen Y, Chen Z, Zhang X, Guo C, Yu Z, Xu P, Sun L, Zhou X, Gong Y, Yu Q, Shi Y (2022) Dysregulated peripheral invariant natural killer T cells in plaque psoriasis patients. Front Cell Dev Biol 9:799560. https://doi.org/10.3389/fcell.2021.799560
Cameron AL, Kirby B, Griffiths CE (2003) Circulating natural killer cells in psoriasis. Br J Dermatol 149:160–164
Bonish B, Jullien D, Dutronc Y, Huang BB, Modlin R, Spada FM et al (2000) Overexpression of CD1d by keratinocytes in psoriasis and CD1d-dependent IFN-gamma production by NK-T cells. J Immunol 165:4076–4085
Bielecki P, Riesenfeld SJ, Hütter J-C, Torlai Triglia E, Kowalczyk MS, Ricardo-Gonzalez RR et al (2021) Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature 592:128–32
Linley H, Ogden A, Jaigirdar S, Buckingham L, Cox J, Priestley M, Saunders A (2023) CD200R1 promotes interleukin-17 production by group 3 innate lymphoid cells by enhancing signal transducer and activator of transcription 3 activation. Mucosal Immunol 16(2):167–179. https://doi.org/10.1016/j.mucimm.2023.01.001. Epub 2023 Jan 7
Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, Abello MV, Novitskaya I, Pierson KC et al (2009) Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Invest Dermatol 129:79–88
Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suárez-Fariñas M, Fuentes-Duculan J et al (2007) Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med 204:3183–3194
Martini E, Wikén M, Cheuk S, Gallais Sérézal I, Baharom F, Ståhle M et al (2017) Dynamic changes in resident and infiltrating epidermal dendritic cells in active and resolved psoriasis. J Invest Dermatol 137:865–73
Hänsel A, Günther C, Ingwersen J, Starke J, Schmitz M, Bachmann M et al (2011) Human slan (6-sulfo LacNAc) dendritic cells are inflammatory dermal dendritic cells in psoriasis and drive strong Th17/Th1 T-cell responses. Journal of Allergy and Clinical Immunology 127:787–94.e9
Cheng JB, Sedgewick AJ, Finnegan AI, Harirchian P, Lee J, Kwon S et al (2018) Transcriptional programming of normal and inflamed human epidermis at single-cell resolution. Cell Rep 25:871–883
Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M (1999) Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 5:919–923
Suzuki T, Hirakawa S, Shimauchi T, Ito T, Sakabe J-I, Detmar M, Tokura Y (2014) VEGF-A promotes IL-17A-producing γδ T cell accumulation in mouse skin and serves as a chemotactic factor for plasmacytoid dendritic cells. J Dermatol Sci 74:116–124
Wohn C, Ober-Blöbaum JL, Haak S, Pantelyushin S, Cheong C, Zahner SP et al (2013) Langerinneg conventional dendritic cells produce IL-23 to drive psoriatic plaque formation in mice. Proc Natl Acad Sci 110:10723–10728
Zhou Y, Xu F, Chen XY, Yan BX, Wang ZY, Chen SQ et al (2022) The epidermal immune microenvironment plays a dominant role in psoriasis development, as revealed by mass cytometry. Cell Mol Immunol 19:1400–1413
Borek I, Köffel R, Feichtinger J, Spies M, Glitzner-Zeis E, Hochgerner M et al (2020) BMP7 aberrantly induced in the psoriatic epidermis instructs inflammation-associated Langerhans cells. J Allergy Clin Immunol 145:1194–207.e11
Yoshiki R, Kabashima K, Honda T, Nakamizo S, Sawada Y, Sugita K et al (2014) IL-23 from Langerhans cells is required for the development of imiquimod-induced psoriasis-like dermatitis by induction of IL-17A-producing γδ T cells. J Invest Dermatol 134:1912–1921
Zheng T, Zhao W, Li H, Xiao S, Hu R, Han M et al (2018) p38α signaling in Langerhans cells promotes the development of IL-17–producing T cells and psoriasiform skin inflammation. Sci Signal 11:eaao1685
Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, Kupper TS (2006) The vast majority of CLA+ T cells are resident in normal skin. J Immunol 176:4431–4439
