
Abstract
Immunoglobulin E (IgE) is a well-known key factor in allergic airway disease; however, its central role in non-allergic airway inflammation is often underestimated. In some airway diseases, IgE is produced as a result of allergic sensitization. However, in others, IgE production occurs despite the lack of a specific allergen. Although multiple pathways contribute to the production of IgE in airway disease, it is its activity in mediating the inflammatory response that is associated with disease. Therefore, an understanding of IgE as the unifying component of upper and lower airway diseases has important implications for both diagnosis and treatment. Understanding the role of IgE in each upper and lower airway disease highlights its potential utility as a diagnostic marker and therapeutic target. Further classification of these diseases by whether they are IgE mediated or non–IgE mediated, rather than by the existence of an underlying allergic component, accounts for both systemic and localized IgE activity. Improvements in diagnostic methodologies and standardization of clinical practices with this classification in mind can help identify patients with IgE-mediated diseases. In doing so, this group of patients can receive optimal care through targeted anti-IgE therapeutics, which have already demonstrated efficacy across numerous IgE-mediated upper and lower airway diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunoglobulin E (IgE) is widely accepted as an integral component of the pathogenesis of many allergic respiratory diseases, with increasing recognition of involvement in the non-allergic forms of disease. This is evident in both upper airway diseases, including allergic rhinitis (AR), non-allergic rhinitis (NAR), and chronic rhinosinusitis with nasal polyps (CRSwNP), and lower airway diseases, including allergic asthma and non-allergic asthma phenotypes (eosinophilic asthma, aspirin-exacerbated respiratory disease [AERD], and non-allergic occupational asthma) [1].
The shared aspects of otherwise discrete airway diseases have led to a continuous or “unified airway” concept for which pathological processes of the upper airway are thought to mirror lower airway events [2, 3]. The primary pathological process in both upper and lower airway disease is driven by inflammation, which can be caused by both allergic (immune response triggered by exposure to an allergen) and non-allergic (triggered by exposure to an environmental factor, resulting in activation of the innate immune pathway) disease mechanisms [4,5,6]. In support of this notion, upper and lower airway diseases often coexist [3, 7] and share common inflammatory processes, including those mediated by IgE [2, 8,9,10]. There are multiple types of IgE that have been linked to airway inflammation: specific IgE antibodies that are produced in response to allergen exposure, and non-specific IgE antibodies that are produced following the activation of the body’s innate immune system or through the action of superantigens [5, 6]. IgE is routinely described in the pathology of both acute and chronic inflammatory allergic diseases [11], and elevated IgE levels are also commonly reported in non-allergic late-onset asthma and nasal polyps (NP), in which the emergence of an IgE response need not be specific [10, 12]. In some cases, Staphylococcus aureus–derived enterotoxins have been proposed to act as superantigens in patients with NP or asthma to produce a local polyclonal IgE response [10, 13].
Historically, the term “allergy” was used to describe the organ-specific or systemic antigen-specific IgE-mediated immune response of the skin and mucosa in response to extrinsic allergen exposures [14]. However, as knowledge of disease pathology has increased, it has become clear that IgE-mediated disease exists regardless of allergen exposure. In NAR, symptomology is identical to AR, despite a negative skin prick test (SPT) for allergic response [14]. Similarly, between 10 and 33% of patients with asthma have an intrinsic, non-allergic form associated with a later, more severe presentation than allergic asthma [14]. It has also been shown that NAR in childhood is associated with a significant risk of developing asthma in adulthood, highlighting the need for exploring non-allergic components of disease [15]. In CRSwNP, IgE plays a major role in disease pathology, which is dominated by T helper 2 (Th2) inflammatory patterns [1, 16]. Although local IgE produced in the NP tissue can be the result of allergic stimulation, NP are also found in patients with no allergic sensitization. Here, the local IgE produced is polyclonal and functional, often as a result of the innate immune response or through the actions of S. aureus enterotoxins [1]. The underlying pathologies of allergic and non-allergic asthma are considerably different. Allergic asthma is defined as elevated systemic specific IgE and/or a positive SPT following an allergen exposure, and non-allergic asthma shows no such response to routine allergy tests [17]. In these patients with non-allergic asthma, production of IgE and local tissue IgE or eosinophil level drive disease, which typically occurs later in life and with more severe presentation compared with allergic asthma [17]. However, despite the lack of allergic sensitization, many diseases of the upper and lower airways are under the influence of local IgE activity [1].
Therefore, applying the learnings surrounding the role of IgE in upper and lower airway diseases in the context of both allergic and non-allergic phenotypes has important implications for treatment.
IgE-Mediated Disease
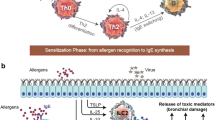
Many diseases of the upper and lower respiratory systems share a common underlying mechanism in the type 2 inflammatory pathway. This pathway is mediated by several key cell types, including eosinophils, mast cells, basophils, Th2 cells, type 2 innate lymphoid cells (ILC2), and IgE-producing B cells. Elevated IgE is a hallmark of type 2 inflammation, where it plays a key effector role in the propagation of the immune response [18]. For simplification, the IgE-mediated process can be broken down into two phases: IgE production and effects of IgE signaling (Fig. 1).

Mechanism of IgE-mediated upper and lower airway disease. In response to allergen exposure, dendritic cells present allergen-specific antigens to naïve T cells, which are activated and differentiate into Th2 cells. These Th2 cells produce key cytokines (IL-4, IL-13), prompting B cells to produce allergen-specific IgE. Alternatively, exposure to external stimuli such as bacteria, fungi, viruses, and particulates promotes epithelial release of IL-25, TSLP, and IL-33. These factors stimulate ILC2 cells to produce IL-5, IL-13, and to a lesser extent, IL-4, which in turn promote B cell production of IgE. Finally, superantigens, including Staphylococcal enterotoxins, can directly cross-link antigen-presenting cells with naïve T cells, bypassing the antigen presentation step, yielding polyclonal IgE. Once produced, local IgE acts on the FcεRI receptors of tissue-resident mast cells and basophils, prompting the release of histamine, leukotrienes, tryptase, and prostaglandin, which manifest as edema, vasodilation, and bronchoconstriction as part of the early response. IgE also binds to FcεRII receptors on B cells for enhanced antigen presentation. Later release of key cytokines recruits proinflammatory cells, including eosinophils and basophils, to the site of inflammation, and additionally promotes the overexpression of mucus-producing goblet cells and contributes to airway hyperresponsiveness. Crosstalk within the inflammatory pathway promotes a self-propagating cycle of chronic inflammation. The lower left side of the figure depicts the fibroblasts and mast cells within the supporting connective tissue of the nasal cavity, while the lower right side depicts the smooth muscle cell layer surrounding the lower respiratory airway. DC, dendritic cell; FcεRI, high-affinity immunoglobulin E receptor; FcεRII, low-affinity immunoglobulin E receptor; IgE, immunoglobulin E; IL, interleukin; ILC, innate lymphoid cell; PGD2, prostaglandin D2; Th2, T helper 2; TSLP, thymic stromal lymphopoietin
IgE Production
Evolutionarily, IgE contributes to the body’s defense against parasites such as helminths [5, 19]. However, environments favoring type 2 inflammation, such as acute and chronic exposures to factors including allergens, bacteria, fungi, viruses, and particulates can lead to B cell class switching and IgE production, which mediates the inflammatory cascade considered to be a major factor in many diseases of the upper and lower airways [1, 11, 13, 20]. B cells are responsible for the generation of these powerful immune responses through the production of a variety of Ig isotypes, including IgE, based on the location and type of exposure [21, 22].
In a localized allergic response, after an initial allergen exposure, antigen-presenting dendritic cells sensitize naïve T cells, promoting their development into Th2 cells. These cells produce the inflammatory cytokines interleukin (IL)-4 and IL-13, leading to B cell activation and production of specific IgE (Fig. 2) [22, 23].

adapted from Akdis M and Akdis CA. Nat Immunol 2012;13(4):312–314 and Davies JM, et al. J Allergy Clin Immunol 2013;131(4):972–976. IgE immunoglobulin E, IgG immunglobulin G, IgM immunoglobulin M, IL interleukin, ILC innate lymphoid cell, MHCII major histocompatibility complex class II, TCR T cell antigen receptor, Th2 T helper 2
B cell activation. In response to Th2-derived IL-4 and IL-13, naïve B cells migrate to B cell follicles for proliferation and formation of germinal centers. In incompletely organized germinal centers, as found in the Th2-centric response, naïve B cells undergo somatic hypermutation and class-switch recombination as part of direct switching to IgE+ B cells. In mature germinal centers, naïve B cells undergo indirect switching, passing through an intermediate IgG4+ B cell phase before transforming into IgE+ B cells. In either case, IgE+ B cells can then leave the germinal center, becoming either memory B cells or long-lived plasma cells. Memory B cells are dividing cells that produce minimal IgE but allow the prompt production of specific IgE-secreting plasma cells following a secondary allergen exposure in the absence of cytokines. It is not well established where IgE memory resides, and this remains a topic under active investigation within the field. Plasma cells do not divide, but produce far more specific IgE. Figure
Meanwhile, in a systemic allergic response, B cells are exposed to antigens in peripheral lymphoid organs, and can then move either into extrafollicular areas for proliferation and differentiation into short-lived plasma cells, or into B cell follicles for proliferation and formation of germinal centers [22, 24]. In the germinal centers, activated B cells undergo genetic modifications to the Ig gene (somatic hypermutation and class-switch recombination), resulting in the production of different Ig isotypes that are highly specific for the antigen [22, 25, 26].
