
Abstract
The respiratory tract is home to a diverse microbial community whose influence on local and systemic immune responses is only beginning to be appreciated. Increasing reports have linked changes in this microbiome to a range of pulmonary and extrapulmonary disorders, including asthma, chronic obstructive pulmonary disease and rheumatoid arthritis. Central to many of these findings is the role of IL-17-type immunity as an important driver of inflammation. Despite the crucial role played by IL-17-mediated immune responses in protection against infection, overt Th17 cell responses have been implicated in the pathogenesis of several chronic inflammatory diseases. However, our knowledge of the influence of bacteria that commonly colonise the respiratory tract on IL-17-driven inflammatory responses remains sparse. In this article, we review the current knowledge on the role of specific members of the airway microbiota in the modulation of IL-17-type immunity and discuss how this line of research may support the testing of susceptible individuals and targeting of inflammation at its earliest stages in the hope of preventing the development of chronic disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The human body is covered at all exposed and barrier surfaces with a panoply of bacteria, archaea, fungi and viruses. However, the true extent of their influence on host health and pathology has only recently been appreciated. Bacteria are the most numerous microbial components of the commensal microbiota and research to date has primarily focused on the bacteria that colonise the gastrointestinal tract. Significant advances have been made in our understanding of the crucial role these microbes perform, from regulating the development of the host immune system to the normal wiring of the brain [1,2,3,4]. Other mucosal sites too are colonised by commensal microorganisms that play critical roles in homeostatic functioning and defence against pathobionts. Once thought a sterile microenvironment, recent advances using culture-independent technologies have identified several bacterial species in healthy human lungs and growing evidence indicates that the upper and lower airways may be an important site for microbial regulation of immune responses, both locally and systemically. In the past decade, connections have been made between the composition of the respiratory tract microbiota and the pathophysiology of chronic inflammatory diseases such as asthma [5], chronic obstructive pulmonary disease (COPD) [6], cystic fibrosis (CF) [7] and rheumatoid arthritis (RA) [8]. In fact, changes in the relative abundance of only a small group of respiratory tract bacteria have repeatedly been associated with a range of chronic inflammatory conditions and a clear finding in many of these investigations is the centrality of IL-17 signalling in response to perturbations of this microbial community. IL-17 expression and neutrophil recruitment are typically associated with early inflammatory events [9], and it might be that altered abundance of these microbes promotes acute IL-17-type inflammation in the airways which, left unchecked, can become chronic and lead to major tissue destruction and fibrosis. Furthermore, IL-23-stimulated Th17 cells exert pleiotropic effects that extend beyond IL-17 expression and may also contribute to inflammatory events in these complex diseases. Here, we highlight the emerging association between IL-17-type immunity and bacteria that commonly colonise the human airways and discuss how this may contribute to the aetiology of chronic inflammatory disease.
The Airway Microbiota
After the gastrointestinal tract, the respiratory tract bears the largest exposure to the external environment. At 70 m2, the surface area of the lungs alone is forty times that of the skin (1.8 m2), owing to the physiological arrangement of the bronchi and alveoli [10]. We breath in approximately 11,000 L of air daily, containing both particulates and microorganisms [11]. As such, the respiratory tract is a major site of microbial exposure and colonisation. Indeed, the respiratory tract microbiota comprises over 600 individual bacterial species that form distinct communities from site-to-site, each adapting to the diverse surfaces and physiological conditions (e.g. temperature and pH) that exist along its length [12, 13].
The anterior nares are lined with keratinizing squamous epithelial cells and, due to the presence of sebum, host a lipophilic bacterial community that closely resembles that found on the skin, including Staphylococcus, Propionibacterium and Corynebacterium species [14]. Lined with columnar ciliated epithelial cells and non-ciliated mucus-secreting goblet cells, the nasopharynx comprises a diverse aero-anaerobic bacterial community comprising Streptococcus, Rothia, Veillonella, Prevotella and Neisseria [15]. The oral cavity is home to a varied microbial community in which Streptococcus, Haemophilus, Neisseria, Actinomyces, Prevotella and Veillonella species are commonly identified in healthy individuals [16]. The oropharynx is lined with a non-keratinizing squamous epithelium and its bacterial community is characterised by the presence of Neisseria, Rothia, Prevotella, Veillonella, Fusobacteria and Leptotrichia species [13, 17,18,19]. Finally, the healthy lungs—once thought to be sterile in the absence of infection—harbor a diverse microbial community, partly resembling that which is found in the mouth [12]. The bronchial tree is lined with ciliated columnar epithelium that soon transitions to the low cuboidal epithelium of the respiratory bronchioles and, ultimately, alveolar epithelium that is specifically adapted for gas exchange [13]. A thin layer of surfactant contributes to alveolar integrity by reducing surface tension at the air–liquid interface. In addition, surfactant displays bacteriostatic effects and surfactant proteins bind to non-host oligosaccharides and promote leukocyte recruitment and phagocytosis [20]. Advanced sequencing technologies have identified Prevotella and Veillonella as the dominant genera in the trachea, bronchi and bronchioles [5]. Streptococcus, Haemophilus, Moraxella, Pseudomonas, Fusobacterium and Porphyromonas taxa are also commonly identified in this dynamic community that results from microaspiration of bacteria and other microbes in oropharyngeal secretions, and ongoing outward movement through mucociliary clearance and the cough reflex [5, 6, 21]. Immune surveillance by alveolar macrophages (AM) also contributes to the turn-over of pulmonary symbionts.
The importance of the microbiota in lung development was revealed by the significantly reduced number and structure of alveoli, and decreased mucus production, in GF mice compared to conventionally-housed mice or their wild counterparts [3]. In addition, the bacterial community in the airways regulates mucosal immune responses against respiratory infection and may have a role in the development of certain forms of lung cancer [22,23,24,25]. We are only beginning to understand, however, the impact of this airway microbiota on resident and circulating host cells, particularly T lymphocytes that orchestrate the pathophysiology of many chronic inflammatory diseases. For a long time, inflammatory diseases were thought to associate with either an IFNγ-driven Th1-type inflammatory response (e.g. multiple sclerosis) or a Th2-type response characterised by the expression of IL-4, IL-5 and IL-13 (e.g. asthma). However, the discovery in 2005 of Th17 cells, a distinct lineage of CD4 T helper cell that preferentially produced the inflammatory cytokine IL-17A, was a significant advance in our understanding of the pathophysiology of many autoimmune inflammatory diseases [26]. Since then, a central pathogenic role for IL-17-type immunity in the development or exacerbation of many chronic respiratory diseases has also become apparent, with a growing body of evidence linking activation of these cells with outgrowth of specific members of the airway microbiota.
The IL-23/Th17 Axis in Chronic Inflammatory Disease
IL-17-producing CD4 T helper (Th17) cells were recognised as a discrete population of CD4+ T cells following the identification of their role in the pathogenesis of experimental autoimmune encephalomyelitis (EAE), a preclinical model of multiple sclerosis (MS) [26, 27]. Subsequently, these cells were found to be key orchestrators of numerous autoimmune and chronic inflammatory diseases [28]. In addition, several innate and adaptive immune cell populations were identified as sources of various IL-17 family members that play important roles in diverse inflammatory settings (Table 1).
Th17 cells develop from naïve precursors following recognition of their cognate antigen and are polarised along the Th17 lineage in response to signalling by the innate cytokines IL-1, IL-6 and TGFβ [56]. These cells are characterised by expression of the “master” transcription factor retinoic acid receptor-related orphan receptor gamma (RORγt) which, in turn, promotes expression of a number of key Th17 cell-associated receptors, including IL-23R and CCR6. Th17 cells produce IL-17A and IL-17F along with a range of other cytokines including IL-21, IL-22, TNFα, and IL-10. IL-17A and IL-17F signal through a heterodimeric receptor complex composed of the ubiquitously expressed IL-17RA and the non-hematopoietic cell-restricted IL-17RC that, in the airways, is expressed on epithelial cells and fibroblasts [59]. IL-17-receptor engagement induces epithelial cell expression of anti-microbial peptides and production of CXC chemokines and granulopoeitic factors that are critical in the recruitment and activation of neutrophils which, collectively, contribute to protection against infection by extracellular bacterial and fungal pathogens [60, 61]. Th17 cells also contribute to the maintenance of barrier function at mucosal sites [62]. It is clear, therefore, that microbial stimulation and Th17 cell function are closely associated and, in fact, Th17 cell development is diminished in germ-free animals, with a concomitant decrease in susceptibility to many preclinical models of complex diseases such as MS, uveitis, RA and psoriasis [63,64,65,66].
Self-antigen-specific Th17 cells that drive many autoimmune and chronic inflammatory diseases develop in the same manner as those elicited upon infection, but appear to be innocuous immediately following differentiation [54, 56, 67, 68]. In order for self-reactive Th17 cells to gain pathogenic potential—the ability to migrate to and access tissue sites where self- or autogenous antigens are expressed and, therein, orchestrate the inflammatory response—these cells require exposure to the cytokine IL-23, which regulates their conversion to pathogenic effectors of chronic inflammation. Upregulation of IFNγ, GM-CSF and other inflammatory mediators in IL-23-stimulated Th17 cells appears to be associated with pathogenicity and suggests the role of Th17 cells extends beyond the actions of IL-17. Hence, these so-called ex-Th17 cells (lately referred to as Th17.1 cells in human studies) are activated and develop differently to those that protect against acute infection with, for example, Candida albicans or Salmonella enterica that do not require IL-23 signalling or deviation of Th17 cell cytokine expression to mediate their effector functions [54, 69, 70]. Whether or not IL-23 signalling is critical for the effector function of Th17 cells that contribute to chronic inflammatory diseases of the airways such as pulmonary fibrosis, CF, COPD and asthma is not known. In fact, very little is known about the influence of the microbiota that colonises the respiratory tract on the expression of IL-23 and other molecules that drive Th17 cell-mediated immunity locally or systemically.
The Centricity of IL-17-Mediated Immunity in the Airways
It is clear that the microbiome is an important determinant of human health and that the host response to its microbial symbionts at several mucosal sites has a significant impact on chronic inflammation. Our understanding of host-microbe interactions in the airways is very much at an early stage, but increasing reports link this microbiota with a range of pulmonary and extra-pulmonary disorders. Central to many of these findings is the role of IL-17-producing immune cells and their activation by innate cytokines, including IL-1, IL-6 and IL-23. In the following sections, we summarise what is known about the induction of IL-17-driven immunity by respiratory tract symbionts, and their potential role in the development and resolution of inflammation in the airways, and beyond.