Cheuk S, Wikén M, Blomqvist L, Nylén S, Talme T, Ståhle M, Eidsmo L (2014) Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 192:3111–3120
Orlik C, Deibel D, Küblbeck J, Balta E, Ganskih S, Habicht J et al (2020) Keratinocytes costimulate naive human T cells via CD2: a potential target to prevent the development of proinflammatory Th1 cells in the skin. Cell Mol Immunol 17:380–394
Conrad C, Boyman O, Tonel G, Tun-Kyi A, Laggner U, de Fougerolles A et al (2007) α1β1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med 13:836–842
Res PC, Piskin G, de Boer OJ, van der Loos CM, Teeling P, Bos JD, Teunissen MB (2010) Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS ONE 5:e14108
Hu P, Wang M, Gao H, Zheng A, Li J, Mu D, Tong J (2021) The role of helper T cells in psoriasis. Front Immunol 12:788940
Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F (2009) Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol 10:857–863
Solberg SM, Aarebrot AK, Sarkar I, Petrovic A, Sandvik LF, Bergum B et al (2021) Mass cytometry analysis of blood immune cells from psoriasis patients on biological therapy. Eur J Immunol 51:694–702
Hirahara K, Ghoreschi K, Laurence A, Yang X-P, Kanno Y, O’Shea JJ (2010) Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev 21:425–434
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ et al (2006) The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Andres-Ejarque R, Ale HB, Grys K, Tosi I, Solanky S, Ainali C et al (2021) Enhanced NF-κB signaling in type-2 dendritic cells at baseline predicts non-response to adalimumab in psoriasis. Nat Commun 12:4741
Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS et al (2008) Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 128:1207–1211
Benham H, Norris P, Goodall J, Wechalekar MD, FitzGerald O, Szentpetery A et al (2013) Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res Ther 15:R136-R
Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L et al (2008) Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol 181:4733–4741
Chang H-W, Yan D, Singh R, Liu J, Lu X, Ucmak D et al (2018) Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome 6:154
Ruiz-Romeu E, Ferran M, de Jesús-Gil C, García P, Sagristà M, Casanova JM et al (2018) Microbe-dependent induction of IL-9 by CLA(+) T cells in psoriasis and relationship with IL-17A. J Invest Dermatol 138:580–587
Kim J, Lee J, Kim HJ, Kameyama N, Nazarian R, Der E, Cohen S, Guttman-Yassky E, Putterman C, Krueger JG (2021) Single-cell transcriptomics applied to emigrating cells from psoriasis elucidate pathogenic versus regulatory immune cell subsets. J Allergy Clin Immunol 148(5):1281–1292. https://doi.org/10.1016/j.jaci.2021.04.021. Epub 2021 Apr 29
Wolk K, Witte K, Witte E, Raftery M, Kokolakis G, Philipp S et al (2013) IL-29 is produced by T(H)17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci Transl Med 5:204ra129
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279
Le ST, Merleev AA, Luxardi G, Shimoda M, Adamopoulos IE, Tsoi LC et al (2019) 2D visualization of the psoriasis transcriptome fails to support the existence of dual-secreting IL-17A/IL-22 Th17 T cells. Front Immunol 10:589
Basu R, O’Quinn Darrell B, Silberger Daniel J, Schoeb Trenton R, Fouser L, Ouyang W et al (2012) Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37:1061–75
Jiang Q, Yang G, Xiao F, Xie J, Wang S, Lu L, Cui D (2021) Role of Th22 cells in the pathogenesis of autoimmune diseases. Front Immunol 12:688066. https://doi.org/10.3389/fimmu.2021.688066
Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE et al (2014) Human TH9 cells are skin-tropic and have autocrine and paracrine proinflammatory capacity. Sci Transl Med 6:219ra8-ra8