B cells primarily produce two Ig isotypes in response to an allergen exposure: IgE and IgG4. IgE production occurs early, before IgG4 levels rise, though after frequent exposures, IgG4 levels may also rise [26]. Once B cells switch to IgE production and leave the germinal center, they become either memory B cells or long-lived plasma cells. Memory B cells are dividing cells, which only secrete a small amount of IgE. Following a secondary exposure, however, memory B cells allow the prompt production of specific IgE–secreting plasma cells. Conversely, long-lived plasma cells do not divide but produce far more specific IgE [1].
More recent evidence has shown that high levels of local IgE and IgE-producing B cells are also present in the nasal mucosa and lungs of patients regardless of allergy status, suggesting that the previously described germinal center reactions (somatic hypermutation and class-switch recombination) can also occur in the local tissue [5, 22, 25, 27].
In healthy individuals, the respiratory epithelium serves as a physical barrier to protect the upper and lower airways from environmental exposures, including viruses, bacteria, fungi, allergens, and contaminants [28]. Epithelial dysfunction due to loss of E-cadherin– and claudin-mediated epithelial tight junctions [24, 28] exposes the airways to these environmental factors, triggering an inflammatory response through the innate immunity pathway, which has been reported in many upper and lower airway diseases, including CRSwNP, asthma, and rhinitis [5, 13, 28]. Epithelial cells are also capable of detecting these exposures through pattern recognition receptors, triggering the release of key mediators, termed “alarmins,” including thymic stromal lymphopoietin (TSLP), IL-25, and IL-33, in response to exposure [24]. These signaling molecules cause ILC2 cells to proliferate and produce IL-5 and IL-13 and, to a lesser extent, IL-4 [29]. Of note, ILC2 cells produce approximately ten-fold more IL-5 and IL-13 compared with activated Th2 cells, and therefore play a vital role in the propagation of the type 2 inflammatory response [24]. ILC2- and Th2-derived IL-5 promotes the differentiation, maturation, mobilization, and survival of eosinophils and supports the development of mast cells and basophils, amplifying the inflammatory response [24]. The alarmins, TSLP, IL-25, and IL-33, can also stimulate dendritic cells to induce naïve T cells into IL-4– and IL-13–producing Th2 cells, further driving the immune response [29] through B cell activation and revision, and the production of IgE [22, 23]. Meanwhile, Th2-derived IL-2 stimulates ILC2 cells, resulting in a positive feedback loop promoting inflammation [29].
Additionally, disruption of the epithelial barrier can allow the entry of bacterial proteins, which can act as superantigens, into the bronchial tissue, leading to a shift toward Th2-mediated inflammation, with increases in eosinophilia, IL-5, and IgE [1, 30]. Superantigens hijack the body’s T cell antigen recognition system by directly cross-linking major histocompatibility complex class II on antigen-presenting cells and T cell receptors on T cells, bypassing the conventional antigen presentation step, and leading to a powerful immune response [30]. This promotes the production of polyclonal IgE, in contrast to the monoclonal IgE produced by the allergic and innate immune responses. Of note, polyclonal IgE may mask the effects of specific IgE by overwhelming the receptors on mast cells [25].
Regardless of whether IgE is produced through allergic sensitization, activation of the innate immune system, or by exposure to superantigens, these pathways are known to converge in the IgE-mediated inflammatory signaling cascade.
Effects of IgE Signaling
IgE is fundamental in the type 2 inflammatory response through its interaction with its two receptors: the high-affinity IgE receptor (FcεRI) and the low-affinity IgE receptor (FcεRII), also referred to as CD23 [24]. IgE has been implicated in a positive feedback mechanism wherein it contributes to the upregulation of both FcεRI and FcεRII, leading to an enhanced hypersensitivity response [31].
Secreted specific IgE binds to FcεRI on mast cells and basophils, sensitizing them to specific allergens ahead of future exposures [23]. Subsequent exposure to an allergen leads to the cross-linking of membrane-bound IgE in mast cells and basophils, inducing cellular degranulation and release of histamine, tryptase, cysteinyl leukotrienes, and platelet-activating factors [23]. The actions of these early response factors can manifest as edema, vasodilation, and bronchoconstriction (Fig. 1) [23].
Following these early events, the later release of cytokines and chemokines (e.g., IL-3, IL-4, IL-5, IL-13, CC chemokine ligand 5, and granulocyte–macrophage colony-stimulating factor) from mast cells and basophils recruits inflammatory cells, including eosinophils, to the site of inflammation (Fig. 1) [23]. Released IL-4 and IL-13 promote the overexpression of mucus-producing goblet cells and contribute to airway hyperresponsiveness [24]. Basophil-derived IL-4 has been shown to directly enhance the function of ILC2 [24]. Importantly, the cross-linking of IgE to FcεRI on mast cells and the subsequent release of IL-4 aids in Th2 differentiation and B cell isotype switching, leading to the production of additional IgE in a positive feedback loop, which can lead to chronic inflammation [24].
Meanwhile, IL-5 promotes the differentiation, maturation, mobilization, and survival of eosinophils and supports the development of mast cells and basophils [24]. The recruitment of eosinophils to sites of inflammation is largely dependent on coordination between IL-5, eotaxin, and IL-13, wherein IL-13 recruits eosinophils to specific tissues under the regulation of IL-5 and eotaxin [32]. Once recruited, eosinophils induce epithelial damage through accumulation, degranulation, and release of toxic proteins, including eosinophil-derived neurotoxin, eosinophil cationic protein, eosinophil peroxidase, and major basic protein [33]. Eosinophils also serve as a source of proinflammatory cytokines (IL-3, IL-4, IL-6, granulocyte–macrophage colony-stimulating factor, tumor necrosis factor α, transforming growth factor β) and chemokines and are able to present antigens to T cells, further propagating the inflammatory cascade [33].
Beyond the expression of FcεRI on mast cells and basophils, its constitutive expression on dendritic cells also plays an important role in the type 2 inflammatory pathway [34, 35]. The role of antigen presentation by dendritic cells is closely linked to the cross-linking of IgE and FcεRI. Studies have shown that FcεRI aids in the presentation of IgE-bound antigens, including allergens, to T cells. These bound antigens are internalized, processed, and presented to T cells, leading to T cell maturation and continuation of the type 2 inflammatory cascade [35]. Of note, signaling following dendritic cell IgE/FcεRI cross-linking has been implicated in the mediation of type 2 inflammation in either a proinflammatory or suppressive manner, depending on the cytokines produced [35].
Another important role of FcεRI in airway disease involves its expression in airway smooth muscle tissue. This tissue has been shown to respond directly to environmental exposures, including dust, microbes, and pollutants, in addition to intrinsic cytokines and Ig [36]. Exposure to IgE, whether it be antigen-bound IgE or independent, and subsequent cross-linking with FcεRI on airway smooth muscle cells, leads to the release of the proinflammatory cytokines IL-4, IL-5, and IL-13, as well as chemokines CC chemokine ligand 11/eotaxin [36].
Meanwhile, the interaction of IgE with FcεRII is involved with several key processes associated with the type 2 immune response, including the regulation of IgE synthesis and allergen transcytosis [37]. Here, epithelial FcεRII contributes to the transport of IgE–allergen immune complexes across the airway epithelium to facilitate allergen sampling [37]. IgE–allergen complexes can be internalized by FcεRII on antigen-presenting cells, leading to allergen processing and presentation on the cell surface to activate T cells, producing additional IgE and amplifying the allergic inflammatory response [37]. Similarly, in B cells, the cross-linking of allergen-bound IgE to FcεRII leads to the internalization, processing, and presentation of antigens to T cells, leading to T cell activation and T cell–mediated allergic inflammation [38]. In response to the binding of IgE to FcεRII on B cells that are currently producing IgE, synthesis can be upregulated or downregulated, depending on the specific form of FcεRII [37].
As described, there are several causative factors of the type 2 inflammatory pathway (allergen exposure, S. aureus superantigen exposure, epithelial dysfunction leading to innate immunity) implicated in upper and lower airway diseases, and several opportunities for positive feedback to enhance the inflammatory response. At the heart of this inflammatory process lies the key mediator: IgE. Understanding the role of IgE in the pathobiology of each disease unifies the upper and lower airways.
Upper Airway Disease
CRSwNP
The long-lasting mucosal inflammation associated with CRSwNP is often Th2 derived and eosinophilic in nature, marked by highly elevated local IgE levels [1]. This elevated local IgE production may be the result of stimulation by extrinsic allergens. However, patients with non-allergic disease also exhibit elevated IgE levels, suggesting that other pathways may contribute to this inflammatory process [1]. Superantigens from viral or bacterial microorganisms can directly activate B cell cytokine release, leading to increased levels of IL-5 and eosinophils [39]. In response to superantigen exposure and subsequent epithelial damage, epithelial cell release alarmins (IL-25, IL-23, and TSLP), which amplify the type 2 immune response by activating ILC2 cells to produce additional IgE [24, 33]. One of the most common contributors to these superantigens is S. aureus, which has been identified in the NP tissue of 63.6% of patients with CRSwNP compared with 33% of controls. Importantly, these observations were independent of serum total or specific IgE levels [40].
One additional trigger of this inflammatory cascade leading to nasal polyposis is through exposure to fungi. Indeed, allergic fungal sinusitis represents a subset of CRSwNP in which patients exhibit eosinophilic mucus in the sinuses, IgE-mediated allergy to fungus, and the presence of fungus in the sinus mucus [41]. In these patients, a localized mucosal allergic response including local IgE synthesis mediates the inflammatory response, ultimately leading to nasal polyposis [41].