Th17-Type Immunity in Chronic Respiratory Diseases
Asthma
Asthma is a chronic inflammatory disease of the airways associated with airway hyperresponsiveness (AHR), leading to bronchoconstriction and airflow obstruction. In asthma patients, exposure to inhaled allergens leads to excessive activation of an inflammatory response in the lungs, typically mediated by allergen-specific Th2 cells that produce IL-4, IL-5 and IL-13, and which coordinate an allergic response centred around eosinophil recruitment and allergen-specific IgE production. Several murine studies have demonstrated a crucial role for commensal microbes in maintaining respiratory homeostasis. In many different models of allergic (or eosinophilic) asthma, AHR, local Th2 cytokine production, IgE concentrations and airway eosinophilia are all elevated in mice housed in germ-free conditions or administered antibiotics in early life, compared to conventionally housed mice or untreated controls [71,72,73,74,75]. Recolonisation of germ-free mice with a normal microbiota appears to suppress systemic IgE production by B cells and downstream expansion of basophilic precursor cells in the bone marrow; otherwise primed to promote a hypersensitive allergic response upon allergen exposure [74]. Numerous studies have demonstrated how stabilisation of the developing microbiota in neonatal mice induces CD4+CD25+Foxp3+ regulatory T (Treg) cells that decrease responsiveness toward aeroallergens; an effect that persists into adulthood [71, 75, 76]. Respiratory symbionts, specifically, have been reported to provide protection against allergic asthma. Adult mice whose lungs were exposed to S. pneumoniae before or after sensitisation with the model allergen ovalbumin (OVA) developed a regulatory-type immune response, characterised by elevated numbers of FoxP3+ Treg cells in lung-draining mediastinal lymph nodes and allergen-specific IL-10 secretion, associated with significantly reduced Th2 cytokine production, eosinophil recruitment and serum IgE [77]. Of note, only exposure to S. pneumoniae at OVA challenge led to a reduction in AHR, perhaps indicating that the mechanisms that suppress this response are transient and depend on recent bacterial stimulation.
Notwithstanding these findings, the adult human asthmatic airway has been associated with increased burden of Haemophilus, Moraxella, Neisseria, Staphylococcus and Streptococcus taxa, and a reduction in Veillonella and Prevotella species [5, 78,79,80,81,82,83]. URT colonisation by S. pneumoniae, H. influenza or M. catarrhalis within the first month of life was associated with increased risk of childhood asthma, and susceptibility to exacerbations in both mild and severe forms of disease [84, 85]. Circulating eosinophil counts and total IgE were significantly increased in children colonised neonatally with any or a combination of these bacteria, by the time they reached 4 years of age [84]. Interestingly, URT colonisation of these neonates with H. influenzae or M. catarrhalis engendered a mixed Th1/Th2/Th17 response, while Staphylococcus aureus colonisation induced a Th17-specific inflammatory profile [86].
Approximately 10% of asthmatics develop a neutrophilic form of disease driven by Th17-type inflammation, which is generally associated with a more severe disease phenotype and resistance to steroids [87, 88]. In these patients, heightened IL-17 expression and neutrophil infiltration can be detected in lung tissue, induced sputum, and serum, and it appears that neutrophils and eosinophils may also be an important source of IL-17 that sustains airway inflammation [29, 30, 87,88,89]. Neutrophilic asthma appears to be associated with a microbial profile quite distinct from that of eosinophilic asthma, with overall reduced microbial diversity and increased presence of opportunistic pathogens [90]. Increased burden of Proteobacteria species, including H. influenza and M. catarrhalis, routinely correlate with neutrophilic asthma, resulting in heightened Th17-related gene expression, CXCL8 production and airway neutrophilia [78, 82, 91, 92]. Patients with poorly controlled neutrophilic asthma exhibited high abundance of Haemophilus species and generally low bacterial diversity [93].
Murine studies have helped delineate how these diverse disease phenotypes may result from the differential induction of Th17-type immune responses. For example, it was found that, unlike the Treg-mediated protective responses elicited by S. pneumoniae in adult mice, neonatal exposure to S. pneumoniae or H. influenzae can trigger exacerbated allergic asthma in adulthood, associated with increased Th17 and Th2 cytokine secretion, and enhanced infiltration of neutrophils and eosinophils into the lungs [94, 95]. Intriguingly, Yang and colleagues used a model of long-term colonisation of the airways by H. influenzae to demonstrate the conversion of a steroid-sensitive Th2 model of eosinophilic inflammation to a steroid-resistant Th17-driven form of neutrophilic disease [96]. A significant increase in expression of IL-17 and RORγt was detected in the lungs of mice exposed to H. influenzae, which was also associated with increased mucin production and airway remodelling. Separately, it was reported that infection with H. influenzae during sensitisation alone can promote this conversion from eosinophilic to neutrophilic inflammation in the airways, underscoring the enduring impact of stimulation by H. influenzae on the development of airway inflammation [97]. Respiratory infection with M. catarrhalis similarly exacerbated a house dust mite (HDM) model of allergic asthma in an IL-17- and TNFα-dependent manner [98]. Airway inflammation was alleviated, and goblet cell hyperplasia and mucus production reduced in IL-17-deficient allergic mice infected with M. catarrhalis, compared to uninfected controls, confirming the crucial role of Th17-type inflammation in this response.
Together, these studies indicate that allergen-specific T cells can be skewed into distinct suppressive or inflammatory phenotypes depending on the bacterial species encountered and the temporality of that exposure (Fig. 1); with individual lymphocyte subpopulations having a significant influence in determining clinical manifestation and disease severity in asthma. Notably, pre-clinical models that rely on peripheral sensitisation may artificially promote a type-2 immune response, whereas allergic-sensitisation through the airways, as occurs in humans, more likely involves Th17-dominant immunity [99].

Inflammatory and suppressive T cell populations and granulocyte involvement in allergic airway inflammation has been associated with acute or long-term exposure to discrete bacterial species. Reduced microbial diversity in the lungs and outgrowth of specific taxa is associated with IL-17-type inflammation in the airways, and systemically. Relative changes in bacterial abundance and their association with cellular mediators of inflammation are indicated
Finally, a role for IL-23 and Th17-type immunity in allergic asthma has recently been described. Upon IL-23 airway exposure, Th2 cytokine production, eosinophil and neutrophil recruitment to the airways, goblet cell hyperplasia and AHR were all enhanced following allergen challenge [100, 101]. However, IL-17A only appeared critical for allergen-induced neutrophil recruitment into the airways. Separately, it was reported that exogenous IL-23 administration to the airways, in the absence of any allergen, can lead to eosinophilia and AHR, mimicking the non-allergic eosinophilic asthma (NAEA) phenotype [102]. This may be relevant given that pathogenic Proteobacteria often enriched in the airways of patients with asthma or COPD, including Haemophilus influenzae and Moraxella catarrhalis, induce significantly more IL-23 by host innate immune cells, compared to Prevotella species commonly found in healthy lungs [103].
Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease (COPD) is characterised by chronic bronchitis, mucociliary dysfunction, emphysema and a progressive decline in lung function. Exposure to tobacco smoke is the main environmental risk factor for the development of COPD, but acute exacerbations commonly result from bacterial and viral infections [104]. While evidence that smoking results in significant changes to the lung microbiome is limited, it does lead to changes in that of the URT [6, 17]. This may play an important etiological role in COPD because it has been demonstrated that microaspiration of disordered URT bacteria to the otherwise healthy lung can lead to an imbalance in the lung microbiome [105]. This, in turn, is associated with increased subclinical lung inflammation characterised by elevated Th17 responses, neutrophil accumulation and altered TLR4 signalling in alveolar macrophages, together with an increase in exhaled nitric oxide [105, 106].
Indeed, changes in the lung microbiota have been associated with the immunopathogenesis of COPD. Notwithstanding inter- and intra-subject heterogeneity, COPD patients, even when clinically stable, have a microbiome that lacks the diversity and richness of that of a healthy individual [5, 107]. A reduction in Prevotella and Veillonella and an overrepresentation of Actinobacteria and Proteobacteria species, in particular Haemophilus, Moraxella and Pseudomonas, have been reported in COPD patients [6, 108, 109]. These shifts in the composition of the microbiome appear to be strongly associated COPD exacerbation [107, 110, 111].
Th17 cells have been implicated as important inflammatory mediators in COPD with increased IL-17-expressing CD4 T cells and concentrations of related cytokines detected in the bronchial submucosa, airway epithelium, lung tissue, BALF and peripheral blood of COPD patients, compared to smokers without COPD and their healthy counterparts [33,34,35,36,37]. Neutrophilic infiltration mediated by IL-1β and IL-17 is prominent in COPD and increased recruitment correlates with a worsening disease course [112, 113]. Interestingly, the degree of neutrophilic inflammation was recently linked to Haemophilus-driven immune responses in the lungs [110]. Genetic predisposition may underscore these responses too, since polymorphisms in the Il17a gene have been associated with the risk of COPD related to tobacco smoking [114]. Notably, in a manner similar to that observed in other inflammatory conditions, conversion of Th17 cells to a more pathogenic IL-17+IFNγ+ double positive ex-Th17-like phenotype has been reported in COPD patients, with a correlation between increasing IFNγ+ CD4 T cell frequency and decreased lung function [38].
Using a murine model of chronic lung inflammation, which mimics many key features of COPD, microbiome dysbiosis involving a decrease in Prevotella and an increase in Pseudomonas, Lactobacillus and Chryseobacterium was observed in mice treated with LPS and elastase [115, 116]. Elevated levels of IL-1β and IL-6 were found in the BALF of diseased mice, coincident with the presence of IL-17A-expressing collagen- and elastin-specific CD4+ and γδ T cells in the lungs. The frequencies of these pulmonary IL-17A+ CD4 and γδ T cells was reduced in germ-free mice and in mice with an antibiotic-depleted microbiota [116]. Crucially, airway inflammation was ameliorated, and lung function improved in these mice, as well as SPF mice treated with an IL-17A-neutralising antibody. Separately, emphysema was attenuated in IL-17A- and IL-17RA-deficient mice exposed to long-term cigarette smoke, compared to wild-type controls, and IL-1RI–dependent IL-17A was found to be critical for pulmonary neutrophilia in H. influenzae-induced acute exacerbation using the same model [117,118,119]. While still preliminary, these studies support the idea that both chronic lung inflammation in COPD and the acute immunopathology linked to exacerbation are modulated by IL-17-centric host immune responses to a dynamic microbial environment.
Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive disorder caused by a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene which results in thickened mucus secretions in the airways of the lungs and the ducts of the pancreas, and impaired mucocillary clearance [120]. Chronic pulmonary disease, associated with excessive neutrophilic inflammation and chronic microbial infection, is the most significant contributor to morbidity and mortality in CF [121]. The secretion of elastase and MMPs, including MMP9, by activated neutrophils contribute to a decline in pulmonary function [122, 123]. Expression of IL-23 and IL-17, which are crucial for neutrophil recruitment, is significantly elevated under steady-state conditions in the airways of CF patients, compared to controls [124,125,126]. Moreover, innate and memory T cell-derived IL-17, as well as neutrophil-derived IL-17, are significantly increased in CF airways during pulmonary exacerbation which, in turn, is associated with increased production of the pro-neutrophilic cytokines CXCL8, IL-6, G-CSF and GM-CSF [39, 125,126,127,128]. Importantly, Th17 cells have been identified in the airway submucosa of children with CF, early in the course of the disease, implicating Th17-driven inflammation in disease etiology [39].
Lung microbial dysbiosis plays a critical role in the pathophysiology of CF. Commencing in the first months and years of life, abnormally viscous secretions and dysregulated inflammation caused by the CFTR mutation are linked with an excessive bacterial biomass, but reduced diversity, in the lower airways [129, 130]. Total bacterial biomass is linked to features of early structural disease such as airway wall thickening, mucus plugging, bronchiectasis and parenchymal disease scores [131]. Conversely, reduced bacterial diversity and increased dominance of certain species, particularly S. aureus and H. influenzae, correlates with antibiotic use, age and heightened inflammation [7, 129, 132, 133]. From late childhood onwards Mycobacterium abscessus, Burkholderia cenocepacia and Pseudomonas aeruginosa begin to dominate and are strongly associated with disease progression [134,135,136,137,138,139]. Nevertheless, recent culture-independent studies have found that both the early stage and adult CF lung is more polymicrobial than previously thought [140, 141]. While interpatient heterogeneity exists, Rothia, Gemella, Actinomyces, Stenotrophomonas, Neisseria and Streptococcus species as well as the obligate anaerobes Prevotella, Veillonella and Fusobacterium have been commonly detected in CF airways [140,141,142,143,144,145,146,147,148]. The functional implications of these diverse and variable non-classical CF bacteria require further study.