Guenova E, Skabytska Y, Hoetzenecker W, Weindl G, Sauer K, Tham M et al (2015) IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci USA 112:2163–2168
Onderdijk AJ, Baerveldt EM, Kurek D, Kant M, Florencia EF, Debets R, Prens EP (2015) IL-4 downregulates IL-1β and IL-6 and induces GATA3 in psoriatic epidermal cells: route of action of a Th2 cytokine. J Immunol 195:1744–1752
Qin L, Waseem TC, Sahoo A, Bieerkehazhi S, Zhou H, Galkina EV, Nurieva R (2018) Insights into the molecular mechanisms of T follicular helper-mediated immunity and pathology. Front Immunol 9:1884. https://doi.org/10.3389/fimmu.2018.01884
Wang Y, Wang L, Yang H, Yuan W, Ren J, Bai Y (2016) Activated circulating T follicular helper cells are associated with disease severity in patients with psoriasis. J Immunol Res 2016:7346030
Niu J, Song Z, Yang X, Zhai Z, Zhong H, Hao F (2015) Increased circulating follicular helper T cells and activated B cells correlate with disease severity in patients with psoriasis. J Eur Acad Dermatol Venereol 29:1791–6
Liu J, Chang H-W, Huang Z-M, Nakamura M, Sekhon S, Ahn R et al (2021) Single-cell RNA sequencing of psoriatic skin identifies pathogenic Tc17 cell subsets and reveals distinctions between CD8+ T cells in autoimmunity and cancer. Journal of Allergy and Clinical Immunology 147:2370–2380
Collier JL, Weiss SA, Pauken KE, Sen DR, Sharpe AH (2021) Not-so-opposite ends of the spectrum: CD8+ T cell dysfunction across chronic infection, cancer and autoimmunity. Nat Immunol 22:809–819
Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn SJ, Kupper TS et al (2013) CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17, and IL-22. J Invest Dermatol 133:973–979
Teunissen MBM, Yeremenko NG, Baeten DLP, Chielie S, Spuls PI, de Rie MA et al (2014) The IL-17A-producing CD8+ T-cell population in psoriatic lesional skin comprises mucosa-associated invariant T cells and conventional T cells. J Invest Dermatol 134:2898–2907
Di Meglio P, Villanova F, Navarini AA, Mylonas A, Tosi I, Nestle FO, Conrad C (2016) Targeting CD8+ T cells prevents psoriasis development. J Allergy Clin Immunol 138:274–6.e6
Ribot JC, Lopes N, Silva-Santos B (2021) γδ T cells in tissue physiology and surveillance. Nat Rev Immunol 21:221–232
Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, Becher B (2012) Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J Clin Invest 122:2252–2256
Qi C, Wang Y, Li P, Zhao J (2021) Gamma delta T cells and their pathogenic role in psoriasis. Front Immunol 12:627139
Matos TR, O’Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS et al (2017) Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing αβ T cell clones. J Clin Invest 127:4031–4041
Laggner U, Di Meglio P, Perera GK, Hundhausen C, Lacy KE, Ali N et al (2011) Identification of a novel proinflammatory human skin-homing Vγ9Vδ2 T cell subset with a potential role in psoriasis. J Immunol 187:2783–2793
Nussbaum L, Chen YL, Ogg GS (2021) Role of regulatory T cells in psoriasis pathogenesis and treatment. Br J Dermatol 184:14–24
Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR et al (2005) Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol 174:164–173
Yang L, Li B, Dang E, Jin L, Fan X, Wang G (2016) Impaired function of regulatory T cells in patients with psoriasis is mediated by phosphorylation of STAT3. J Dermatol Sci 81:85–92
Sivasami P, Elkins C, Diaz-Saldana PP, Goss K, Peng A, Hamersky MIV et al (2023) Obesity-induced dysregulation of skin-resident PPAR&γ+ Treg cells promotes IL-17A-mediated psoriatic inflammation. Immunity 56:1844–61.e6
Yan K, Xu W, Huang Y, Zhang Z, Huang Q, Xin KZ et al (2018) Methotrexate restores the function of peripheral blood regulatory T cells in psoriasis vulgaris via the CD73/AMPK/mTOR pathway. Br J Dermatol 179:896–905