As part of the inflammatory response associated with nasal polyposis, activation of eosinophils, in addition to basophils and some T cells, leads to the overproduction of the galectin-10 protein and subsequent auto-crystallization, forming what is known as Charcot-Leyden crystals. These crystals have been observed in the mucosa and mucus of patients with CRSwNP, particularly in more severe cases [42]. Charcot-Leyden crystals can stimulate the innate immune response or adaptive immunity in the presence of allergens, leading to increased IgE production, enhancing the type 2 inflammatory response [42, 43]. Additionally, the presence of Charcot-Leyden crystals has been linked to epithelial recruitment of neutrophils, leading to persistent, severe airway disease, which is non-responsive to current therapies [42].
Unsurprisingly, similarities in disease pathology contribute to considerable overlap between patients with CRSwNP and asthma. Between 30 and 70% of patients with CRSwNP have comorbid asthma, and ~ 70% of patients with asthma have an elevated risk of CRSwNP [44,45,46]. In patients with CRSwNP, elevated total IgE levels are predictive of asthma comorbidity [23]. The upper and lower airways share many of the same histological structures, such as the basement membrane, lamina propria, and ciliary epithelium [47]. CRSwNP and asthma often present together [47]. Further, many of the prominent underlying pathological features of CRSwNP, including high expression levels of type 2 cytokines (IL-4, IL-5, and IL-13, among others) and elevated IgE, are also key contributors to the pathology of asthma, as described below [47]. Moreover, the polyclonal IgE produced following S. aureus superantigen exposure can contribute to the persistent type 2 inflammation associated with CRSwNP through continuous mast cell activation, and the elevated specific polyclonal IgE to S. aureus enterotoxins in the serum of these patients is a known risk factor for asthma severity [23].
AERD
AERD, also called non-steroidal anti-inflammatory drug (NSAID)-exacerbated respiratory disease (N-ERD), is a form of asthma defined by three key factors: severe asthma, recurrent and severe NP, and acute respiratory response to NSAIDs [48]. Hallmarks of the disease include the overproduction of leukotrienes and prostaglandin D2, which are involved in the inflammatory and immune response pathways [49, 50]. Leukotrienes are potent bronchoconstrictors responsible for many of the symptoms in AERD, while prostaglandin D2 induces vasodilation and increases vascular permeability [24].
Although many patients with AERD have comorbid allergy, AERD also often occurs in non-allergic hosts with elevated eosinophilic inflammation and is sometimes associated with elevated total serum IgE, indicating that a pathway distinct from the antigen-specific type 2 allergic response is likely involved [51]. In one proposed disease mechanism, leukotriene-induced IL-33 expression activates the release of cytokines (IL-5 and IL-13) by ILC2 cells, leading to IgE production, activation, and degranulation of mast cells, and, ultimately, the eosinophilic inflammation commonly associated with AERD [51].
Similarly to other IgE-mediated airway diseases, the presence of specific IgE to S. aureus superantigens is observed in the local nasal tissue of patients with AERD [52]. It is suggested that this specific local IgE plays a role in modifying the severity of airway inflammation in patients with AERD [52]. This is in contrast to the limited reports linking serum-specific S. aureus IgE to AERD, emphasizing the role of local IgE in disease [52].
Allergic Rhinitis
AR is defined as rhinitis or rhinoconjunctivitis where symptoms, including nasal congestion, nasal itch, rhinorrhea, and sneezing, present following exposure to an aeroallergen, most commonly dander, mold, pollen, or residues from cockroaches and dust mites, with evidence of specific IgE to that allergen detected by SPT or serum-specific IgE testing [53]. In patients with AR, allergen exposure leads to a rise in local and systemic specific IgE production [1]. Observed elevation in serum IgE in these patients is believed to be due to spillover from the affected organ, rather than being produced in the blood [1]. Upon allergen exposure, the IgE-mediated type 2 inflammatory cascade is initiated, as previously described [1]. Additionally, the IgE-mediated activation of airway epithelial cells can further amplify the allergic reaction through the innate immune response [1]. Studies have shown that the disease severity of AR can be modified by environmental factors, including temperature, humidity, and air pollution. AR also leads to a higher colonization rate of S. aureus and superantigen exposure, leading to increased Th2 cytokine production and inflammation and worsening disease severity [54].
Non-allergic Rhinitis
A subset of patients with rhinitis are classified as having NAR, accounting for roughly 25% of all patients with rhinitis [55]. In these patients, no systemic markers of allergy, such as elevated antigen-specific IgE, are detected, despite the presence of rhinitis symptoms [1]. NAR can be induced by environmental irritants, such as change in weather, barometric pressure, temperature, or irritants, which cause abnormal vasomotor responses leading to mucus production and congestion. One common contributor to the pathophysiology of NAR is endonasal infections, which, along with exposure to exogenous irritants, may trigger mucosal inflammation through activation of T cell–mediated delayed-type hypersensitivity and activation of IgE-bearing mast cells via Ig-free light chains (FLCs) [1]. FLCs are a byproduct of receptor revision, wherein an ~ 10–40% excess of Ig light chains is produced versus the heavy-chain variant [56]. These FLCs have conventionally been thought of as inconsequential, contributing neither to specific receptor binding nor complement activation [56, 57]. More recently, however, these FLCs have been believed to serve as an alternative for IgE in the inflammatory pathway in several disorders by inducing hypersensitivity through mast cell activation. This mechanism has been implicated in both AR and NAR [1].
A further classification of rhinitis, local allergic rhinitis (LAR), describes the onset of disease symptoms with a localized allergic response in the nasal mucosa similar to that of AR; however, diagnostic tests, including SPT or serum IgE, are negative [58]. Despite negative systemic allergic testing, it is suspected that some patients with idiopathic rhinitis produce local specific IgE, indicating the presence of allergic disease [59]. This is further supported by evidence of the presence of Th2 inflammatory pathway components within the local tissue of patients with AR [60,61,62,63], suggesting the possibility of localized IgE synthesis and secretion [1]. Of note, asthma is a common comorbidity of rhinitis, regardless of allergic status, further suggesting a link between upper and lower airway disease [64].
Lower Airway Disease
Asthma comprises a heterogeneous spectrum of lower airway diseases, rather than a single disease. Most commonly, asthma is defined by allergy status based on whether the clinical symptoms were precipitated by exposure to a common aeroallergen, as confirmed by a positive SPT or serum-specific IgE [65].
Allergic Asthma
It is widely accepted that in response to an allergen exposure, susceptible patients experience elevated local specific clonal IgE, which is believed to be the cause of allergic airway inflammation in asthma through activation of the type 2 inflammatory cascade leading to eosinophilia [33, 66]. In support of this, type 2–high asthma is characterized by elevated levels of IgE and Th2 cytokines, including IL-4, IL-5, and IL-13 [66]. As previously described, these factors are key to the cyclic amplification of IgE production and subsequent inflammation associated with disease.
Non-allergic Asthma
There are a considerable number of classifications of non-allergic asthma, making it more difficult to diagnose and manage [67, 68]. The variety of asthma phenotypes includes early onset, eosinophilic, obese, and neutrophilic [68]. Early-onset allergic asthma is typically most prevalent in childhood and early adulthood, with a switch toward non-allergic asthma in adulthood [24]. However, across these phenotypes, there is considerable overlap, and they are not mutually exclusive [68]. Disease presentation tends to be more severe in patients with non-allergic asthma versus allergic asthma and can present highly heterogeneously in terms of eosinophilia, fixed airflow limitation, association with chronic rhinosinusitis, and presence of neutrophilic inflammation [67]. Typically, patient age, age at onset, and female to male ratio are higher in patients with non-allergic asthma versus allergic asthma [65].
Despite this variability in non-allergic asthma subtypes, many of the underlying characteristics are similar to those in allergic asthma, with the same treatment approaches suggested for both [67]. These similarities include increased activation of the type 2 inflammatory cascade, as evidenced by elevated levels of IL-4, IL-5, and IL-13; the presence of eosinophilic inflammation, elevated local IgE, and airway remodeling, including epithelial denudation; and thickening of the basement membrane and bronchial smooth muscle, which is said to be indistinguishable between allergic and non-allergic asthma subtypes [69, 70]. Although a biomarker for early-onset allergic asthma, IgE levels are often elevated in non-allergic late-onset asthma, but this IgE is often polyclonal and, as in CRSwNP, attributed to S. aureus enterotoxins [13].
Other Lower Airway Diseases
Additional diseases of the lower airway have been linked to IgE, namely allergic bronchopulmonary aspergillosis (ABPA) [71]. ABPA is a distinct endotype of allergic asthma characterized by markedly elevated IgE levels, allergic sensitization to Aspergillus, and mucus plugging. Inhalation of Aspergillus spores has been shown to cause ABPA in allergic individuals [71]. Repeated inhalation primarily elicits a type 1 allergic response in these patients, though type 3 and type 4 reactions also occur [71]. Exposure induces a polyclonal antibody response, leading to elevated total IgE levels as well as Aspergillus-specific IgE and IgG [71].
Diagnostic Value of IgE
The central effector role of IgE in the type 2 inflammatory pathway across various upper and lower airway diseases supports the utility of IgE as a diagnostic marker. Proper testing is required to determine whether a symptomatic individual’s serum versus local IgE levels are elevated in order to provide an accurate diagnosis, because these details may directly impact the categorization of disease [1].