Elevated IL-23 and IL-17—in steady-state conditions and during exacerbations—are found in the sputum of CF patients colonised with P. aeruginosa, but not S. aureus, and decrease upon antibiotic use [124, 126, 149, 150]. Neutrophils in the sputum and blood of CF patients experiencing P. aeruginosa exacerbation express IL-17A and the inducible IL-17RC receptor subunit, in an IL-23-dependent fashion, and are associated with elevated elastase and MMP9 activity, compared to post-antibiotic treatment neutrophils [150]. In mice challenged with P. aeruginosa, deficiency in the Cftr gene results in elevated IL-17 concentrations in BALF, associated with enhanced expression of IL-6, IL-8 and CXCL2 and increased neutrophil counts, compared to P. aeruginosa-infected WT controls [151]. Neutralisation of IL-17A leads to reduced neutrophilia and lung pathology. Similarly, IL-23-deficient mice challenged with a clinical, mucoid isolate of P. aeruginosa developed significantly less airway inflammation than WT mice, associated with reduced IL-17, IL-6 and CXCL1 and decreased airway neutrophilia and MMP-9 expression [152]. Interestingly, bacterial clearance was not diminished in these mice, indicating that IL-23 may be a promising therapeutic target to reduce airway damage in patients with CF.
Non-CF Bronchiectasis
Chronic pulmonary infection and sustained neutrophilic inflammation also feature strongly in non-CF bronchiectasis (henceforth bronchiectasis), which varies in etiology but is characterised by abnormal, thick-walled and dilated bronchi, reduced lung function and the production of purulent sputum [153, 154]. While the majority of cases of bronchiectasis are idiopathic, a significant number present as a smoking-related COPD co-pathology [155]. Bronchiectasis is less well defined than CF but studies of the microbiome suggests that a core airway microbiome is shared between pediatric CF and bronchiectasis patients, which diverges in adulthood [156]. Haemophilus, Pseudomonas, Streptococcus, Staphylococcus, Moraxella, Veillonella, Prevotella and Achromobacter, as well as non-tuberculous Mycobacterium, are amongst the most abundant bacterial taxa in the lungs of patients with bronchiectasis [154, 157,158,159,160]. Moreover, as with CF, significantly elevated IL-23 and IL-17 production is seen in children and adults with bronchiectasis, which is reduced following treatment with the antibiotic clarithromycin [39, 41, 161]. IL-1α, IL-6 and CXCL8 levels were also significantly elevated in adults with established bronchiectasis and concurrent airway infection [41].
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive and usually fatal fibrotic lung disease of unknown etiology that is associated with chronic inflammation and dysregulated wound-healing [162]. It appears that IL-1β and IL-23-driven IL-17 signalling is instrumental early in the pathogenesis of pulmonary inflammation and fibrosis, likely through promotion of neutrophilic inflammation and MMP expression by fibroblasts [32, 163,164,165]. Indeed, elevated neutrophil counts in BAL fluid is a predictor of early mortality in IPF patients [166].
Recent studies have supported a role for the lung microbiota in IPF pathogenesis, and germ-free mice are protected against mortality in preclinical models of disease [167, 168]. In addition, a number of anti-microbials have entered clinical trials in human patients with IPF [169, 170]. Characterisation of bronchoalveolar lavage fluid (BALF) from IPF patients has demonstrated that disruption of the airway microbiota is associated with alveolar inflammation and significantly increased risk of disease progression and mortality [167, 168, 171]. Specifically, increased abundance of Neisseria, Haemophilus, Veillonella, Staphylococcus and Streptococcus taxa have been associated with worsening IPF [171,172,173].
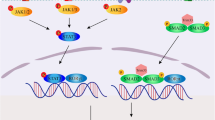
The precise mechanisms by which the lung microbiota influences IPF pathogenesis, and the role of IL-17 is only beginning to be revealed. Of note, it was recently demonstrated that microbiota-induced IL-17R signalling directly induces production of proinflammatory and profibrotic genes in lung epithelial cells and promotes lung fibrosis in a bleomycin-induced mouse model of IPF [31]. In that study, IL-17B was produced by alveolar macrophages correlating with the significantly elevated IL-17A and IL-17B expression by human BALF cells from IPF patients, compared to controls [31, 32]. Germ-free or antibiotic-treated mice, as well as mice that are deficient in IL-17A, IL-17B or IL-17E/IL-25, are resistant to bleomycin-induced fibrosis [31, 32, 163]. Importantly, Yang and colleagues identified outer membrane vesicles (OMVs) expressed by respiratory tract-associated Gram-negative anaerobes, including Bacteroides ovatus, Bacteroides stercoris and P. melaninogenica, that induce IL-17A and IL-17B expression, Th17 cell development and neutrophil recruitment to the lungs [31]. Evidence indicates that expansion of these specific species may result from altered host metabolic pathways following lung injury. Together, these findings support the idea that dysregulated growth of specific respiratory symbionts could initiate or perpetuate the immunopathology of IPF and opens a new area of investigation in the etiology of this poorly understood disease. If confirmed by independent investigators, the mechanistic discoveries by Yang and colleagues may point to novel microbial and metabolic targets for early therapeutic intervention in IPF.
Systemic Impact of the Airway Microbiota
Oral and Lung Bacteria Promote Joint Inflammation
Rheumatoid arthritis (RA) is a systemic autoimmune disease that is particularly associated with chronic inflammation at synovial joints. Pathogenic Th17 cells orchestrate the destruction of articular cartilage and underlying bone via expression of the pro-osteoclastogenic cytokines IL-1, IL-6, IL-17 and TNFα and B cells produce autoantibodies against a variety of endogenous antigens [42,43,44, 174,175,176]. IL-23 is critically required for pathogenicity of collagen-specific Th17 cells in the pre-clinical model collagen-induced arthritis (CIA) and pathogenic human Th17.1 cells (CD161+ Th17 cells that have converted to an IFNγ-producing phenotype) are important in the early phase of RA [177,178,179]. IL-23 also promotes expression of IL-1, IL-6, IL-17, GM-CSF and TNFα in synovial tissues and has been shown to stimulate osteoclast differentiation in human PBMCs in an IL-17-dependent manner [177, 178, 180].
Th17 cells also promote humoral immunity through germinal center formation, B cell activation and isotype class switching [181]. RA is characterised by increased levels of circulating autoantibodies including rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA) [42]. Systemic RF and ACPA can be identified several years before the onset of synovial pathology, suggesting that immunity against these proteins is initiated outside the joint [182]. A concordance rate as low as 15% in monozygotic twins indicates a strong environmental influence in RA development and IgA-dominant autoantibody responses in individuals with preclinical and early RA support that the initiation of RA-related autoimmunity might occur at mucosal sites [65, 183,184,185]. Indeed, growing data indicates that the microbiome may be an important factor. Disease is attenuated in numerous pre-clinical models when mice are housed in germ-free conditions [186,187,188]. Antimicrobial drugs have proven an effective treatment in some RA patients for decades [189]. And, it has recently been reported that active disease correlates with increased intestinal burden of Prevotella copri, Lactobacillus salivarius and the Actinobacteria genius Collinsella [190,191,192,193]. Similarly, in the lower airways, microbiome analysis of BAL samples from preclinical or early, untreated RA patients found significantly reduced microbial diversity compared to healthy controls, with a decrease in the relative abundance of the genera Actinomyces, Burkholderia, Prevotella and Porphyromonas [8, 194] (Fig. 2).
Autoantibodies have been detected in sputum from preclinical high-risk individuals—even in the absence of seropositivity [185]. Elevated ACPA prior to RA diagnosis has also been linked to bronchiectasis and asthma, while cigarette smoke and other bronchial stressors—perhaps including microbial factors—can increase ACPA titers and susceptibility to RA in persons with disease-associated HLA alleles [185, 195,196,197,198]. Significantly more ACPA-positive early RA patients had germinal center formation in bronchial tissue and histopathologic evidence of airway inflammation, compared to ACPA-negative RA patients or healthy controls, regardless of smoking status [199]. And, inducible bronchus-associated lymphoid tissue (iBALT)—not present in healthy human lung tissue—containing RA autoantibody-producing plasma cells, was increased in lung biopsies from RA patients with related lung disease [200]. These studies strongly implicate the lungs as a critical mucosal site in the etiology of autoimmune arthritis.
Periodontitis and RA share similar pathophysiological mechanisms including chronic inflammation with adjacent bone resorption. Not surprisingly then, a link has also emerged between oral microbiome perturbations, oral inflammation and arthritogenesis. Rothia, Lactobacillus and Prevotella species are enriched in the oral microbiome of pre-articular high-risk individuals, while Haemophilus and Neisseria species were depleted [192, 201]. Oral pathobionts including P. nigrescens or Porphyromonas gingivalis have also been associated with increased ACPA titers in patients with RA. Importantly, P. gingivalis was demonstrated to preferentially drive the generation of Th17-type immunity in human peripheral blood mononuclear cells (PBMC) by stimulating production of IL-1β, IL-6 and IL-23, but not the Th1-associated cytokine IL-12 [202]. And, in mice, oral administration of either P. nigrescens or P. gingivalis exacerbated CIA severity via IL-1-driven collagen-specific Th17 responses [203,204,205]. Finally, DNA fragments from P. nigrescens, P. gingivalis and Prevotella intermedia, and antibodies against periodontal bacteria, have been identified in serum and synovial fluid from RA patients with periodontitis [206, 207]. Collectively, these findings support the involvement of Th17-promoting URT pathobionts in the pathogenesis of RA. It may be that, in genetically susceptible individuals, bacteria residing in diverse regions of the respiratory tract can provide the environmental trigger leading to airway inflammation and, ultimately, systemic autoimmunity.
Regulation of CNS Autoimmune Inflammation in the Airways
Multiple Sclerosis (MS) is a chronic, progressive inflammatory disease of the central nervous system (CNS). MS is instigated by the infiltration of autoreactive T cells and other immune cell subsets into the CNS which attack the myelin sheath surrounding nerve axons, resulting in the formation of an inflammatory plaque and reduced signal conductance [208]. The etiology of MS is not fully understood and much of our understanding of the pathophysiology of the disease has arisen from studies of experimental autoimmune encephalomyelitis (EAE), an animal model that recapitulates many of the clinical and pathological features of the human disease. These investigations have revealed that Th17 cells are the critical pathogenic effector cells in EAE and that exposure to IL-23 is a critical step in their acquisition of pathogenic Th1-like properties [209, 210]. In MS patients too, CD161+ Th17.1 cells that express IFNγ and GM-CSF have been demonstrated to be the main pathogenic T cell subset associated with relapse and disease progression [211].