Zhao M, Wang LT, Liang GP, Zhang P, Deng XJ, Tang Q et al (2014) Up-regulation of microRNA-210 induces immune dysfunction via targeting FOXP3 in CD4(+) T cells of psoriasis vulgaris. Clin Immunol 150:22–30
Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C et al (2015) Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med 7:279ra39-ra39
Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ et al (2017) Human Tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep 20:2921–2934
Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN et al (2012) Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 109:7037–7042
Tokura Y, Phadungsaksawasdi P, Kurihara K, Fujiyama T, Honda T (2021) Pathophysiology of skin resident memory T cells. Front Immunol 11:618897. https://doi.org/10.3389/fimmu.2020.618897
Leijten EF, van Kempen TS, Olde Nordkamp MA, Pouw JN, Kleinrensink NJ, Vincken NL et al (2021) Tissue-resident memory CD8+ T cells from skin differentiate psoriatic arthritis from psoriasis. Arthritis Rheumatol 73:1220–32
Kurihara K, Fujiyama T, Phadungsaksawasdi P, Ito T, Tokura Y (2019) Significance of IL-17A-producing CD8(+)CD103(+) skin resident memory T cells in psoriasis lesion and their possible relationship to clinical course. J Dermatol Sci 95:21–27
Masson Regnault M, Konstantinou M-P, Khemis A, Poulin Y, Bourcier M, Amelot F et al (2017) Early relapse of psoriasis after stopping brodalumab: a retrospective cohort study in 77 patients. J Eur Acad Dermatol Venereol 31:1491–6
Clark RA (2011) Gone but not forgotten: lesional memory in psoriatic skin. J Invest Dermatol 131:283–285
Merola JF, Landewé R, McInnes IB, Mease PJ, Ritchlin CT, Tanaka Y et al (2023) Bimekizumab in patients with active psoriatic arthritis and previous inadequate response or intolerance to tumour necrosis factor-α inhibitors: a randomised, double-blind, placebo-controlled, phase 3 trial (BE COMPLETE). The Lancet 401:38–48
Fujiyama T, Umayahara T, Kurihara K, Shimauchi T, Ito T, Aoshima M et al (2020) Skin infiltration of pathogenic migratory and resident T cells is decreased by secukinumab treatment in psoriasis. J Invest Dermatol 140:2073–6.e6
Hartwig T, Pantelyushin S, Croxford AL, Kulig P, Becher B (2015) Dermal IL-17-producing γδ T cells establish long-lived memory in the skin. Eur J Immunol 45:3022–3033
Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NVC, Jenisch S et al (2006) Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet 78:827–851
Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C et al (2014) The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun 5:5621
Minns D, Smith KJ, Alessandrini V, Hardisty G, Melrose L, Jackson-Jones L et al (2021) The neutrophil antimicrobial peptide cathelicidin promotes Th17 differentiation. Nat Commun 12:1285
Fuentes-Duculan J, Bonifacio KM, Hawkes JE, Kunjravia N, Cueto I, Li X et al (2017) Autoantigens ADAMTSL5 and LL37 are significantly upregulated in active psoriasis and localized with keratinocytes, dendritic cells and other leukocytes. Exp Dermatol 26:1075–1082
Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL et al (2016) Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med 213:2399–2412
Bourgeois EA, Subramaniam S, Cheng TY, De Jong A, Layre E, Ly D et al (2015) Bee venom processes human skin lipids for presentation by CD1a. J Exp Med 212:149–163
Kim JH, Hu Y, Yongqing T, Kim J, Hughes VA, Le Nours J et al (2016) CD1a on Langerhans cells controls inflammatory skin disease. Nat Immunol 17:1159–1166
Yoo HJ, Kim NY, Kim JH (2021) Current understanding of the roles of CD1a-restricted T cells in the immune system. Mol Cells 44:310–317
Roesner LM, Farag AK, Pospich R, Traidl S, Werfel T (2022) T-cell receptor sequencing specifies psoriasis as a systemic and atopic dermatitis as a skin-focused, allergen-driven disease. Allergy 77(9):2737–2747. https://doi.org/10.1111/all.15272. Epub 2022 Mar 14