Testing for Allergic Disease
The current diagnostic paradigm for upper and lower airway disease focuses on first differentiating between allergic and non-allergic disease. Symptomatic patients are initially assessed for patient history, with follow-up diagnostic testing geared toward identifying the presence of allergy [72, 73] through assessment of specific IgE levels, rather than local production [74].
An SPT represents a fairly common, relatively inexpensive test that provides immediate results and often serves as the first choice in confirmatory allergic testing [72, 74]. Despite its utility in identifying specific allergens in IgE-mediated allergic disease, the SPT has a few inherent limitations. SPT fails to examine the local production of IgE, such as in the nasal mucosa or lungs. In patients with only local IgE production, a SPT will yield a negative result, suggesting the absence of an allergic component [1]. Additionally, the use of SPT is highly variable by location, with a distinct lack of standardization in testing practices. Moreover, the use of allergy medications, including antihistamines, is known to interfere with the results and interpretation of SPT [75]. Scoring the SPT relies on the interpretation of a patient’s response in relation to their symptoms, which requires an experienced physician [76]. Ultimately, the utility of an SPT is bound by the specific panel of allergens used [76]. Often, a panel of eight to ten of the most common, locally relevant allergens is capable of identifying allergy in many patients, though a larger allergen panel may be required to fully assess a population [76].
Serum testing identifies the level of specific IgE in the blood of patients to specific selected allergens, and can be seen as complementary to SPT [77, 78]. Overall, serum-specific IgE testing is advantageous in that it can be performed in patients with skin disease and those who are receiving allergy medications such as antihistamines, which could interfere with SPT; it can identify sensitivity to potential cross-reacting allergens; and it eliminates the possibility of systemic reactions [75, 77]. However, these tests also come with their own limitations. First, they are time consuming and require a high volume of allergens and a blood sample to complete [79]. There is also currently no standardized reporting established for serum-specific IgE results, which can contribute to variability between tests [77]. Further, this form of testing may be more expensive than SPT methods, preventing its routine use [77]. The results obtained from serum-specific IgE testing may not correlate well with other testing methodologies, and may not be fully informative of clinical status, including disease severity, presence of tolerance through blocking IgG4 antibodies, or total IgE levels [77].
For patients in whom diagnostic uncertainty remains after routine methodologies are exhausted, more expansive testing, such as an immuno-solid-phase allergen chip (ISAC), may be performed. ISAC tests for specific IgE against multiple allergen components using a multiplex assay [74]. As ISAC testing assesses serum-specific IgE, it does not provide information regarding local IgE levels [74]. This methodology is highly expensive, so is often reserved until after other tests are performed, but it may yield more conclusive results [74]. Moreover, ISAC test results should be examined carefully because the associated benefit of testing multiple allergen components can inadvertently identify allergic sensitizations that are irrelevant to a patient or that do not contribute to symptoms [80].
Taken together, routine IgE testing methodologies come with their own advantages, yet they are also impacted by numerous practical limitations [74, 76,77,78,79, 81]. Additionally, these routine allergy testing methodologies must be further confirmed by the presence of symptoms or through allergen provocation assessments if a patient’s clinical history and sensitization disagree or are unavailable [82]. It is important to note, however, that many upper and lower airway diseases only present with local IgE increases rather than an allergic response, which cannot be diagnosed by routine SPT or elevated serum IgE methodologies. These situations require additional, localized testing to determine the role of IgE mediation.
Testing for Local IgE: Unmet Need
As described, the current diagnostic paradigm in patients presenting symptoms of upper or lower airway disease relies on the use of routine allergy tests, which examine circulating specific or total IgE to identify the presence of an allergic component of the disease. In many upper and lower airway diseases, however, it is local IgE activity that drives symptomology through the type 2 inflammatory pathway [1, 72, 73].
After initial suspicion of asthma from patient history, routine allergic testing methodologies are often the first employed [67]. However, total IgE levels of patients with asthma often fall within the normal range, whereas other patients without asthma may present with elevated IgE outside of this range [83]. Additionally, patients with non-allergic asthma may only present with a localized polyclonal IgE response [13], despite similarities in underlying type 2 inflammatory disease pathobiology [67]. Failure to identify a specific allergen by conventional means does not indicate the absence of IgE-mediated asthma, because a local reaction may be the cause of symptomology [67].
Traditionally, the distinction between AR and NAR is based on an SPT and serum IgE analysis [1]. However, in the subgroup of patients with LAR, accumulating evidence suggests that local specific IgE production may occur in response to allergen exposure despite negative systemic tests [1]. Diagnostic tools in patients with rhinitis often fail to distinguish AR, NAR, and LAR because the measurement of local IgE by means of nasal provocation testing or nasal cytology is typically not performed in patients with rhinitis outside of clinical research, and its use in the routine evaluation of rhinitis is not recognized [1, 84]. Therefore, NAR is typically diagnosed by exclusion, because this group of patients has a poorly defined pathogenesis [1]. Some patients with rhinitis may exhibit elevated serum IgE levels; however, it has been shown that this rise is largely due to spillover from a local increase in IgE, further suggesting the need for screening at the local level [1].
In patients with CRSwNP, testing for specific and total IgE in the serum may predict asthma comorbidity and severity [23]. However, the high aeroallergen sensitivity and localized allergic response associated with CRSwNP has shown no differences in imaging, symptomatic severity, or disease recurrence in allergic versus non-allergic individuals [85]. In many patients with CRSwNP, the disease is highlighted by the presence of S. aureus–specific IgE in the local NP tissue [39], suggesting that the severe inflammatory response associated with contributing to CRSwNP stems from local polyclonal IgE involvement [1].
Together, these observations suggest that the role of IgE in some upper and lower airway diseases may be underestimated by initial testing of systemic total or specific IgE, further highlighting the need for diagnosis by a localized allergic response. In support of this, it has been suggested that in patients with a negative SPT or no evidence of elevated serum-specific IgE, additional testing should be utilized to identify patients with localized allergic responses [86, 87].
Testing for Local IgE: Methodologies
The nasal allergen challenge has been used in combination with routine allergen tests to differentiate between patients with AR (positive SPT or serum-specific IgE), NAR (negative nasal allergy challenge), and LAR (negative serum-specific IgE/SPT, positive nasal allergy challenge) [86]. Despite this utility in AR, it has been demonstrated that the use of nasal provocation testing to determine allergic sensitization in patients with CRSwNP is limited, likely due to the polyclonality of local IgE- or IgG4-blocking activity in these patients [88]. Additionally, nasal allergen provocation tests or challenges must be performed under clinical supervision, are labor and resource intensive, have variable specificity, and are often stressful for the patient [81].
The most direct method for investigating the pathology of disease tissue, including accurate measurements of local IgE levels, involves the collection of tissue via biopsy [1]. Indeed, studies have demonstrated elevated polyclonal IgE, S. aureus–specific IgE, colonization with S. aureus, and tissue eosinophilia within the NP tissue of patients with CRSwNP [89]. However, because tissue biopsy can be invasive, it is only performed when a patient already requires surgical intervention [1]. Therefore, a tissue biopsy is of limited utility in routine diagnosis.
One technique used to sample local airway IgE is through the collection of nasal secretions by nasal lavage and absorption techniques, although sample collection from the lower airway via bronchosorption is more challenging [1]. Studies have suggested that cytokine detection in samples obtained by nasosorption and sputosorption techniques reflects lower airway eosinophilia, although these methodologies still require extensive validation [90]. These techniques are performed under the assumption that protein levels in these secretions are representative of those in the underlying mucosa [1]. In one study, patients from three trials were examined to determine the impact of four treatment methodologies (omalizumab, mepolizumab, methylprednisolone, and doxycycline) on periostin levels in either the nasal secretions or serum of patients with CRSwNP to determine its utility as a biomarker of response to treatment. Improvements in eosinophilic inflammation and clinical outcomes corresponded with periostin levels, indicating that treatment disrupts the inflammatory cascade at both local and systemic levels [91]. However, it is important to note that in measuring IgE levels there exists a disconnect between local and serum total IgE when treating with omalizumab, as omalizumab-IgE complexes form with an extended half-life, falsely increasing total IgE measures [91].
Another study pioneered the use of an allergen microarray chip in assessing levels of local specific IgE to house dust mites (HDM) in the nasal secretions of patients with AR. Here, total and specific IgE levels to HDM were measured in the serum and nasal secretions of patients with confirmed allergic sensitization to HDM using ImmunoCAP fluorescent enzyme immunoassay and customized microarray assays to determine whether these measurements correlated. Overall findings in this study suggested good sensitivity using the microarray approach, superior to that of ImmunoCAP fluorescent enzyme immunoassay, in the detection of local specific IgE to HDM in nasal secretions of patients with AR. This suggests the utility of microarray technology as a non-invasive alternative to measuring IgE levels in the nasal secretions of patients with AR [92]. Finally, the utility of the ISAC chip in assessing response to allergen-specific immunotherapy in patients with allergic sensitization to HDM has previously been examined. Here, the ISAC test was used to examine the protection exhibited by treatment on a variety of HDM allergens through analysis of patient serum and nasal secretions. Ultimately, it was found that patients with varying HDM allergic sensitization profiles responded differently to treatment, and therefore, stratifying patients by their specific sensitization profile may better guide treatment [93].