Epidemiological studies have long associated relapses in MS patients with systemic infection. Correale and colleagues demonstrated that bystander activation and increased sensitisation of autoreactive myelin-specific T cells resulted in elevated numbers of circulating IFNγ+ CD4 T cells and exacerbated disease in relapsing–remitting MS (RRMS) patients shortly following systemic infection [212]. Originally identified as Th1 cells, it is possible that these cells are Th17.1 cells that have converted to a pathogenic phenotype following microbial stimulation. In mice, over 90% of IFNγ-producing CD4 T cells in the CNS at the height of disease are actually ex-Th17 cells that upregulated Tbet following exposure to IL-23 and now exhibit Th1-like properties [54]. Myelin-reactive Th17 cells only upregulate IL-23R following differentiation in peripheral lymphoid tissues, however, and are innocuous immediately following their differentiation in vitro and in vivo [56, 67]. When and where these cells are exposed to this pathogenic signal is unknown.
Studies in pre-clinical models have implicated the respiratory tract in the pathogenicity of CNS autoimmune inflammation. Using intra-vital microscopy in a rat model of disease, Odoardi and colleagues demonstrated that transferred myelin-reactive CD4 T cells migrate to the lungs and BALT prior to their appearance in the CNS and the onset of neurological deficits [213]. These cells appeared to reside transiently in bronchi and alveoli before accumulating in BALT and draining lymph nodes, during which time they under-went functional reprogramming that allowed them to enter the CNS and instigate neuroinflammation. Separately, it was found that expansion of a pro-inflammatory population of granulocytic myeloid-derived suppressor cells (MDSCs) in the lungs during EAE promotes Th17 cell pathogenicity in an IL-6-dependent fashion [214].
Conversely, the respiratory pathogen Bordetella pertussis was found to suppress the severity of disease in a mouse model of EAE [215]. Infection with B. pertussis resulted in increased numbers of IL-10-producing Treg cells that suppressed expression of the adhesion molecules VLA-4 and LFA-1 on myelin-specific Th17 cells, impairing their migration to the CNS. In addition, subclinical pulmonary inflammation induced by LPS exposure or in an autophagy-defective mouse model appears to stall the trafficking of CCR6+ Th17 cells in the lungs, through enhanced expression of the chemokine CCL20 [216].

Relative changes in bacterial taxa and inflammatory mediators during chronic inflammatory disease. Reduced microbial diversity in the lungs and outgrowth of specific taxa is associated with IL-17-type inflammation in the airways, and systemically. Relative changes in bacterial abundance and their association with cellular mediators of inflammation are indicated. Abbreviations: COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis; ILC3, type-3 innate lymphoid cell; NKT, natural killer T cell; RA, rheumatoid arthritis
Regardless of the clinical outcome, these studies identify the lungs as a critical site that impacts Th17 cell encephalitogenicity and disease exacerbation in MS. Germ-free or antibiotic-treated mice are relatively resistant to EAE and reconstitution of these mice with bacteria that specifically colonise the murine gut can promote Th17 cell pathogenicity; although a systematic review of relevant human studies failed to find major differences in the gut microbiomes of MS patients and healthy controls [217, 218]. Studies on an etiological connection between human respiratory tract bacteria and CNS autoimmune inflammation have not been reported but should now be a vital new avenue of MS research.
Concluding Remarks
An axis exists between the airway microbiome and IL-17-type host responses that contribute to the immunopathology of many chronic inflammatory diseases. Advanced sequencing technologies have expanded our awareness of the respiratory tract microbiota which, during health, is low in biomass but high in diversity, and is dominated by Prevotella and Veillonella species [12, 13, 15, 17]. Reduced bacterial diversity is a common feature during exacerbation of chronic lung diseases such as asthma, CF, IPF and COPD, and often correlates with increased burden of discrete Proteobacteria species. Furthermore, whether by molecular mimicry, shared antigenic targets or epitope spreading, or through bystander activation and migration of self-reactive T cells, it appears that local inflammation at distinct sites along the respiratory tract can modulate extra-pulmonary diseases including RA and perhaps MS. Studies of the gastrointestinal tract demonstrate the strong influence that various mucosal symbionts exert over Th17 cell development and effector function, locally and systemically. Yet, we have scant understanding of the role played by individual respiratory tract bacteria, or those that contribute to a collective outcome, in shaping Th17-mediated chronic inflammation. It is a fascinating thought that our poor grasp of the early pathogenesis of multi-factorial diseases such as asthma, IPF and RA may lie in insufficient appreciation of the impact of the bacteria that colonise the respiratory tract and that, perhaps, these processes will become better elucidated with more detailed knowledge of how discrete respiratory symbionts promote pathogenic Th17-type immune responses locally in the airways, and systemically.
As our understanding of the respiratory tract microbiota progresses from description of this community to defining it’s impact on human health, it will be crucial to determine a causal role for specific bacterial strains in driving IL-17-signalling, neutrophil infiltration and resultant immunopathology (Fig. 3). Conversely, the metabolic changes associated with inflammation may create an environmental niche that some Gammaproteobacteria have developed the capacity to exploit, perpetuating a cycle of inflammation [219]. Hence, production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) during an inflammatory response may allow adaptable strains to bloom and outcompete resident bacteria that lack this capacity but, alas, these bacteria may not be the primary instigators of inflammation. Either way, it is imperative we understand the earliest inflammatory events in order to prevent the initial development of these chronic inflammatory diseases.

The centricity of IL-17-mediated immunity in chronic inflammation associated with the airways. Abbreviations: MDSC, myeloid-derived suppressor cells; AM, alveolar macrophage; \(\gamma\delta\), gamma delta T cell; ILC3, type-3 innate lymphoid cell; CF, cystic fibrosis; COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis; MS, multiple sclerosis; RA, rheumatoid arthritis RA. Created with BioRender.com
If certain strains of bacteria that have a fundamental involvement in the aetiology of a disease can be identified, then early microbiome sequencing of susceptible individuals and therapeutic targeting of the IL-17 pathway may present a novel preventative approach. Several exciting new drugs targeting the IL-17 axis have proven efficacious in the treatment of inflammatory disorders such as psoriasis and inflammatory arthritis [28]. However, Th17 cells act in pleiotropic ways to orchestrate immunity at diverse sites of inflammation. IL-1β and IL-23 stimulate expression of numerous inflammatory molecules, in addition to guiding Th17 cell development and effector function. There is redundancy amongst members of the IL-17 family; for example, IL-17A and IL-17F use the same receptor complex to stimulate expression of chemokines and various myelopoietic and granulopoeitic factors. And, Th17 cells support B cell activation, isotype class switching and high-affinity antibody production. Hence, the effects of Th17 cells extend beyond the actions of IL-17A and may explain why IL-17-blockade during exacerbations in some chronic inflammatory diseases often fail or deliver only partial responses. It may be, too, that we miss the acute IL-17-driven pathophysiology in some of these diseases which has given way to a cycle of inflammation and dysbiosis by the time symptoms present clinically.
Identification of individual strains of bacteria that drive the initial inflammatory events, or those that collectively engender chronic inflammation, may permit prophylactic targeting at the very earliest stages of disease development. Airway microbiome composition can act as a biomarker of potential early disease activity and allow for stratification of at-risk individuals. Altered microbiota structure and increased burden of causal strains may signal for interventional approaches including the use of novel small molecule drugs, aerosol delivery of probiotics or narrow-spectrum antibiotics, the use of vaccines or administration of synthetic bacteriophages that reduce harmful populations of bacteria. Ongoing research in this area should also elucidate critical endogenous signalling pathways and new targets for drug therapies. Importantly, recent research has focused on non-canonical pathways involved in the acquisition of pathogenicity by Th17 cells. These include the RNA helicase DDX5 that controls aspects of RORγt-mediated Th17 cell development and CDL5, a scavenger receptor with pattern recognition receptor (PRR) activity that is expressed by lung epithelial cells, Th17 cells and macrophages in inflamed tissues and acts as a negative regulator of the transition from a non-pathogenic to a pathogenic Th17 phenotype [220,221,222]. In individuals at risk of developing chronic inflammatory disease, prophylactically targeting molecules that regulate progression to a pathogenic Th17 cell phenotype, while sparing protective Th17 capacity, should maintain a balanced immune response without increased susceptibility to infection and, perhaps, lead to improved patient outcomes compared to those achieved therapeutically.
References
Pickard JM, Zeng MY, Caruso R, Nunez G (2017) Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev 279(1):70–89
Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G (2008) Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456(7221):507–510
Yun Y, Srinivas G, Kuenzel S, Linnenbrink M, Alnahas S, Bruce KD, Steinhoff U, Baines JF, Schaible UE (2014) Environmentally determined differences in the murine lung microbiota and their relation to alveolar architecture. PLoS One 9(12):e113466
Cryan JF, Dinan TG (2012) Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13(10):701–712
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO (2010) Disordered microbial communities in asthmatic airways. PLoS One 5(1):e8578
Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, Martinez FJ, Huffnagle GB (2011) Analysis of the lung microbiome in the healthy smoker and in COPD. PLoS One 6(2):e16384
Cox MJ, Allgaier M, Taylor B, Baek MS, Huang YJ, Daly RA, Karaoz U, Andersen GL, Brown R, Fujimura KE, Wu B, Tran D, Koff J, Kleinhenz ME, Nielson D, Brodie EL, Lynch SV (2010) Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS One 5(6):e11044