Harden JL, Hamm D, Gulati N, Lowes MA, Krueger JG (2015) Deep sequencing of the T-cell Receptor repertoire demonstrates polyclonal T-cell infiltrates in psoriasis. F1000Res 4:460
Debes GF, McGettigan SE (2019) Skin-associated B cells in health and inflammation. J Immunol 202:1659–1666
Lu J, Ding Y, Yi X, Zheng J (2016) CD19+ B cell subsets in the peripheral blood and skin lesions of psoriasis patients and their correlations with disease severity. Braz J Med Biol Res 49:e5374-e
Czarnowicki T, Gonzalez J, Bonifacio KM, Shemer A, Xiangyu P, Kunjravia N et al (2016) Diverse activation and differentiation of multiple B-cell subsets in patients with atopic dermatitis but not in patients with psoriasis. Journal of Allergy and Clinical Immunology 137:118–29.e5
Guarneri C, Aguennouz M, Guarneri F, Polito F, Benvenga S, Cannavò SP (2018) Autoimmunity to heterogeneous nuclear ribonucleoprotein A1 in psoriatic patients and correlation with disease severity. J Dtsch Dermatol Ges 16:1103–1107
Jones DA, Yawalkar N, Suh KY, Sadat S, Rich B, Kupper TS (2004) Identification of autoantigens in psoriatic plaques using expression cloning. J Invest Dermatol 123:93–100
Yuan Y, Qiu J, Lin ZT, Li W, Haley C, Mui UN et al (2019) Identification of novel autoantibodies associated with psoriatic arthritis. Arthritis Rheumatol 71:941–951
Mizumaki K, Horii M, Kano M, Komuro A, Matsushita T (2021) Suppression of IL-23-mediated psoriasis-like inflammation by regulatory B cells. Sci Rep 11:2106
Yanaba K, Kamata M, Ishiura N, Shibata S, Asano Y, Tada Y et al (2013) Regulatory B cells suppress imiquimod-induced, psoriasis-like skin inflammation. J Leukoc Biol 94:563–573
Benhadou F, Mintoff D, Del Marmol V (2019) Psoriasis: keratinocytes or immune cells - which is the trigger? Dermatology 235:91–100
Albanesi C, Madonna S, Gisondi P, Girolomoni G (2018) The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front Immunol 9:1549
Zhou X, Chen Y, Cui L, Shi Y, Guo C (2022) Advances in the pathogenesis of psoriasis: from keratinocyte perspective. Cell Death Dis 13:81
Kim TG, Jee H, Fuentes-Duculan J, Wu WH, Byamba D, Kim DS et al (2014) Dermal clusters of mature dendritic cells and T cells are associated with the CCL20/CCR6 chemokine system in chronic psoriasis. J Invest Dermatol 134:1462–1465
Pène J, Chevalier S, Preisser L, Vénéreau E, Guilleux MH, Ghannam S et al (2008) Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol 180:7423–7430
Mabuchi T, Takekoshi T, Hwang ST (2011) Epidermal CCR6+ γδ T cells are major producers of IL-22 and IL-17 in a murine model of psoriasiform dermatitis. J Immunol 187:5026–5031
Campbell JJ, Ebsworth K, Ertl LS, McMahon JP, Newland D, Wang Y et al (2017) IL-17-Secreting γδ T cells are completely dependent upon CCR6 for homing to inflamed skin. J Immunol 199:3129–3136
Müller A, Hennig A, Lorscheid S, Grondona P, Schulze-Osthoff K, Hailfinger S, Kramer D (2018) IκBζ is a key transcriptional regulator of IL-36–driven psoriasis-related gene expression in keratinocytes. Proc Natl Acad Sci 115:10088–10093
Ekman AK, Bivik Eding C, Rundquist I, Enerbäck C (2019) IL-17 and IL-22 promote keratinocyte stemness in the germinative compartment in psoriasis. J Invest Dermatol 139:1564–73.e8
Ni X, Xu Y, Wang W, Kong B, Ouyang J, Chen J et al (2022) IL-17D-induced inhibition of DDX5 expression in keratinocytes amplifies IL-36R-mediated skin inflammation. Nat Immunol 23:1577–1587
Srivastava A, Luo L, Lohcharoenkal W, Meisgen F, Pasquali L, Pivarcsi A, Sonkoly E (2021) Cross-talk between IFN-γ and TWEAK through miR-149 amplifies skin inflammation in psoriasis. J Allergy Clin Immunol 147:2225–2235