The basophil activation test serves as a useful ex vivo test of allergic response, in which basophils from whole blood or isolated peripheral blood mononuclear cells are exposed to allergens of interest, and the subsequent expression of basophil activation markers in response to exposure is measured by flow cytometry [81, 94]. Although the analysis of whole and isolated peripheral blood samples is more typically used in the detection of systemic allergen response, recent studies have shown that this technique may hold a utility as a non-invasive analysis of localized response [81]. In patients with LAR, the basophil activation test has demonstrated sensitivity sufficient to diagnose an IgE-mediated response where an SPT and serum-specific IgE testing were found to be negative [95]. Moreover, studies have shown that the use of antihistamines does not impact the results of the basophil activation test, and they therefore do not need to be discontinued before testing [78]. One potential drawback of these tests, however, is that they rely on the prior confirmation of an allergen of interest by SPT or measure of serum-specific IgE [94]. Additionally, there is currently limited standardization regarding the technique and interpretation of basophil activation test outcomes [78]. With these established, the basophil activation test serves as a promising methodology for determining IgE activity at both the local and systemic levels.
Although there are many available diagnostic tools to determine the role of IgE in disease, there is still room for improvement. Negative results on the most routinely used tests (SPT, serum testing for allergen-specific IgE) do not conclusively indicate the lack of an allergic response, and neither do they identify local innate immunity. However, more localized tests can be invasive, not broadly applicable, and are limited by the lack of routine use in practice. As technology and standardization of practice continue to improve, so too will the diagnosis of IgE-mediated disease. In the meantime, the use of a treatment that addresses the common, underlying factor in these diseases can serve to improve patient outcomes.
Anti-IgE as an Effective Treatment
As previously described, the common underlying pathophysiology of many upper and lower airway diseases is suggestive of a continuous or “unified” airway [2, 3]. An IgE-mediated inflammatory process is the key driver, leading to epithelial injury and airway remodeling [2, 3]. Targeting this process with anti-IgE therapy is effective in both allergic and non-allergic diseases, expanding the prior notion of IgE involvement beyond allergic asthma [33]. The efficacy of the anti-IgE therapy omalizumab has led to its approval in the treatment of allergic asthma, severe AR, and CRSwNP in a variety of countries [96,97,98].
In upper airway disease, anti-IgE therapy for NP has shown significant improvements in both clinical and patient-reported outcomes [97]. Similarly, in patients with AR, anti-IgE therapy demonstrated safe control of symptoms, improved quality of life, minimized use of rescue medications, and prevented asthma exacerbations in patients with comorbid asthma [98,99,100,101,102]. In patients with CRSwNP, anti-IgE therapy led to reductions in inflammation as measured by imaging [103]. Patients with asthma with comorbidities from the PROSPERO and EXTRA asthma trials showed reductions in the number of exacerbations experienced comparable with patients with asthma without comorbidities, whereas improvements in forced expiratory volume in 1 s were greater in patients with comorbidities than without [104].
In patients with lower airway disease, similar efficacy has been demonstrated in response to anti-IgE therapy. In patients with moderate-to-severe allergic asthma inadequately controlled by medium- to high-dose inhaled corticosteroids, anti-IgE therapy demonstrated a significant reduction in asthma exacerbations [105]. Interestingly, patients whose asthma responded well to treatment from the SOLAR trial saw a significant likelihood of achieving improvements in comorbid rhinitis as well [106]. Finally, anti-IgE therapy has shown efficacy in a number of non-allergic asthma subtypes, including non-atopic asthma [107,108,109,110], severe eosinophilic asthma [111,112,113], AERD [114,115,116,117,118,119], and severe occupational asthma [120].
Taken together, the clinical efficacy of anti-IgE therapy across numerous airway diseases challenges the traditional allergen-centric definition of upper and lower airway disease. Rather than distinguishing between allergic/atopic versus non-allergic/non-atopic disease, it may be more appropriate to think of disease as responses driven by the type (i.e., monoclonal vs. polyclonal) and location (i.e., upper vs. lower airway) of IgE-mediated versus non–IgE-mediated disease (Table 1).
In defining disease by the role of IgE (e.g., IgE mediated vs. non–IgE mediated), physicians can diagnose based on local or systemic IgE involvement as the underlying cause of disease and effectively treat at the source. Prompt reduction in IgE levels can disrupt the cascade of inflammation, including eosinophilic involvement, by using a targeted therapeutic, regardless of allergic status. Because IgE mediates the inflammatory response underlying many upper and lower airway diseases, halting its activity quickly and completely may lead to efficient symptom reduction in patients [96]. Targeting IgE directly using the anti-IgE antibody omalizumab provides a prompt reduction in free IgE levels, with patients with allergic asthma reporting > 95% reduction within days after starting therapy [121]. This is in comparison with other treatment options targeting other drivers of type 2 inflammation, such as mepolizumab, which blocks IL-5 signaling and has no effect on IgE levels in patients with severe asthma [122], and dupilumab, which blocks IL-4 and IL-13 signaling [123] and gradually decreases serum total IgE levels (70% reduction from baseline at 52 weeks) in patients with allergic asthma [124]. The differences in timing and magnitude of IgE reduction between treatment options are likely due to their different mechanisms of action, with omalizumab targeting IgE directly.
For omalizumab, additional studies have investigated the complexities of treating patients with anti-IgE. Although serum total IgE levels are useful, they are not predictive of clinical response to omalizumab [97, 125,126,127]. Additionally, serum total IgE levels are known to increase following omalizumab treatment initiation due to IgE–anti-IgE complexes forming, and it is unclear how this can affect dosing [128]. Further, both total IgE and specific-allergen IgE have been associated with omalizumab treatment response in asthma [129]. These findings further emphasize the need to accurately assess different IgE parameters (for example, serum IgE, specific IgE, local IgE) to provide an accurate diagnosis and guide choice of treatment with anti-IgE drugs, such as omalizumab, or with other treatments that lower IgE levels, such as dupilumab.
Summary
There is considerable evidence supporting the role of IgE in many upper and lower airway diseases as the underlying cause of damaging inflammation independent of allergy [1, 33]. Further, this evidence suggests that in many of these airway diseases, there exists some degree of both local allergic IgE production and a local non-allergic IgE response through innate immunity or superantigens [8]. This convergence on the unifying component in the pathobiology of these diseases highlights the utility of IgE as a diagnostic tool. However, current methodologies focus primarily on distinguishing allergic and non-allergic disease by measuring systemic IgE via SPT or serum-specific IgE testing, rather than investigating the disease-modifying role of local IgE.
This diagnostic paradigm poses a particular problem in the treatment of diseases lacking an allergic component, because evidence has accumulated in support of the efficacy of anti-IgE therapeutics in treating both upper and lower airway disease, regardless of allergy status or total IgE levels. The routine testing for the presence of specific IgE to diagnose allergy status rather than assessing local IgE levels prevents physicians from identifying the disease for which local IgE causes symptomology and which will respond to anti-IgE therapy. Similarly, when utilizing anti-IgE therapy, evidence suggests clinical efficacy regardless of total IgE levels.
Rather than distinguishing between allergic/atopic versus non-allergic/non-atopic disease, it may be more appropriate to think of disease as responses driven by the type (i.e., monoclonal vs. polyclonal) and location (i.e., upper vs. lower airway) of IgE-mediated versus non–IgE-mediated disease. When interpreting test results, it is important to consider the involvement and diagnosis of local as well as systemic IgE. Doing so would allow for the effective, targeted treatment of IgE-mediated upper and lower airway disease by addressing the root cause of the disease, regardless of allergic status.