Demoruelle MK, Norris J, Holers V, Harris J, Deane K (2014) The lung microbiome differs in asymptomatic subjects at elevated risk of future rheumatoid arthritis compared with healthy control subjects. Ann Am Thorac Soc 11:S74
Veldhoen M (2017) Interleukin 17 is a chief orchestrator of immunity. Nat Immunol 18(6):612–621
Effros RM (2006) Anatomy, development, and physiology of the lungs. GI Motility
Gusareva ES, Acerbi E, Lau KJX, Luhung I, Premkrishnan BNV, Kolundzija S, Purbojati RW, Wong A, Houghton JNI, Miller D, Gaultier NE, Heinle CE, Clare ME, Vettath VK, Kee C, Lim SBY, Chenard C, Phung WJ, Kushwaha KK, Nee AP, Putra A, Panicker D, Yanqing K, Hwee YZ, Lohar SR, Kuwata M, Kim HL, Yang L, Uchida A, Drautz-Moses DI, Junqueira ACM, Schuster SC (2019) Microbial communities in the tropical air ecosystem follow a precise diel cycle. Proc Natl Acad Sci U S A 116(46):23299–23308
Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB (2016) The Microbiome and the Respiratory Tract. Annu Rev Physiol 78:481–504
Man WH, de Steenhuijsen Piters WA, Bogaert D (2017) The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Micro 15(5):259–270
Zhou Y, Mihindukulasuriya KA, Gao H, La Rosa PS, Wylie KM, Martin JC, Kota K, Shannon WD, Mitreva M, Sodergren E, Weinstock GM (2014) Exploration of bacterial community classes in major human habitats. Genome Biol 15(5):R66
Edouard S, Million M, Bachar D, Dubourg G, Michelle C, Ninove L, Charrel R, Raoult D (2018) The nasopharyngeal microbiota in patients with viral respiratory tract infections is enriched in bacterial pathogens. Eur J Clin Microbiol Infect Dis 37(9):1725–1733
Le Bars P, Matamoros S, Montassier E, Le Vacon F, Potel G, Soueidan A, Jordana F, de La Cochetiere MF (2017) The oral cavity microbiota: between health, oral disease, and cancers of the aerodigestive tract. Can J Microbiol 63(6):475–492
Charlson ES, Chen J, Custers-Allen R, Bittinger K, Li H, Sinha R, Hwang J, Bushman FD, Collman RG (2010) Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One 5(12):e15216
Pettigrew MM, Laufer AS, Gent JF, Kong Y, Fennie KP, Metlay JP (2012) Upper respiratory tract microbial communities, acute otitis media pathogens, and antibiotic use in healthy and sick children. Appl Environ Microbiol 78(17):6262–6270
Teo SM, Mok D, Pham K, Kusel M, Serralha M, Troy N, Holt BJ, Hales BJ, Walker ML, Hollams E, Bochkov YA, Grindle K, Johnston SL, Gern JE, Sly PD, Holt PG, Holt KE, Inouye M (2015) The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe 17(5):704–715
LeVine AM, Whitsett JA, Gwozdz JA, Richardson TR, Fisher JH, Burhans MS, Korfhagen TR (2000) Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol 165(7):3934–3940
Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, Beck JM, Curtis JL, Huffnagle GB (2015) Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio 6(2):e00037
Huffnagle GB, Dickson RP, Lukacs NW (2017) The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol 10(2):299–306
Cameron SJS, Lewis KE, Huws SA, Hegarty MJ, Lewis PD, Pachebat JA, Mur LAJ (2017) A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS One 12(5):e0177062-e
Ren Y, Su H, She Y, Dai C, Xie D, Narrandes S, Huang S, Chen C, Xu W (2019) Whole genome sequencing revealed microbiome in lung adenocarcinomas presented as ground-glass nodules. Transl Lung Cancer Res 8(3):235–246
Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, Ameh S, Sandel D, Liang XS, Mazzilli S, Whary MT, Meyerson M, Germain R, Blainey PC, Fox JG, Jacks T (2019) Commensal microbiota promote lung cancer development via gammadelta T cells. Cell 176(5):998-1013.e16
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6(11):1123–1132
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6(11):1133–1141
McGeachy MJ, Cua DJ, Gaffen SL (2019) The IL-17 family of cytokines in health and disease. Immunity 50(4):892–906
Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, Olivenstein R, Elias J, Chakir J (2001) IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol 108(3):430–438
Halwani R, Sultana A, Vazquez-Tello A, Jamhawi A, Al-Masri AA, Al-Muhsen S (2017) Th-17 regulatory cytokines IL-21, IL-23, and IL-6 enhance neutrophil production of IL-17 cytokines during asthma. J Asthma 54(9):893–904
Yang D, Chen X, Wang J, Lou Q, Lou Y, Li L, Wang H, Chen J, Wu M, Song X, Qian Y (2019) Dysregulated lung commensal bacteria drive interleukin-17B production to promote pulmonary fibrosis through their outer membrane vesicles. Immunity 50(3):692-706.e7
Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA (2010) Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207(3):535–552
Paats MS, Bergen IM, Hoogsteden HC, van der Eerden MM, Hendriks RW (2012) Systemic CD4+ and CD8+ T-cell cytokine profiles correlate with GOLD stage in stable COPD. Eur Respir J 40(2):330–337
Di Stefano A, Caramori G, Gnemmi I, Contoli M, Vicari C, Capelli A, Magno F, D’Anna SE, Zanini A, Brun P, Casolari P, Chung KF, Barnes PJ, Papi A, Adcock I, Balbi B (2009) T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol 157(2):316–324
Eustace A, Smyth LJC, Mitchell L, Williamson K, Plumb J, Singh D (2011) Identification of cells expressing IL-17A and IL-17F in the lungs of patients with COPD. Chest 139(5):1089–1100
Vargas-Rojas MI, Ramírez-Venegas A, Limón-Camacho L, Ochoa L, Hernández-Zenteno R, Sansores RH (2011) Increase of Th17 cells in peripheral blood of patients with chronic obstructive pulmonary disease. Respir Med 105(11):1648–1654
Zhang J, Chu S, Zhong X, Lao Q, He Z, Liang Y (2013) Increased expression of CD4+IL-17+ cells in the lung tissue of patients with stable chronic obstructive pulmonary disease (COPD) and smokers. Int Immunopharmacol 15(1):58–66
Xu W, Li R, Sun Y (2019) Increased IFN-γ-producing Th17/Th1 cells and their association with lung function and current smoking status in patients with chronic obstructive pulmonary disease. BMC Pulm Med 19(1):137
Tan H-L, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC (2011) The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med 184(2):252–258
Brodlie M, McKean MC, Johnson GE, Anderson AE, Hilkens CM, Fisher AJ, Corris PA, Lordan JL, Ward C (2011) Raised interleukin-17 is immunolocalised to neutrophils in cystic fibrosis lung disease. The Eur Respir J 37(6):1378–1385
Chen ACH, Martin ML, Lourie R, Rogers GB, Burr LD, Hasnain SZ, Bowler SD, McGuckin MA, Serisier DJ (2015) Adult non-cystic fibrosis bronchiectasis is characterised by airway luminal Th17 pathway activation. PLoS One 10(3):e0119325-e
McInnes IB, Schett G (2011) The pathogenesis of rheumatoid arthritis. N Engl J Med 365(23):2205–2219
Gaffen SL (2009) The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr Rheumatol Rep 11(5):365–370
Van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, Dolhain RJ, Lubberts E (2011) Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum 63(1):73–83
Chalan P, Kroesen B-J, van der Geest KSM, Huitema MG, Abdulahad WH, Bijzet J, Brouwer E, Boots AMH (2013) Circulating CD4+CD161+ T lymphocytes are increased in seropositive arthralgia patients but decreased in patients with newly diagnosed rheumatoid arthritis. PLoS One 8(11):e79370
Kouri V-P, Olkkonen J, Ainola M, Li T-F, Björkman L, Konttinen YT, Mandelin J (2013) Neutrophils produce interleukin-17B in rheumatoid synovial tissue. Rheumatology 53(1):39–47
Liu D, Cao T, Wang N, Liu C, Ma N, Tu R, Min X (2016) IL-25 attenuates rheumatoid arthritis through suppression of Th17 immune responses in an IL-13-dependent manner. Sci Rep 6(1):36002
Akitsu A, Ishigame H, Kakuta S, Chung SH, Ikeda S, Shimizu K, Kubo S, Liu Y, Umemura M, Matsuzaki G, Yoshikai Y, Saijo S, Iwakura Y (2015) IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vgamma6(+)gammadelta T cells. Nat Commun 6:7464
Cornelissen F, Mus AM, Asmawidjaja PS, van Hamburg JP, Tocker J, Lubberts E (2009) Interleukin-23 is critical for full-blown expression of a non-autoimmune destructive arthritis and regulates interleukin-17A and RORgammat in gammadelta T cells. Arthritis Res Ther 11(6):R194
Ito Y, Usui T, Kobayashi S, Iguchi-Hashimoto M, Ito H, Yoshitomi H, Nakamura T, Shimizu M, Kawabata D, Yukawa N, Hashimoto M, Sakaguchi N, Sakaguchi S, Yoshifuji H, Nojima T, Ohmura K, Fujii T, Mimori T (2009) Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheum 60(8):2294–2303
Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O’Brien RL (2007) Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J Immunol 179(8):5576–5583
McGinley AM, Edwards SC, Raverdeau M, Mills KHG (2018) Th17 cells, γδ T cells and their interplay in EAE and multiple sclerosis. J Autoimmun 87:97–108
Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, Farrelly C, Tubridy N, Mills KHG (2009) CD39<sup>+</sup>Foxp3<sup>+</sup> regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol 183(11):7602
Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B (2011) Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol 12(3):255–263
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L (2008) Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172(1):146–155
McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ (2007) TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 8(12):1390–1397
Hatfield JK, Brown MA (2015) Group 3 innate lymphoid cells accumulate and exhibit disease-induced activation in the meninges in EAE. Cell Immunol 297(2):69–79
Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH (2009) Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31(2):331–341
Li J, Casanova JL, Puel A (2018) Mucocutaneous IL-17 immunity in mice and humans: host defense vs. excessive inflammation. Mucosal Immunol 11(3):581–9
Peck A, Mellins ED (2010) Precarious balance: Th17 cells in host defense. Infect Immun 78(1):32–38
Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK (2001) Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194(4):519–527
Guglani L, Khader SA (2010) Th17 cytokines in mucosal immunity and inflammation. Curr Opin HIV AIDS 5(2):120–127
Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G (2011) Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479(7374):538–541
Horai R, Silver PB, Chen J, Agarwal RK, Chong WP, Jittayasothorn Y, Mattapallil MJ, Nguyen S, Natarajan K, Villasmil R, Wang P, Karabekian Z, Lytton SD, Chan CC, Caspi RR (2013) Breakdown of immune privilege and spontaneous autoimmunity in mice expressing a transgenic T cell receptor specific for a retinal autoantigen. J Autoimmun 44:21–33
Maeda Y, Takeda K (2019) Host-microbiota interactions in rheumatoid arthritis. Exp Mol Med 51(12):1–6
Zakostelska Z, Malkova J, Klimesova K, Rossmann P, Hornova M, Novosadova I, Stehlikova Z, Kostovcik M, Hudcovic T, Stepankova R, Juzlova K, Hercogova J, Tlaskalova-Hogenova H, Kverka M (2016) Intestinal microbiota promotes psoriasis-like skin inflammation by enhancing Th17 response. PLoS One 11(7):e0159539
McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, Cua DJ (2009) The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol 10(3):314–324
Feng T, Qin H, Wang L, Benveniste EN, Elson CO, Cong Y (2011) Th17 cells induce colitis and promote Th1 cell responses through IL-17 induction of innate IL-12 and IL-23 production. J Immunol 186(11):6313–6318
Godinez I, Keestra AM, Spees A, Baumler AJ (2011) The IL-23 axis in Salmonella gastroenteritis. Cell Microbiol 13(11):1639–1647
Lee SJ, McLachlan JB, Kurtz JR, Fan D, Winter SE, Baumler AJ, Jenkins MK, McSorley SJ (2012) Temporal expression of bacterial proteins instructs host CD4 T cell expansion and Th17 development. PLoS Pathog 8(1):e1002499
Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, Nicod LP, Lloyd CM, Marsland BJ (2014) Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med 20(6):642–647
Herbst T, Sichelstiel A, Schär C, Yadava K, Bürki K, Cahenzli J, McCoy K, Marsland BJ, Harris NL (2011) Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Respir Crit Care Med 184(2):198–205
Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, Blumberg RS (2012) Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336(6080):489–493
Hill DA, Siracusa MC, Abt MC, Kim BS, Kobuley D, Kubo M, Kambayashi T, Larosa DF, Renner ED, Orange JS, Bushman FD, Artis D (2012) Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med 18(4):538–546
Russell SL, Gold MJ, Hartmann M, Willing BP, Thorson L, Wlodarska M, Gill N, Blanchet MR, Mohn WW, McNagny KM, Finlay BB (2012) Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep 13(5):440–447
Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, Panzer AR, LaMere B, Rackaityte E, Lukacs NW, Wegienka G, Boushey HA, Ownby DR, Zoratti EM, Levin AM, Johnson CC, Lynch SV (2016) Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med 22(10):1187–1191
Preston JA, Thorburn AN, Starkey MR, Beckett EL, Horvat JC, Wade MA, Sullivan BJ, Thomas R, Beagley KW, Gibson PG, Foster PS, Hansbro PM (2011) Streptococcus pneumoniae infection suppresses allergic airways disease by inducing regulatory T-cells. Eur Respir J 37(1):53
Huang YJ, Nariya S, Harris JM, Lynch SV, Choy DF, Arron JR, Boushey H (2015) The airway microbiome in patients with severe asthma: Associations with disease features and severity. J Allergy Clin Immunol 136(4):874–884
Zhang Q, Cox M, Liang Z, Brinkmann F, Cardenas PA, Duff R, Bhavsar P, Cookson W, Moffatt M, Chung KF (2016) Airway microbiota in severe asthma and relationship to asthma severity and phenotypes. PLoS One 11(4):e0152724
Huang YJ, Marsland BJ, Bunyavanich S, O’Mahony L, Leung DY, Muraro A, Fleisher TA (2017) The microbiome in allergic disease: Current understanding and future opportunities-2017 PRACTALL document of the American Academy of Allergy, Asthma & Immunology and the European Academy of Allergy and Clinical Immunology. J Allergy Clin Immunol 139(4):1099–1110
Jounio U, Juvonen R, Bloigu A, Silvennoinen-Kassinen S, Kaijalainen T, Kauma H, Peitso A, Saukkoriipi A, Vainio O, Harju T, Leinonen M (2010) Pneumococcal carriage is more common in asthmatic than in non-asthmatic young men. Clin Respir J 4(4):222-9
Marri PR, Stern DA, Wright AL, Billheimer D, Martinez FD. Asthma-associated differences in microbial composition of induced sputum (2013) J Allergy Clin Immunol 131(2):346–52.e1–3
Green BJ, Wiriyachaiporn S, Grainge C, Rogers GB, Kehagia V, Lau L, Carroll MP, Bruce KD, Howarth PH (2014) Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLoS One 9(6):e100645-e
Bisgaard H, Hermansen MN, Buchvald F, Loland L, Halkjaer LB, Bonnelykke K, Brasholt M, Heltberg A, Vissing NH, Thorsen SV, Stage M, Pipper CB (2007) Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med 357(15):1487–1495
Larsen JM, Brix S, Thysen AH, Birch S, Rasmussen MA, Bisgaard H (2014) Children with asthma by school age display aberrant immune responses to pathogenic airway bacteria as infants. J Allergy Clin Immunol 133(4):1008–1013
Folsgaard NV, Schjorring S, Chawes BL, Rasmussen MA, Krogfelt KA, Brix S, Bisgaard H (2013) Pathogenic bacteria colonizing the airways in asymptomatic neonates stimulates topical inflammatory mediator release. Am J Respir Crit Care Med 187(6):589–595
Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, Barnes PJ (1999) Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med 160(5 Pt 1):1532–1539
Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID (2002) Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax 57(10):875–879
Agache I, Ciobanu C, Agache C, Anghel M (2010) Increased serum IL-17 is an independent risk factor for severe asthma. Respir Med 104(8):1131–1137
Taylor SL, Leong LEX, Choo JM, Wesselingh S, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Jenkins C, Peters MJ, Baraket M, Marks GB, Gibson PG, Simpson JL, Rogers GB (2018) Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol 141(1):94-103.e15
Bisgaard H, Hermansen MN, Bonnelykke K, Stokholm J, Baty F, Skytt NL, Aniscenko J, Kebadze T, Johnston SL (2010) Association of bacteria and viruses with wheezy episodes in young children: prospective birth cohort study. BMJ (Clinical research ed) 341:c4978
Wood LG, Simpson JL, Hansbro PM, Gibson PG (2010) Potentially pathogenic bacteria cultured from the sputum of stable asthmatics are associated with increased 8-isoprostane and airway neutrophilia. Free Radic Res 44(2):146–154
Simpson JL, Daly J, Baines KJ, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, Hugenholtz P, Willner D, Gibson PG (2016) Airway dysbiosis: Haemophilus influenzae and Tropheryma in poorly controlled asthma. Eur Respir J 47(3):792–800
Yang B, Liu R, Yang T, Jiang X, Zhang L, Wang L, Wang Q, Luo Z, Liu E, Fu Z (2015) Neonatal Streptococcus pneumoniae infection may aggravate adulthood allergic airways disease in association with IL-17A. PLoS One 10(3):e0123010
McCann JR, Mason SN, Auten RL, St. Geme III JW, Seed PC (2016) Early-life intranasal colonization with nontypeable Haemophilus influenzae exacerbates juvenile airway disease in mice. Infect Immun 84(7):2022-2030
Yang X, Wang Y, Zhao S, Wang R, Wang C (2018) Long-term exposure to low-dose Haemophilus influenzae during allergic airway disease drives a steroid-resistant neutrophilic inflammation and promotes airway remodeling. Oncotarget 9(38):24898–24913
Essilfie A-T, Simpson JL, Horvat JC, Preston JA, Dunkley ML, Foster PS, Gibson PG, Hansbro PM (2011) Haemophilus influenzae infection drives IL-17-mediated neutrophilic allergic airways disease. PLoS Pathog 7(10):e1002244
Alnahas S, Hagner S, Raifer H, Kilic A, Gasteiger G, Mutters R, Hellhund A, Prinz I, Pinkenburg O, Visekruna A, Garn H, Steinhoff U (2017) IL-17 and TNF-α are key mediators of Moraxella catarrhalis triggered exacerbation of allergic airway inflammation. Front Immunol 8(1562)
Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN (2009) Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med 180(8):720–730
Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, Saito Y, Hatano M, Tokuhisa T, Iwakura Y, Puccetti P, Iwamoto I, Nakajima H (2008) IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med 178(10):1023–1032
Lee HS, Park DE, Lee JW, Chang Y, Kim HY, Song WJ, Kang HR, Park HW, Chang YS, Cho SH (2017) IL-23 secreted by bronchial epithelial cells contributes to allergic sensitization in asthma model: role of IL-23 secreted by bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 312(1):L13-L21
Lee HS, Park DE, Lee JW, Sohn KH, Cho SH, Park HW (2020) Role of interleukin-23 in the development of nonallergic eosinophilic inflammation in a murine model of asthma. Exp Mol Med 52(1):92–104
Larsen JM, Steen-Jensen DB, Laursen JM, Sondergaard JN, Musavian HS, Butt TM, Brix S (2012) Divergent pro-inflammatory profile of human dendritic cells in response to commensal and pathogenic bacteria associated with the airway microbiota. PLoS One 7(2):e31976
Mammen MJ, Sethi S (2016) COPD and the microbiome. Respirology 21(4):590–599
Segal LN, Alekseyenko AV, Clemente JC, Kulkarni R, Wu B, Chen H, Berger KI, Goldring RM, Rom WN, Blaser MJ, Weiden MD (2013) Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome 1:19
Segal LN, Clemente JC, Tsay JC, Koralov SB, Keller BC, Wu BG, Li Y, Shen N, Ghedin E, Morris A, Diaz P, Huang L, Wikoff WR, Ubeda C, Artacho A, Rom WN, Sterman DH, Collman RG, Blaser MJ, Weiden MD (2016) Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol 1:16031
Dy R, Sethi S (2016) The lung microbiome and exacerbations of COPD. Curr Opin Pulm Med 22(3):196–202
Mayhew D, Devos N, Lambert C, Brown JR, Clarke SC, Kim VL, Magid-Slav M, Miller BE, Ostridge KK, Patel R, Sathe G, Simola DF, Staples KJ, Sung R, Tal-Singer R, Tuck AC, Van Horn S, Weynants V, Williams NP, Devaster JM, Wilkinson TMA (2018) Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax 73(5):422–430
Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, Erb-Downward JR, Huffnagle GB, Hayashi S, Elliott WM, Cooper J, Sin DD, Lenburg ME, Spira A, Mohn WW, Hogg JC (2015) Host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 192(4):438–445
Wang Z, Maschera B, Lea S, Kolsum U, Michalovich D, Van Horn S, Traini C, Brown JR, Hessel EM, Singh D (2019) Airway host-microbiome interactions inchronic obstructive pulmonary disease. Respir Res 20(1):113
Murphy TF, Brauer AL, Grant BJ, Sethi S (2005) Moraxella catarrhalis in chronic obstructive pulmonary disease: burden of disease and immune response. Am J Respir Crit Care Med 172(2):195–199
Zou Y, Chen X, Liu J, Zhou DB, Kuang X, Xiao J, Yu Q, Lu X, Li W, Xie B, Chen Q (2017) Serum IL-1β and IL-17 levels in patients with COPD: associations with clinical parameters. Int J Chron Obstruct Pulmon Dis 12:1247–1254
Zhang L, Cheng Z, Liu W, Wu K (2013) Expression of interleukin (IL)-10, IL-17A and IL-22 in serum and sputum of stable chronic obstructive pulmonary disease patients. COPD 10(4):459–465
Ponce-Gallegos MA, Perez-Rubio G, Ambrocio-Ortiz E, Partida-Zavala N, Hernandez-Zenteno R, Flores-Trujillo F, Garcia-Gomez L, Hernandez-Perez A, Ramirez-Venegas A, Falfan-Valencia R (2020) Genetic variants in IL17A and serum levels of IL-17A are associated with COPD related to tobacco smoking and biomass burning. Sci Rep 10(1):784
Park H, Shin JW, Park S-G, Kim W (2014) Microbial communities in the upper respiratory tract of patients with asthma and chronic obstructive pulmonary disease. PLoS One 9(10):e109710
Yadava K, Pattaroni C, Sichelstiel AK, Trompette A, Gollwitzer ES, Salami O, von Garnier C, Nicod LP, Marsland BJ (2015) Microbiota promotes chronic pulmonary inflammation by enhancing IL-17A and autoantibodies. Am J Respir Crit Care Med 193(9):975–987
Xiong J, Tian J, Zhou L, Le Y, Sun Y (2020) Interleukin-17A Deficiency Attenuated Emphysema and Bone Loss in Mice Exposed to Cigarette Smoke. Int J Chron Obstruct Pulmon Dis 15:301–310
Chen K, Pociask DA, McAleer JP, Chan YR, Alcorn JF, Kreindler JL, Keyser MR, Shapiro SD, Houghton AM, Kolls JK, Zheng M (2011) IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS One 6(5):e20333