Sidler D, Wu P, Herro R, Claus M, Wolf D, Kawakami Y et al (2017) TWEAK mediates inflammation in experimental atopic dermatitis and psoriasis. Nat Commun 8:15395
Chen X, Takai T, Xie Y, Niyonsaba F, Okumura K, Ogawa H (2013) Human antimicrobial peptide LL-37 modulates proinflammatory responses induced by cytokine milieus and double-stranded RNA in human keratinocytes. Biochem Biophys Res Commun 433:532–537
Lou F, Sun Y, Xu Z, Niu L, Wang Z, Deng S et al (2020) Excessive polyamine generation in keratinocytes promotes self-RNA sensing by dendritic cells in psoriasis. Immunity 53:204–16.e10
Funakoshi A, Tatsuno K, Shimauchi T, Fujiyama T, Ito T, Tokura Y (2019) Cholecystokinin downregulates psoriatic inflammation by its possible self-regulatory effect on epidermal keratinocytes. J Immunol 202:2609–2615
Lizzul PF, Aphale A, Malaviya R, Sun Y, Masud S, Dombrovskiy V, Gottlieb AB (2005) Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J Invest Dermatol 124:1275–1283
Li Q, Ke F, Zhang W, Shen X, Xu Q, Wang H et al (2011) Plasmin plays an essential role in amplification of psoriasiform skin inflammation in mice. PLoS ONE 6:e16483
Goldminz AM, Au SC, Kim N, Gottlieb AB, Lizzul PF (2013) NF-κB: an essential transcription factor in psoriasis. J Dermatol Sci 69:89–94
Pauls K, Schön M, Kubitza RC, Homey B, Wiesenborn A, Lehmann P et al (2001) Role of integrin alphaE(CD103)beta7 for tissue-specific epidermal localization of CD8+ T lymphocytes. J Invest Dermatol 117:569–575
Saiag P, Coulomb B, Lebreton C, Bell E, Dubertret L (1985) Psoriatic fibroblasts induce hyperproliferation of normal keratinocytes in a skin equivalent model in vitro. Science 230:669–672
Miura H, Sano S, Higashiyama M, Yoshikawa K, Itami S (2000) Involvement of insulin-like growth factor-I in psoriasis as a paracrine growth factor: dermal fibroblasts play a regulatory role in developing psoriatic lesions. Arch Dermatol Res 292:590–597
Gubán B, Vas K, Balog Z, Manczinger M, Bebes A, Groma G et al (2016) Abnormal regulation of fibronectin production by fibroblasts in psoriasis. Br J Dermatol 174:533–541
Angiolilli C, Leijten EFA, Bekker CPJ, Eeftink E, Giovannone B, Nordkamp MO et al (2022) ZFP36 family members regulate the proinflammatory features of psoriatic dermal fibroblasts. J Investig Dermatol 142:402–413
Glowacka E, Lewkowicz P, Rotsztejn H, Zalewska A (2010) IL-8, IL-12 and IL-10 cytokines generation by neutrophils, fibroblasts and neutrophils- fibroblasts interaction in psoriasis. Adv Med Sci 55:254–260
Lee HJ, Hong YJ, Kim M (2021) Angiogenesis in chronic inflammatory skin disorders. Int J Mol Sci 22:12035
Mercurio L, Failla CM, Capriotti L, Scarponi C, Facchiano F, Morelli M et al (2020) Interleukin (IL)-17/IL-36 axis participates to the crosstalk between endothelial cells and keratinocytes during inflammatory skin responses. PLoS ONE 15:e0222969
Gangwar RS, Gudjonsson JE, Ward NL (2022) Mouse models of psoriasis: a comprehensive review. J Investig Dermatol 142:884–897
Guo R, Zhang T, Meng X, Lin Z, Lin J, Gong Y, Liu X, Yu Y, Zhao G, Ding X, Chen X, Lu L (2019) Lymphocyte mass cytometry identifies a CD3-CD4+ cell subset with a potential role in psoriasis. JCI Insight 4(6):e125306. https://doi.org/10.1172/jci.insight.125306
Farrera C, Melchiotti R, Petrov N, Weng Teng KW, Wong MT, Loh CY et al (2020) T-cell phenotyping uncovers systemic features of atopic dermatitis and psoriasis. J Allergy Clin Immunol 145:1021–5.e15
Fyhrquist N, Muirhead G, Prast-Nielsen S, Jeanmougin M, Olah P, Skoog T et al (2019) Microbe-host interplay in atopic dermatitis and psoriasis. Nat Commun 10:4703
Nattkemper LA, Tey HL, Valdes-Rodriguez R, Lee H, Mollanazar NK, Albornoz C et al (2018) The genetics of chronic itch: gene expression in the skin of patients with atopic dermatitis and psoriasis with severe itch. J Invest Dermatol 138:1311–1317