References
De Schryver E, Devuyst L, Derycke L et al (2015) Local immunoglobulin E in the nasal mucosa: clinical implications. Allergy Asthma Immunol Res 7(4):321–331. https://doi.org/10.4168/aair.2015.7.4.321
Meltzer EO, Szwarcberg J, Pill MW (2004) Allergic rhinitis, asthma, and rhinosinusitis: diseases of the integrated airway. J Manag Care Pharm 10(4):310–317. https://doi.org/10.18553/jmcp.2004.10.4.310
Samitas K, Carter A, Kariyawasam HH, Xanthou G (2018) Upper and lower airway remodelling mechanisms in asthma, allergic rhinitis and chronic rhinosinusitis: the one airway concept revisited. Allergy 73(5):993–1002. https://doi.org/10.1111/all.13373
Giavina-Bianchi P, Aun MV, Takejima P, Kalil J, Agondi RC (2016) United airway disease: current perspectives. J Asthma Allergy 9:93–100. https://doi.org/10.2147/JAA.S81541
Gould HJ, Sutton BJ, Beavil AJ et al (2003) The biology of IgE and the basis of allergic disease. Annu Rev Immunol 21:579–628. https://doi.org/10.1146/annurev.immunol.21.120601.141103
Zhang N, Holtappels G, Gevaert P et al (2011) Mucosal tissue polyclonal IgE is functional in response to allergen and SEB. Allergy 66(1):141–148. https://doi.org/10.1111/j.1398-9995.2010.02448.x
Xia S, Zhu Z, Guan WJ et al (2018) Correlation between upper and lower airway inflammations in patients with combined allergic rhinitis and asthma syndrome: a comparison of patients initially presenting with allergic rhinitis and those initially presenting with asthma. Exp Ther Med 15(2):1761–1767. https://doi.org/10.3892/etm.2017.5536
Bachert C, Zhang N (2012) Chronic rhinosinusitis and asthma: novel understanding of the role of IgE above atopy. J Intern Med 272(2):133–143. https://doi.org/10.1111/j.1365-2796.2012.02559.x
de Benedictis FM, Bush A (2020) Janus looks both ways: how do the upper and lower airways interact? Paediatr Respir Rev 34:59–66. https://doi.org/10.1016/j.prrv.2019.06.004
Verbruggen K, Van Cauwenberge P, Bachert C (2009) Anti-IgE for the treatment of allergic rhinitis - and eventually nasal polyps? Int Arch Allergy Immunol 148(2):87–98. https://doi.org/10.1159/000155739
Navinés-Ferrer A, Serrano-Candelas E, Molina-Molina GJ, Martín M (2016) IgE-related chronic diseases and anti-IgE-based treatments. J Immunol Res 2016:8163803. https://doi.org/10.1155/2016/8163803
Beeh K, Ksoll M, Buhl R (2000) Elevation of total serum immunoglobulin E is associated with asthma in nonallergic individuals. Eur Respir J 16(4):609–614. https://doi.org/10.1034/j.1399-3003.2000.16d07.x
Bachert C, Humbert M, Hanania NA et al (2020) Staphylococcus aureus and its IgE-inducing enterotoxins in asthma: current knowledge. Eur Respir J 55(4):1901592. https://doi.org/10.1183/13993003.01592-2019
Novak N, Bieber T (2003) Allergic and nonallergic forms of atopic diseases. J Allergy Clin Immunol 112(2):252–262. https://doi.org/10.1067/mai.2003.1595
Carr TF, Stern DA, Halonen M, Wright AL, Martinez FD (2019) Non-atopic rhinitis at age 6 is associated with subsequent development of asthma. Clin Exp Allergy 49(1):35–43. https://doi.org/10.1111/cea.13276
Tomassen P, Vandeplas G, Van Zele T et al (2016) Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol 137(5):1449–1456.e4. https://doi.org/10.1016/j.jaci.2015.12.1324
Loureiro CC, Amaral L, Ferreira JA et al (2018) Omalizumab for severe asthma: beyond allergic asthma. Biomed Res Int 2018:3254094. https://doi.org/10.1155/2018/3254094
Dunican EM, Fahy JV (2015) The role of type 2 inflammation in the pathogenesis of asthma exacerbations. Ann Am Thorac Soc 12(Suppl 2):S144–S149. https://doi.org/10.1513/AnnalsATS.201506-377AW
Wu LC, Zarrin AA (2014) The production and regulation of IgE by the immune system. Nat Rev Immunol 14(4):247–259. https://doi.org/10.1038/nri3632
Burrows B, Martinez FD, Halonen M, Barbee RA, Cline MG (1989) Association of asthma with serum IgE levels and skin-test reactivity to allergens. N Engl J Med 320(5):271–277. https://doi.org/10.1056/NEJM198902023200502
Cerutti A, Puga I, Cols M (2011) Innate control of B cell responses. Trends Immunol 32(5):202–211. https://doi.org/10.1016/j.it.2011.02.004
Dullaers M, De Bruyne R, Ramadani F, Gould HJ, Gevaert P, Lambrecht BN (2012) The who, where, and when of IgE in allergic airway disease. J Allergy Clin Immunol 129(3):635–645. https://doi.org/10.1016/j.jaci.2011.10.029
Humbert M, Bousquet J, Bachert C et al (2019) IgE-mediated multimorbidities in allergic asthma and the potential for omalizumab therapy. J Allergy Clin Immunol Pract 7(5):1418–1429. https://doi.org/10.1016/j.jaip.2019.02.030
Kuruvilla ME, Lee FE-H, Lee GB (2019) Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol 56(2):219–233. https://doi.org/10.1007/s12016-018-8712-1
Gevaert P, Nouri-Aria KT, Wu H et al (2013) Local receptor revision and class switching to IgE in chronic rhinosinusitis with nasal polyps. Allergy 68(1):55–63. https://doi.org/10.1111/all.12054
Aalberse RC, Platts-Mills TA, Rispens T (2016) The developmental history of IgE and IgG4 antibodies in relation to atopy, eosinophilic esophagitis, and the modified TH2 response. Curr Allergy Asthma Rep 16(6):45. https://doi.org/10.1007/s11882-016-0621-x
Gatto D, Brink R (2010) The germinal center reaction. J Allergy Clin Immunol 126(5):898–907; quiz 908–909. https://doi.org/10.1016/j.jaci.2010.09.007
Yuksel H, Turkeli A (2017) Airway epithelial barrier dysfunction in the pathogenesis and prognosis of respiratory tract diseases in childhood and adulthood. Tissue Barriers 5(4):e1367458. https://doi.org/10.1080/21688370.2017.1367458
Li BWS, Hendriks RW (2013) Group 2 innate lymphoid cells in lung inflammation. Immunology 140(3):281–287. https://doi.org/10.1111/imm.12153
Fraser JD (2011) Clarifying the mechanism of superantigen toxicity. PLoS Biol 9(9):e1001145. https://doi.org/10.1371/journal.pbio.1001145
Oettgen HC, Geha RS (1999) IgE in asthma and atopy: cellular and molecular connections. J Clin Invest 104(7):829–835. https://doi.org/10.1172/JCI8205
Pope SM, Brandt EB, Mishra A et al (2001) IL-13 induces eosinophil recruitment into the lung by an IL-5– and eotaxin-dependent mechanism. J Allergy Clin Immunol 108(4):594–601. https://doi.org/10.1067/mai.2001.118600
Matucci A, Vultaggio A, Maggi E, Kasujee I (2018) Is IgE or eosinophils the key player in allergic asthma pathogenesis? Are we asking the right question? Respir Res 19:113. https://doi.org/10.1186/s12931-018-0813-0
Platzer B, Stout M, Fiebiger E (2015) Functions of dendritic-cell-bound IgE in allergy. Mol Immunol 68(2 Pt A):116–119. https://doi.org/10.1016/j.molimm.2015.05.016
Shin J-S, Greer AM (2015) The role of FcεRI expressed in dendritic cells and monocytes. Cell Mol Life Sci 72(12):2349–2360. https://doi.org/10.1007/s00018-015-1870-x
Redhu NS, Gounni AS (2013) The high affinity IgE receptor (FcεRI) expression and function in airway smooth muscle. Pulm Pharmacol Ther 26(1):86–94. https://doi.org/10.1016/j.pupt.2012.04.004
Dhaliwal B, Pang MO, Keeble AH et al (2017) IgE binds asymmetrically to its B cell receptor CD23. Sci Rep 7:45533. https://doi.org/10.1038/srep45533
Selb R, Eckl-Dorna J, Neunkirchner A et al (2017) CD23 surface density on B cells is associated with IgE levels and determines IgE-facilitated allergen uptake, as well as activation of allergen-specific T cells. J Allergy Clin Immunol 139(1):290–299.e4. https://doi.org/10.1016/j.jaci.2016.03.042
Bachert C, Gevaert P, Holtappels G, Johansson SG, van Cauwenberge P (2001) Total and specific IgE in nasal polyps is related to local eosinophilic inflammation. J Allergy Clin Immunol 107(4):607–614. https://doi.org/10.1067/mai.2001.112374
Van Zele T, Gevaert P, Watelet JB et al (2004) Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J Allergy Clin Immunol 114(4):981–983. https://doi.org/10.1016/j.jaci.2004.07.013
Pant H, Schembri MA, Wormald PJ, Macardle PJ (2009) IgE-mediated fungal allergy in allergic fungal sinusitis. Laryngoscope 119(6):1046–1052. https://doi.org/10.1002/lary.20170
Gevaert E, Delemarre T, De Volder J et al (2020) Charcot-Leyden crystals promote neutrophilic inflammation in patients with nasal polyposis. J Allergy Clin Immunol 145(1):427–430.e4. https://doi.org/10.1016/j.jaci.2019.08.027
Persson EK, Verstraete K, Heyndrickx I et al (2019) Protein crystallization promotes type 2 immunity and is reversible by antibody treatment. Science 364(6442):eaaw4295. https://doi.org/10.1126/science.aaw4295
Langdon C, Mullol J (2016) Nasal polyps in patients with asthma: prevalence, impact, and management challenges. J Asthma Allergy 9:45–53. https://doi.org/10.2147/JAA.S86251
Larsen K (1996) The clinical relationship of nasal polyps to asthma. Allergy Asthma Proc 17(5):243–249