Roos AB, Sethi S, Nikota J, Wrona CT, Dorrington MG, Sanden C, Bauer CM, Shen P, Bowdish D, Stevenson CS, Erjefalt JS, Stampfli MR (2015) IL-17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 192(4):428–437
O’Sullivan BP, Freedman SD (2009) Cystic fibrosis. Lancet 373(9678):1891–1904
Davis PB (2006) Cystic fibrosis since 1938. Am J Respir Crit Care Med 173(5):475–482
Gaggar A, Hector A, Bratcher PE, Mall MA, Griese M, Hartl D (2011) The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur Respir J 38(3):721–727
Meyer KC, Lewandoski JR, Zimmerman JJ, Nunley D, Calhoun WJ, Dopico GA (1991) Human neutrophil elastase and elastase/alpha 1-antiprotease complex in cystic fibrosis. Comparison with interstitial lung disease and evaluation of the effect of intravenously administered antibiotic therapy. Am Rev Respir Dis 144(3 Pt 1):580–5
Decraene A, Willems-Widyastuti A, Kasran A, De Boeck K, Bullens DM, Dupont LJ (2010) Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable Cystic Fibrosis patients. Respir Res 11(1):177
Brodlie M, McKean MC, Johnson GE, Anderson AE, Hilkens CMU, Fisher AJ, Corris PA, Lordan JL, Ward C (2011) Raised interleukin-17 is immunolocalised to neutrophils in cystic fibrosis lung disease. Eur Respir J 37(6):1378–1385
McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C, Finder JD, Pilewski JM, Carreno BM, Goldman SJ, Pirhonen J, Kolls JK (2005) Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol 175(1):404–412
Dubin PJ, McAllister F, Kolls JK (2007) Is cystic fibrosis a TH17 disease? Inflamm Res 56(6):221–227
Chan YR, Chen K, Duncan SR, Lathrop KL, Latoche JD, Logar AJ, Pociask DA, Wahlberg BJ, Ray P, Ray A, Pilewski JM, Kolls JK (2013) Patients with cystic fibrosis have inducible IL-17+IL-22+ memory cells in lung draining lymph nodes. J Allergy Clin Immunol 131(4):1117–29, 29.e1–5
Frayman KB, Wylie KM, Armstrong DS, Carzino R, Davis SD, Ferkol TW, Grimwood K, Storch GA, Ranganathan SC (2019) Differences in the lower airway microbiota of infants with and without cystic fibrosis. J Cyst Fibros 18(5):646–652
Muhlebach MS, Zorn BT, Esther CR, Hatch JE, Murray CP, Turkovic L, Ranganathan SC, Boucher RC, Stick SM, Wolfgang MC (2018) Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog 14(1):e1006798-e
Taylor SL, Leong LEX, Ivey KL, Wesselingh S, Grimwood K, Wainwright CE, Rogers GB (2020) Total bacterial load, inflammation, and structural lung disease in paediatric cystic fibrosis. J Cyst Fibros
McGuigan L, Callaghan M (2015) The evolving dynamics of the microbial community in the cystic fibrosis lung. Environ Microbiol 17(1):16–28
Acosta N, Heirali A, Somayaji R, Surette MG, Workentine ML, Sibley CD, Rabin HR, Parkins MD (2018) Sputum microbiota is predictive of long-term clinical outcomes in young adults with cystic fibrosis. Thorax 73(11):1016–1025
Martiniano SL, Nick JA, Daley CL (2016) Nontuberculous Mycobacterial Infections in Cystic Fibrosis. Clin Chest Med 37(1):83–96
Gilligan PH (2014) Infections in Patients with Cystic Fibrosis: Diagnostic Microbiology Update. Clin Lab Med 34(2):197–217
Breuer O, Schultz A, Turkovic L, de Klerk N, Keil AD, Brennan S, Harrison J, Robertson C, Robinson PJ, Sly PD, Ranganathan S, Stick SM, Caudri D (2019) Changing prevalence of lower airway infections in young children with cystic fibrosis. Am J Respir Crit Care Med 200(5):590–599
Garcia-Nuñez M, Garcia-Gonzalez M, Pomares X, Montón C, Millares L, Quero S, Prina E, Asensio O, Bosque M, Capilla S, Cuevas O, Monsó E (2020) The respiratory microbiome in cystic fibrosis: compartment patterns and clinical relationships in early stage disease. Front Microbiol 11(1463)
Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, Tunney MM, Wolfgang MC (2012) The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One 7(9):e45001
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET (2012) Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 186(9):857–865
Surette MG (2014) The cystic fibrosis lung microbiome. Ann Am Thorac Soc 11(Supplement 1):S61–S65
Cuthbertson L, Walker AW, Oliver AE, Rogers GB, Rivett DW, Hampton TH, Ashare A, Elborn JS, De Soyza A, Carroll MP, Hoffman LR, Lanyon C, Moskowitz SM, O'Toole GA, Parkhill J, Planet PJ, Teneback CC, Tunney MM, Zuckerman JB, Bruce KD, van der Gast CJ (2020) Lung function and microbiota diversity in cystic fibrosis. Microbiome 8(1):45
Zemanick ET, Wagner BD, Robertson CE, Ahrens RC, Chmiel JF, Clancy JP, Gibson RL, Harris WT, Kurland G, Laguna TA, McColley SA, McCoy K, Retsch-Bogart G, Sobush KT, Zeitlin PL, Stevens MJ, Accurso FJ, Sagel SD, Harris JK (2017) Airway microbiota across age and disease spectrum in cystic fibrosis. Eur Respir J 50(5):1700832
Armougom F, Bittar F, Stremler N, Rolain JM, Robert C, Dubus JC, Sarles J, Raoult D, La Scola B (2009) Microbial diversity in the sputum of a cystic fibrosis patient studied with 16S rDNA pyrosequencing. Eur J Clin Microbiol Infect Dis 28(9):1151–1154
Guss AM, Roeselers G, Newton IL, Young CR, Klepac-Ceraj V, Lory S, Cavanaugh CM (2011) Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J 5(1):20–29
Acosta N, Whelan FJ, Somayaji R, Poonja A, Surette MG, Rabin HR, Parkins MD (2017) The evolving cystic fibrosis microbiome: a comparative cohort study spanning 16 years. Ann Am Thorac Soc 14(8):1288–1297
Rogers GB, Hart CA, Mason JR, Hughes M, Walshaw MJ, Bruce KD (2003) Bacterial diversity in cases of lung infection in cystic fibrosis patients: 16S ribosomal DNA (rDNA) length heterogeneity PCR and 16S rDNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol 41(8):3548–3558
Hogan DA, Willger SD, Dolben EL, Hampton TH, Stanton BA, Morrison HG, Sogin ML, Czum J, Ashare A (2016) Analysis of lung microbiota in bronchoalveolar lavage, protected brush and sputum samples from subjects with mild-to-moderate cystic fibrosis lung disease. PLoS One 11(3):e0149998
Berdah L, Taytard J, Leyronnas S, Clement A, Boelle PY, Corvol H (2018) Stenotrophomonas maltophilia: a marker of lung disease severity. Pediatr Pulmonol 53(4):426–430
Bayes HK, Ritchie ND, Evans TJ (2016) Interleukin-17 is required for control of chronic lung infection caused by Pseudomonas aeruginosa. Infect Immun 84(12):3507–3516
Taylor PR, Bonfield TL, Chmiel JF, Pearlman E (2016) Neutrophils from F508del cystic fibrosis patients produce IL-17A and express IL-23 - dependent IL-17RC. Clin Immunol 170:53–60
Hsu D, Taylor P, Fletcher D, van Heeckeren R, Eastman J, van Heeckeren A, Davis P, Chmiel JF, Pearlman E, Bonfield TL (2016) Interleukin-17 pathophysiology and therapeutic intervention in cystic fibrosis lung infection and inflammation. Infect Immun 84(9):2410–2421
Dubin PJ, Kolls JK (2007) IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol 292(2):L519–28
Magis-Escurra C, Reijers MH (2015) Bronchiectasis. BMJ. Clin Evid 2015:1507
Schäfer J, Griese M, Chandrasekaran R, Chotirmall SH, Hartl D (2018) Pathogenesis, imaging and clinical characteristics of CF and non-CF bronchiectasis. BMC Pulm Med 18(1):79
O’Brien C, Guest PJ, Hill SL, Stockley RA (2000) Physiological and radiological characterisation of patients diagnosed with chronic obstructive pulmonary disease in primary care. Thorax 55(8):635–642
Van der Gast CJ, Cuthbertson L, Rogers GB, Pope C, Marsh RL, Redding GJ, Bruce KD, Chang AB, Hoffman LR (2014) Three clinically distinct chronic pediatric airway infections share a common core microbiota. Ann Am Thorac Soc 11(7):1039–1048
Cox MJ, Turek EM, Hennessy C, Mirza GK, James PL, Coleman M, Jones A, Wilson R, Bilton D, Cookson WO, Moffatt MF, Loebinger MR (2017) Longitudinal assessment of sputum microbiome by sequencing of the 16S rRNA gene in non-cystic fibrosis bronchiectasis patients. PLoS One 12(2):e0170622
Rogers GB, van der Gast CJ, Cuthbertson L, Thomson SK, Bruce KD, Martin ML, Serisier DJ (2013) Clinical measures of disease in adult non-CF bronchiectasis correlate with airway microbiota composition. Thorax 68(8):731–737
Fowler SJ, French J, Screaton NJ, Foweraker J, Condliffe A, Haworth CS, Exley AR, Bilton D (2006) Nontuberculous mycobacteria in bronchiectasis: prevalence and patient characteristics. Eur Respir J 28(6):1204–1210
Tunney MM, Einarsson GG, Wei L, Drain M, Klem ER, Cardwell C, Ennis M, Boucher RC, Wolfgang MC, Elborn JS (2013) Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respir Crit Care Med 187(10):1118–1126
Fouka E, Lamprianidou E, Arvanitidis K, Filidou E, Kolios G, Miltiades P, Paraskakis E, Antoniadis A, Kotsianidis I, Bouros D (2014) Low-dose clarithromycin therapy modulates Th17 response in non-cystic fibrosis bronchiectasis patients. Lung 192(6):849–855
Maher TM, Wells AU, Laurent GJ (2007) Idiopathic pulmonary fibrosis: multiple causes and multiple mechanisms? Eur Respir J 30(5):835–839
Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, Fahy RJ, Crotty TB, Hirani N, Flynn RJ, Voehringer D, McKenzie ANJ, Donnelly SC, Fallon PG (2014) IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci U S A 111(1):367–372
Gasse P, Riteau N, Vacher R, Michel ML, Fautrel A, di Padova F, Fick L, Charron S, Lagente V, Eberl G, Le Bert M, Quesniaux VF, Huaux F, Leite-de-Moraes M, Ryffel B, Couillin I (2011) IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS One 6(8):e23185
Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M, Barnes JL, Chandrasekar B (2007) IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ Physiol 293(6):H3356–H3365
Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE Jr (2008) Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest 133(1):226–232
O’Dwyer DN, Ashley SL, Gurczynski SJ, Xia M, Wilke C, Falkowski NR, Norman KC, Arnold KB, Huffnagle GB, Salisbury ML, Han MK, Flaherty KR, White ES, Martinez FJ, Erb-Downward JR, Murray S, Moore BB, Dickson RP (2019) Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am J Respir Crit Care Med 199(9):1127–1138
Invernizzi R, Barnett J, Rawal B, Nair A, Ghai P, Kingston S, Chua F, Wu Z, Wells AU, Renzoni ER, Nicholson AG, Rice A, Lloyd CM, Byrne AJ, Maher TM, Devaraj A, Molyneaux PL (2020) Bacterial burden in the lower airways predicts disease progression in idiopathic pulmonary fibrosis and is independent of radiological disease extent. Eur Respir J 1901519
Shulgina L, Cahn AP, Chilvers ER, Parfrey H, Clark AB, Wilson EC, Twentyman OP, Davison AG, Curtin JJ, Crawford MB, Wilson AM (2013) Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: a randomised controlled trial. Thorax 68(2):155–162
Kawamura K, Ichikado K, Yasuda Y, Anan K, Suga M (2017) Azithromycin for idiopathic acute exacerbation of idiopathic pulmonary fibrosis: a retrospective single-center study. BMC Pulm Med 17(1):94
Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, Moore BB, White ES, Flaherty KR, Huffnagle GB, Martinez FJ (2014) Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med 2(7):548–556
Molyneaux PL, Cox MJ, Willis-Owen SAG, Mallia P, Russell KE, Russell A-M, Murphy E, Johnston SL, Schwartz DA, Wells AU, Cookson WOC, Maher TM, Moffatt MF (2014) The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 190(8):906–913