He H, Bissonnette R, Wu J, Diaz A, Saint-Cyr Proulx E, Maari C et al (2021) Tape strips detect distinct immune and barrier profiles in atopic dermatitis and psoriasis. J Allergy Clin Immunol 147:199–212
Tsoi LC, Patrick MT, Shuai S, Sarkar MK, Chi S, Ruffino B et al (2022) Cytokine responses in nonlesional psoriatic skin as clinical predictor to anti-TNF agents. J Allergy Clin Immunol 149:640–9.e5
Swindell WR, Sarkar MK, Liang Y, Xing X, Gudjonsson JE (2016) Cross-disease transcriptomics: unique IL-17A signaling in psoriasis lesions and an autoimmune PBMC signature. J Invest Dermatol 136:1820–1830
Nakamizo S, Dutertre CA, Khalilnezhad A, Zhang XM, Lim S, Lum J, Koh G, Foong C, Yong PJA, Tan KJ, Sato R, Tomari K, Yvan-Charvet L, He H, Guttman-Yassky E, Malleret B, Shibuya R, Iwata M, Janela B, Goto T, Lucinda TS, Tang MBY, Theng C, Julia V, Hacini-Rachinel F, Kabashima K, Ginhoux F (2021) Single-cell analysis of human skin identifies CD14+ type 3 dendritic cells co-producing IL1B and IL23A in psoriasis. J Exp Med 218(9):e20202345. https://doi.org/10.1084/jem.20202345. Epub 2021 Jul 19
Gao Y, Yao X, Zhai Y, Li L, Li H, Sun X et al (2021) Single cell transcriptional zonation of human psoriasis skin identifies an alternative immunoregulatory axis conducted by skin resident cells. Cell Death Dis 12:450
Kim SH, Oh J, Roh WS, Park J, Chung KB, Lee GH, Lee YS, Kim JH, Lee HK, Lee H, Park CO, Kim DY, Lee MG, Kim TG (2023) Pellino-1 promotes intrinsic activation of skin-resident IL-17A-producing T cells in psoriasis. J Allergy Clin Immunol 151(5):1317–1328. https://doi.org/10.1016/j.jaci.2022.12.823. Epub 2023 Jan 13
Zhang B, Roesner LM, Traidl S, Koeken V, Xu CJ, Werfel T, Li Y (2023) Single-cell profiles reveal distinctive immune response in atopic dermatitis in contrast to psoriasis. Allergy 78:439–453
Liu Y, Wang H, Taylor M, Cook C, Martínez-Berdeja A, North JP et al (2022) Classification of human chronic inflammatory skin disease based on single-cell immune profiling. Sci Immunol 7:eabl9165
Bellinato F, Gisondi P, Girolomoni G (2022) Risk of lymphohematologic malignancies in patients with chronic plaque psoriasis: a systematic review with meta-analysis. J Am Acad Dermatol 86:86–96
Grzywa TM, Sosnowska A, Rydzynska Z, Lazniewski M, Plewczynski D, Klicka K et al (2021) Potent but transient immunosuppression of T-cells is a general feature of CD71(+) erythroid cells. Commun Biol 4:1384
Grzywa TM, Nowis D, Golab J (2021) The role of CD71(+) erythroid cells in the regulation of the immune response. Pharmacol Ther 228:107927
Chen L, Li J, Zhu W, Kuang Y, Liu T, Zhang W et al (2020) Skin and gut microbiome in psoriasis: gaining insight into the pathophysiology of it and finding novel therapeutic strategies. Front Microbiol 11:589726
Acknowledgements
T.M.G. is supported by PRELUDIUM grant funded by National Center of Science (UMO-2021/41/N/NZ6/02774) and the Foundation for Polish Science (FNP). Figures were created with BioRender.com.
Funding
T.M.G. is supported by the National Centre of Science (2021/41/N/NZ6/02774) and Foundation for Polish Science.
Author information
Authors and Affiliations
Contributions
I.S. and M.P. performed literature search and edited the manuscript. T.M.G. conceived, designed, and wrote the manuscript and performed literature search. All authors provided critical feedback, reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sieminska, I., Pieniawska, M. & Grzywa, T.M. The Immunology of Psoriasis—Current Concepts in Pathogenesis. Clinic Rev Allerg Immunol 66, 164–191 (2024). https://doi.org/10.1007/s12016-024-08991-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-024-08991-7