Philpott CM, Erskine S, Hopkins C et al (2018) Prevalence of asthma, aspirin sensitivity and allergy in chronic rhinosinusitis: data from the UK National Chronic Rhinosinusitis Epidemiology Study. Respir Res 19(1):129. https://doi.org/10.1186/s12931-018-0823-y
Massoth L, Anderson C, McKinney KA (2019) Asthma and chronic rhinosinusitis: diagnosis and medical management. Med Sci (Basel) 7(4):53. https://doi.org/10.3390/medsci7040053
Carr TF, Kraft M (2018) Use of biomarkers to identify phenotypes and endotypes of severe asthma. Ann Allergy Asthma Immunol 121(4):414–420. https://doi.org/10.1016/j.anai.2018.07.029
Feng X, Ramsden MK, Negri J et al (2016) Eosinophil production of prostaglandin D2 in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol 138(4):1089–1097.e3. https://doi.org/10.1016/j.jaci.2016.04.042
Liu M, Yokomizo T (2015) The role of leukotrienes in allergic diseases. Allergol Int 64(1):17–26. https://doi.org/10.1016/j.alit.2014.09.001
Liu T, Kanaoka Y, Barrett NA et al (2015) Aspirin-exacerbated respiratory disease involves a cysteinyl leukotriene–driven IL-33–mediated mast cell activation pathway. J Immunol 195(8):3537–3545. https://doi.org/10.4049/jimmunol.1500905
Yoo HS, Shin YS, Liu JN, Kim M-A, Park H-S (2013) Clinical significance of immunoglobulin E responses to staphylococcal superantigens in patients with aspirin-exacerbated respiratory disease. Int Arch Allergy Immunol 162(4):340–345. https://doi.org/10.1159/000353976
Small P, Kim H (2011) Allergic rhinitis. Allergy Asthma. Clin Immunol 7:S3. https://doi.org/10.1186/1710-1492-7-S1-S3
Shiomori T, Yoshida S, Miyamoto H, Makishima K (2000) Relationship of nasal carriage of Staphylococcus aureus to pathogenesis of perennial allergic rhinitis. J Allergy Clin Immunol 105(3):449–454. https://doi.org/10.1067/mai.2000.104256
James LK, Durham SR (2009) Rhinitis with negative skin tests and absent serum allergen-specific IgE: more evidence for local IgE? J Allergy Clin Immunol 124(5):1012–1013. https://doi.org/10.1016/j.jaci.2009.09.029
Nakano T, Matsui M, Inoue I, Awata T, Katayama S, Murakoshi T (2011) Free immunoglobulin light chain: its biology and implications in diseases. Clin Chim Acta 412(11–12):843–849. https://doi.org/10.1016/j.cca.2011.03.007
Thio M, Groot Kormelink T, Fischer MJ, Blokhuis BR, Nijkamp FP, Redegeld FA (2012) Antigen binding characteristics of immunoglobulin free light chains: crosslinking by antigen is essential to induce allergic inflammation. PLoS One 7(7):e40986. https://doi.org/10.1371/journal.pone.0040986
Rondón C, Bogas G, Barrionuevo E, Blanca M, Torres MJ, Campo P (2017) Nonallergic rhinitis and lower airway disease. Allergy 72(1):24–34. https://doi.org/10.1111/all.12988
Rondón C, Doña I, López S et al (2008) Seasonal idiopathic rhinitis with local inflammatory response and specific IgE in absence of systemic response. Allergy 63(10):1352–1358. https://doi.org/10.1111/j.1398-9995.2008.01695.x
Cameron LA, Durham SR, Jacobson MR et al (1997) Expression of IL-4, Cϵ RNA, and Iϵ RNA in the nasal mucosa of patients with seasonal rhinitis: effect of topical corticosteroids. J Allergy Clin Immunol 101(3):330–336. https://doi.org/10.1016/s0091-6749(98)70244-1
KleinJan A, Godthelp T, van Toornenenbergen AW, Fokkens WJ (1997) Allergen binding to specific IgE in the nasal mucosa of allergic patients. J Allergy Clin Immunol 99(4):515–521. https://doi.org/10.1016/s0091-6749(97)70079-4
Pawankar R, Okuda M, Yssel H, Okumura K, Ra C (1997) Nasal mast cells in perennial allergic rhinitics exhibit increased expression of the Fc epsilonRI, CD40L, IL-4, and IL-13, and can induce IgE synthesis in B cells. J Clin Invest 99(7):1492–1499. https://doi.org/10.1172/JCI119311
Cameron L, Hamid Q, Wright E et al (2000) Local synthesis of epsilon germline gene transcripts, IL-4, and IL-13 in allergic nasal mucosa after ex vivo allergen exposure. J Allergy Clin Immunol 106(1 Pt 1):46–52. https://doi.org/10.1067/mai.2000.107398
Chawes BLK (2011) Upper and lower airway pathology in young children with allergic- and non-allergic rhinitis. Dan Med Bull 58(5):B4278
Pillai P, Corrigan CJ, Ying S (2011) Airway epithelium in atopic and nonatopic asthma: similarities and differences. ISRN Allergy 2011:195846. https://doi.org/10.5402/2011/195846
Kupryś-Lipińska I, Molińska K, Kuna P (2016) The effect of omalizumab on eosinophilic inflammation of the respiratory tract in patients with allergic asthma. Pneumonol Alergol Pol 84(4):232–243. https://doi.org/10.5603/PiAP.2016.0029
Vennera MDC, Picado C (2014) Novel diagnostic approaches and biological therapeutics for intrinsic asthma. Int J Gen Med 7:365–371. https://doi.org/10.2147/IJGM.S45259
Wenzel SE (2012) Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 18(5):716–725. https://doi.org/10.1038/nm.2678
Froidure A, Mouthuy J, Durham SR, Chanez P, Sibille Y, Pilette C (2016) Asthma phenotypes and IgE responses. Eur Respir J 47(1):304–319. https://doi.org/10.1183/13993003.01824-2014
Shifren A, Witt C, Christie C, Castro M (2012) Mechanisms of remodeling in asthmatic airways. J Allergy (Cairo) 2012:316049. https://doi.org/10.1155/2012/316049
Shah A, Panjabi C (2014) Allergic aspergillosis of the respiratory tract. Eur Respir Rev 23(131):8–29. https://doi.org/10.1183/09059180.00007413
Hellings PW, Scadding G, Alobid I et al (2012) Executive summary of European Task Force document on diagnostic tools in rhinology. Rhinology 50(4):339–352. https://doi.org/10.4193/Rhino11.252
Kaplan AG, Balter MS, Bell AD, Kim H, McIvor RA (2009) Diagnosis of asthma in adults. CMAJ 181(10):E210–E220. https://doi.org/10.1503/cmaj.080006
Griffiths RLM, El-Shanawany T, Jolles SRA et al (2017) Comparison of the performance of skin prick, ImmunoCAP, and ISAC tests in the diagnosis of patients with allergy. Int Arch Allergy Immunol 172(4):215–223. https://doi.org/10.1159/000464326
Heinzerling L, Mari A, Bergmann KC et al (2013) The skin prick test – European standards. Clin Transl Allergy 3(1):3. https://doi.org/10.1186/2045-7022-3-3
Haahtela T, Burbach GJ, Bachert C et al (2014) Clinical relevance is associated with allergen-specific wheal size in skin prick testing. Clin Exp Allergy 44(3):407–416. https://doi.org/10.1111/cea.12240
Fatteh S, Rekkerth DJ, Hadley JA (2014) Skin prick/puncture testing in North America: a call for standards and consistency. Allergy Asthma Clin Immunol 10:44. https://doi.org/10.1186/1710-1492-10-44
Ansotegui IJ, Melioli G, Canonica GW et al (2020) IgE allergy diagnostics and other relevant tests in allergy, a World Allergy Organization position paper. World Allergy Organ J 13(2):100080. https://doi.org/10.1016/j.waojou.2019.100080
Fall BI, Niessner R (2009) Detection of known allergen-specific IgE antibodies by immunological methods. Methods Mol Biol 509:107–122. https://doi.org/10.1007/978-1-59745-372-1_7
van Hage M, Hamsten C, Valenta R (2017) ImmunoCAP assays: pros and cons in allergology. J Allergy Clin Immunol 140(4):974–977. https://doi.org/10.1016/j.jaci.2017.05.008
Hemmings O, Kwok M, McKendry R, Santos AF (2018) Basophil activation test: old and new applications in allergy. Curr Allergy Asthma Rep 18(12):77. https://doi.org/10.1007/s11882-018-0831-5
Agache I, Bilò M, Braunstahl GJ et al (2015) In vivo diagnosis of allergic diseases—allergen provocation tests. Allergy 70(4):355–365. https://doi.org/10.1111/all.12586
Mahesh PA (2017) Evaluation of asthma severity: relevance of total serum IgE, sputum and peripheral eosinophilia. Lung India 34(3):290–291. https://doi.org/10.4103/lungindia.lungindia_109_17
Quillen DM, Feller DB (2006) Diagnosing rhinitis: allergic vs. nonallergic. Am Fam Physician 73(9):1583–1590
Brook CD, Agarwal P (2020) Updates to the current understanding of the relationship between allergy and chronic sinusitis. Curr Otorhinolaryngol Rep 8:191–197. https://doi.org/10.1007/s40136-020-00287-6
Eguiluz-Gracia I, Pérez-Sánchez N, Bogas G, Campo P, Rondón C (2019) How to diagnose and treat local allergic rhinitis: a challenge for clinicians. J Clin Med 8(7):1062. https://doi.org/10.3390/jcm8071062
Incorvaia C, Fuiano N, Canonica GW (2013) Seeking allergy when it hides: which are the best fitting tests? World Allergy Organ J 6(1):11. https://doi.org/10.1186/1939-4551-6-11
Calus L, Devuyst L, De Ruyck N, Van Zele T, Bachert C, Gevaert P (2013) Nasal allergen provocation test in nasal polyposis with and without allergy. Clin Transl Allergy 3(Suppl 2):O14. https://doi.org/10.1186/2045-7022-3-s2-o14
Gevaert P, Holtappels G, Johansson SGO, Cuvelier C, Cauwenberge P, Bachert C (2005) Organization of secondary lymphoid tissue and local IgE formation to Staphylococcus aureus enterotoxins in nasal polyp tissue. Allergy 60(1):71–79. https://doi.org/10.1111/j.1398-9995.2004.00621.x