Tong X, Su F, Xu X, Xu H, Yang T, Xu Q, Dai H, Huang K, Zou L, Zhang W, Pei S, Xiao F, Li Y, Wang C (2019) Alterations to the lung microbiome in idiopathic pulmonary fibrosis patients. Front Cell Infect Microbiol 9:149
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T (1999) IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103(9):1345–1352
Ruscitti P, Di Benedetto P, Berardicurti O, Liakouli V, Carubbi F, Cipriani P, Giacomelli R (2018) Adipocytokines in rheumatoid arthritis: the hidden link between inflammation and cardiometabolic comorbidities. J Immunol Res 2018:8410182
Sato K, Takayanagi H (2006) Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr Opin Rheumatol 18(4):419–426
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ (2003) Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med 198(12):1951–1957
Guo W, Yu D, Wang X, Luo C, Chen Y, Lei W, Wang C, Ge Y, Xue W, Tian Q, Gao X, Yao W (2016) Anti-inflammatory effects of interleukin-23 receptor cytokine-binding homology region rebalance T cell distribution in rodent collagen-induced arthritis. Oncotarget 7(22):31800–31813
Chalan P, Kroesen BJ, van der Geest KS, Huitema MG, Abdulahad WH, Bijzet J, Brouwer E, Boots AM (2013) Circulating CD4+CD161+ T lymphocytes are increased in seropositive arthralgia patients but decreased in patients with newly diagnosed rheumatoid arthritis. PLoS One 8(11):e79370
Yago T, Nanke Y, Kawamoto M, Furuya T, Kobashigawa T, Kamatani N, Kotake S (2007) IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res Ther 9(5):R96
Mitsdoerffer M, Lee Y, Jager A, Kim HJ, Korn T, Kolls JK, Cantor H, Bettelli E, Kuchroo VK (2010) Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A 107(32):14292–14297
Deane KD, Norris JM, Holers VM (2010) Preclinical rheumatoid arthritis: identification, evaluation, and future directions for investigation. Rheum Dis Clin North Am 36(2):213–241
MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, Silman AJ (2000) Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 43(1):30–37
Svard A, Kastbom A, Reckner-Olsson A, Skogh T (2008) Presence and utility of IgA-class antibodies to cyclic citrullinated peptides in early rheumatoid arthritis: the Swedish TIRA project. Arthritis Res Ther 10(4):R75
Willis VC, Demoruelle MK, Derber LA, Chartier-Logan CJ, Parish MC, Pedraza IF, Weisman MH, Norris JM, Holers VM, Deane KD (2013) Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum 65(10):2545–2554
Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D (2010) Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32(6):815–827
Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, Teng F, Pasman L, Ortiz-Lopez A, Jupp R, Wu HJ, Kasper DL, Benoist C, Mathis D (2016) Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A 113(50):E8141–E8150
Evans-Marin H, Rogier R, Koralov SB, Manasson J, Roeleveld D, van der Kraan PM, Scher JU, Koenders MI, Abdollahi-Roodsaz S (2018) Microbiota-dependent involvement of Th17 cells in murine models of inflammatory arthritis. Arthritis Rheumatol 70(12):1971–1983
O’Dell JR, Blakely KW, Mallek JA, Eckhoff PJ, Leff RD, Wees SJ, Sems KM, Fernandez AM, Palmer WR, Klassen LW, Paulsen GA, Haire CE, Moore GF (2001) Treatment of early seropositive rheumatoid arthritis: a two-year, double-blind comparison of minocycline and hydroxychloroquine. Arthritis Rheum 44(10):2235–2241
Maeda Y, Kumanogoh A, Takeda K (2016) Altered composition of gut microbiota in rheumatoid arthritis patients. Nihon Rinsho Meneki Gakkai Kaishi 39(1):59–63
Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, Huttenhower C, Littman DR (2013) Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2:e01202
Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, Lan Z, Chen B, Li Y, Zhong H, Xie H, Jie Z, Chen W, Tang S, Xu X, Wang X, Cai X, Liu S, Xia Y, Li J, Qiao X, Al-Aama JY, Chen H, Wang L, Wu QJ, Zhang F, Zheng W, Li Y, Zhang M, Luo G, Xue W, Xiao L, Li J, Chen W, Xu X, Yin Y, Yang H, Wang J, Kristiansen K, Liu L, Li T, Huang Q, Li Y, Wang J (2015) The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 21(8):895–905
Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, Nelson H, Matteson EL, Taneja V (2016) An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med 8(1):43
Scher JU, Joshua V, Artacho A, Abdollahi-Roodsaz S, Öckinger J, Kullberg S, Sköld M, Eklund A, Grunewald J, Clemente JC, Ubeda C, Segal LN, Catrina AI (2016) The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome 4(1):60
Quirke AM, Perry E, Cartwright A, Kelly C, De Soyza A, Eggleton P, Hutchinson D, Venables PJ (2015) Bronchiectasis is a model for chronic bacterial infection inducing autoimmunity in rheumatoid arthritis. Arthritis Rheumatol 67(9):2335–2342
Zaccardelli A, Liu X, Ford JA, Cui J, Lu B, Chu SH, Schur PH, Speyer CB, Costenbader KH, Robinson WH, Sokolove J, Karlson EW, Camargo CA Jr, Sparks JA (2019) Asthma and elevation of anti-citrullinated protein antibodies prior to the onset of rheumatoid arthritis. Arthritis Res Ther 21(1):246
Symmons DP, Bankhead CR, Harrison BJ, Brennan P, Barrett EM, Scott DG, Silman AJ (1997) Blood transfusion, smoking, and obesity as risk factors for the development of rheumatoid arthritis: results from a primary care-based incident case-control study in Norfolk. England Arthritis Rheum 40(11):1955–1961
Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, Ronnelid J, Harris HE, Ulfgren AK, Rantapaa-Dahlqvist S, Eklund A, Padyukov L, Alfredsson L (2006) A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 54(1):38–46
Reynisdottir G, Olsen H, Joshua V, Engstrom M, Forsslund H, Karimi R, Skold CM, Nyren S, Eklund A, Grunewald J, Catrina AI (2016) Signs of immune activation and local inflammation are present in the bronchial tissue of patients with untreated early rheumatoid arthritis. Ann Rheum Dis 75(9):1722–1727
Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD (2006) Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest 116(12):3183–3194
Tong Y, Zheng L, Qing P, Zhao H, Li Y, Su L, Zhang Q, Zhao Y, Luo Y, Liu Y (2019) Oral microbiota perturbations are linked to high risk for rheumatoid arthritis. Front Cell Infect Microbiol 9:475
Moutsopoulos NM, Kling HM, Angelov N, Jin W, Palmer RJ, Nares S, Osorio M, Wahl SM (2012) Porphyromonas gingivalis promotes Th17 inducing pathways in chronic periodontitis. J Autoimmun 39(4):294–303
Mikuls TR, Payne JB, Yu F, Thiele GM, Reynolds RJ, Cannon GW, Markt J, McGowan D, Kerr GS, Redman RS, Reimold A, Griffiths G, Beatty M, Gonzalez SM, Bergman DA, Hamilton BC 3rd, Erickson AR, Sokolove J, Robinson WH, Walker C, Chandad F, O’Dell JR (2014) Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol 66(5):1090–1100
Quirke AM, Lugli EB, Wegner N, Hamilton BC, Charles P, Chowdhury M, Ytterberg AJ, Zubarev RA, Potempa J, Culshaw S, Guo Y, Fisher BA, Thiele G, Mikuls TR, Venables PJ (2014) Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: a potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann Rheum Dis 73(1):263–269
de Aquino SG, Abdollahi-Roodsaz S, Koenders MI, van de Loo FAJ, Pruijn GJM, Marijnissen RJ, Walgreen B, Helsen MM, van den Bersselaar LA, de Molon RS, Campos MJA, Cunha FQ, Cirelli JA, van den Berg WB (2014) Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1–driven Th17 response. J Immunol 192(9):4103
Moen K, Brun JG, Valen M, Skartveit L, Eribe EK, Olsen I, Jonsson R (2006) Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial DNAs. Clin Exp Rheumatol 24(6):656–663
Martinez-Martinez RE, Abud-Mendoza C, Patino-Marin N, Rizo-Rodriguez JC, Little JW, Loyola-Rodriguez JP (2009) Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J Clin Periodontol 36(12):1004–1010
McDonald WI, Sears TA (1970) The effects of experimental demyelination on conduction in the central nervous system. Brain 93(3):583–598
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421(6924):744–748
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201(2):233–240
Van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, Dankers W, Verjans GM, De Vries HE, Lubberts E, Hintzen RQ (2018) T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain 141(5):1334–49
Correale J, Fiol M, Gilmore W (2006) The risk of relapses in multiple sclerosis during systemic infections. Neurology 67(4):652–659
Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schlager C, Lodygin D, Heckelsmiller K, Nietfeld W, Ellwart J, Klinkert WE, Lottaz C, Nosov M, Brinkmann V, Spang R, Lehrach H, Vingron M, Wekerle H, Flugel-Koch C, Flugel A (2012) T cells become licensed in the lung to enter the central nervous system. Nature 488(7413):675-9
Glenn JD, Liu C, Whartenby KA (2019) Frontline Science: Induction of experimental autoimmune encephalomyelitis mobilizes Th17-promoting myeloid derived suppressor cells to the lung. J Leukoc Biol 105(5):829–841
Edwards SC, Higgins SC, Mills KHG (2015) Respiratory infection with a bacterial pathogen attenuates CNS autoimmunity through IL-10 induction. Brain Behav Immun 50:41–46
Kanayama M, Danzaki K, He Y-W, Shinohara ML (2016) Lung inflammation stalls Th17-cell migration en route to the central nervous system during the development of experimental autoimmune encephalomyelitis. Int Immunol 28(9):463–469
Chu F, Shi M, Lang Y, Shen D, Jin T, Zhu J, Cui L (2018) Gut Microbiota in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis: Current Applications and Future Perspectives. Mediators Inflamm 2018:8168717
Mirza A, Forbes JD, Zhu F, Bernstein CN, Van Domselaar G, Graham M, Waubant E, Tremlett H (2020) The multiple sclerosis gut microbiota: a systematic review. Mult Scler Relat Disord 37:101427
Winter SE, Baumler AJ (2014) Dysbiosis in the inflamed intestine: chance favors the prepared microbe. Gut Microbes 5(1):71–73
Huang W, Thomas B, Flynn RA, Gavzy SJ, Wu L, Kim SV, Hall JA, Miraldi ER, Ng CP, Rigo F, Meadows S, Montoya NR, Herrera NG, Domingos AI, Rastinejad F, Myers RM, Fuller-Pace FV, Bonneau R, Chang HY, Acuto O, Littman DR (2015) DDX5 and its associated lncRNA Rmrp modulate TH17 cell effector functions. Nature 528(7583):517–522
Sanjurjo L, Aran G, Roher N, Valledor AF, Sarrias MR (2015) AIM/CD5L: a key protein in the control of immune homeostasis and inflammatory disease. J Leukoc Biol 98(2):173–184
Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Meyer Zu Horste G, Pawlak M, Kishi Y, Joller N, Karwacz K, Zhu C, Ordovas-Montanes M, Madi A, Wortman I, Miyazaki T, Sobel RA, Park H, Regev A, Kuchroo VK (2015) CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 163(6):1413–27
Funding
Open Access funding provided by the IReL Consortium. This work was supported by a Science Foundation Ireland (SFI) Starting Investigator Research Grant (15/SIRG/3426) to SJL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mannion, J.M., McLoughlin, R.M. & Lalor, S.J. The Airway Microbiome-IL-17 Axis: a Critical Regulator of Chronic Inflammatory Disease. Clinic Rev Allerg Immunol 64, 161–178 (2023). https://doi.org/10.1007/s12016-022-08928-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-022-08928-y