Melo JT Jr, Tunstall T, Pizzichini MMM et al (2019) IL-5 levels in nasosorption and sputosorption correlate with sputum eosinophilia in allergic asthma. Am J Respir Crit Care Med 199(2):240–243. https://doi.org/10.1164/rccm.201807-1279LE
De Schryver E, Derycke L, Calus L et al (2017) The effect of systemic treatments on periostin expression reflects their interference with the eosinophilic inflammation in chronic rhinosinusitis with nasal polyps. Rhinology 55(2):152–160. https://doi.org/10.4193/Rhin16.314
Berings M, Arasi S, De Ruyck N et al (2017) Reliable mite-specific IgE testing in nasal secretions by means of allergen microarray. J Allergy Clin Immunol 140(1):301–303.e8. https://doi.org/10.1016/j.jaci.2016.11.047
Rodríguez-Domínguez A, Berings M, Rohrbach A et al (2020) Molecular profiling of allergen-specific antibody responses may enhance success of specific immunotherapy. J Allergy Clin Immunol 146(5):1097–1108. https://doi.org/10.1016/j.jaci.2020.03.029
Hoffmann HJ, Santos AF, Mayorga C et al (2015) The clinical utility of basophil activation testing in diagnosis and monitoring of allergic disease. Allergy 70(11):1393–1405. https://doi.org/10.1111/all.12698
Gómez E, Campo P, Rondón C et al (2013) Role of the basophil activation test in the diagnosis of local allergic rhinitis. J Allergy Clin Immunol 132(4):975–976.e1–e5. https://doi.org/10.1016/j.jaci.2013.07.016
Busse W, Corren J, Lanier BQ et al (2001) Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 108(2):184–190. https://doi.org/10.1067/mai.2001.117880
Gevaert P, Omachi TA, Corren J et al (2020) Efficacy and safety of omalizumab in nasal polyposis: 2 randomized phase 3 trials. J Allergy Clin Immunol 146(3):595–605. https://doi.org/10.1016/j.jaci.2020.05.032
Okubo K, Okano M, Sato N et al (2020) Add-on omalizumab for inadequately controlled severe pollinosis despite standard-of-care: a randomized study. J Allergy Clin Immunol Pract 8(9):3130–3140.e2. https://doi.org/10.1016/j.jaip.2020.04.068
Casale TB, Condemi J, LaForce C et al; Omalizumab Seasonal Allergic Rhinitis Trial Group (2001) Effect of omalizumab on symptoms of seasonal allergic rhinitis: a randomized controlled trial. JAMA 286(23):2956–2967. https://doi.org/10.1001/jama.286.23.2956
Chervinsky P, Casale T, Townley R et al (2003) Omalizumab, an anti-IgE antibody, in the treatment of adults and adolescents with perennial allergic rhinitis. Ann Allergy Asthma Immunol 91(2):160–167. https://doi.org/10.1016/S1081-1206(10)62171-0
Kamin W, Kopp MV, Erdnuess F, Schauer U, Zielen S, Wahn U (2010) Safety of anti-IgE treatment with omalizumab in children with seasonal allergic rhinitis undergoing specific immunotherapy simultaneously. Pediatr Allergy Immunol 21(1 Pt 2):e160–e165. https://doi.org/10.1111/j.1399-3038.2009.00900.x
Vignola AM, Humbert M, Bousquet J et al (2004) Efficacy and tolerability of anti-immunoglobulin E therapy with omalizumab in patients with concomitant allergic asthma and persistent allergic rhinitis: SOLAR. Allergy 59(7):709–717. https://doi.org/10.1111/j.1398-9995.2004.00550.x
Pinto JM, Mehta N, DiTineo M, Wang J, Baroody FM, Naclerio RM (2010) A randomized, double-blind, placebo-controlled trial of anti-IgE for chronic rhinosinusitis. Rhinology 48(3):318–324. https://doi.org/10.4193/Rhin09.144
Chen M, Choo E, Haselkorn T et al (2019) Effect of omalizumab in asthma patients by number of asthma-related and allergic comorbidities in PROSPERO. Presented at the American College of Allergy, Asthma & Immunology Annual Scientific Meeting, November 7–11, 2019, Houston, TX
Lanier B, Bridges T, Kulus M, Taylor AF, Berhane I, Vidaurre CF (2009) Omalizumab for the treatment of exacerbations in children with inadequately controlled allergic (IgE-mediated) asthma. J Allergy Clin Immunol 124(6):1210–1216. https://doi.org/10.1016/j.jaci.2009.09.021
Humbert M, Boulet LP, Niven RM, Panahloo Z, Blogg M, Ayre G (2009) Omalizumab therapy: patients who achieve greatest benefit for their asthma experience greatest benefit for rhinitis. Allergy 64(1):81–84. https://doi.org/10.1111/j.1398-9995.2008.01846.x
Çelebi Sözener Z, Aydın Ö, Mısırlıgil Z et al (2018) Omalizumab in non-allergic asthma: a report of 13 cases. J Asthma 55(7):756–763. https://doi.org/10.1080/02770903.2017.1362427
de Llano LP, Vennera MDC, Álvarez FJ, et al; Spanish Registry (2013) Effects of omalizumab in non-atopic asthma: results from a Spanish multicenter registry. J Asthma 50(3):296–301. https://doi.org/10.3109/02770903.2012.757780
Domingo C, Pomares X, Angril N, Rudi N, Amengual MJ, Mirapeix RM (2013) Effectiveness of omalizumab in non-allergic severe asthma. J Biol Regul Homeost Agents 27(1):45–53
Pillai P, Chan Y-C, Wu S-Y et al (2016) Omalizumab reduces bronchial mucosal IgE and improves lung function in non-atopic asthma. Eur Respir J 48(6):1593–1601. https://doi.org/10.1183/13993003.01501-2015
Agache I, Beltran J, Akdis C et al (2020) Efficacy and safety of treatment with biologicals (benralizumab, dupilumab, mepolizumab, omalizumab and reslizumab) for severe eosinophilic asthma. A systematic review for the EAACI guidelines - recommendations on the use of biologicals in severe asthma. Allergy 75(5):1023–1042. https://doi.org/10.1111/all.14221
Kurokawa M, Koya T, Takeuchi H et al (2020) Association of upper and lower airway eosinophilic inflammation with response to omalizumab in patients with severe asthma. J Asthma 57(1):71–78. https://doi.org/10.1080/02770903.2018.1541357
Shoda Y, Watanabe M, Wada K et al (2019) Successful management of severe asthma in a young boy with eosinophilic chronic rhinosinusitis who received omalizumab: a case report. Allergy Asthma Clin Immunol 15:55. https://doi.org/10.1186/s13223-019-0369-7
Cameli P, Perruzza M, Salvini M et al (2019) Omalizumab treatment in Samter’s triad: case series and review of the literature. Eur Rev Med Pharmacol Sci 23(18):8124–8129. https://doi.org/10.26355/eurrev_201909_19031
Forster-Ruhrmann U, Stergioudi D, Pierchalla G, Fluhr JW, Bergmann KC, Olze H (2020) Omalizumab in patients with NSAIDs-exacerbated respiratory disease. Rhinology 58(3):226–232. https://doi.org/10.4193/Rhin19.318
Hayashi H, Fukutomi Y, Mitsui C et al (2020) Omalizumab for aspirin-hypersensitivity and leukotriene overproduction in aspirin-exacerbated respiratory disease: a randomized trial. Am J Respir Crit Care Med 201(12):1488–1498. https://doi.org/10.1164/rccm.201906-1215OC
Jean T, Eng V, Sheikh J et al (2019) Effect of omalizumab on outcomes in patients with aspirin-exacerbated respiratory disease. Allergy Asthma Proc 40(5):316–320. https://doi.org/10.2500/aap.2019.40.4241
Lang DM, Aronica MA, Maierson ES, Wang XF, Vasas DC, Hazen SL (2018) Omalizumab can inhibit respiratory reaction during aspirin desensitization. Ann Allergy Asthma Immunol 121(1):98–104. https://doi.org/10.1016/j.anai.2018.05.007
Türk M, Bahçecioğlu SN, Tutar N, Oymak FS, Gülmez I, Yılmaz I (2018) Omalizumab treatment for atopic severe persistant asthma: a single-center, long-term, real-life experience with 38 patients. Turk Thorac J 19(4):187–192. https://doi.org/10.5152/TurkThoracJ.2018.17109
Lavaud F, Bonniaud P, Dalphin JC et al (2013) Usefulness of omalizumab in ten patients with severe occupational asthma. Allergy 68(6):813–815. https://doi.org/10.1111/all.12149
Busse WW (2001) Anti-immunoglobulin E (omalizumab) therapy in allergic asthma. Am J Respir Crit Care Med 164(8 Pt 2):S12–S17. https://doi.org/10.1164/ajrccm.164.supplement_1.2103026
Kurosawa M, Sutoh E (2019) Prospective open-label study of 48-week subcutaneous administration of mepolizumab in Japanese patients with severe eosinophilic asthma. J Investig Allergol Clin Immunol 29(1):40–45. https://doi.org/10.18176/jiaci.0285
Bachert C, Han JK, Desrosiers M et al (2019) Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet 394(10209):1638–1650. https://doi.org/10.1016/S0140-6736(19)31881-1
Corren J, Castro M, O’Riordan T et al (2020) Dupilumab efficacy in patients with uncontrolled, moderate-to-severe allergic asthma. J Allergy Clin Immunol Pract 8(2):516–526. https://doi.org/10.1016/j.jaip.2019.08.050
Bousquet J, Rabe K, Humbert M et al (2007) Predicting and evaluating response to omalizumab in patients with severe allergic asthma. Respir Med 101(7):1483–1492. https://doi.org/10.1016/j.rmed.2007.01.011
Korn S, Haasler I, Fliedner F et al (2012) Monitoring free serum IgE in severe asthma patients treated with omalizumab. Respir Med 106(11):1494–1500. https://doi.org/10.1016/j.rmed.2012.07.010
Tamer F, Gulru Erdogan F, Dincer Rota D, Yildirim D, Akpinar Kara Y (2019) Efficacy of omalizumab in patients with chronic spontaneous urticaria and its association with serum IgE levels and eosinophil count. Acta Dermatovenerol Croat 27(2):101–106
Hamilton RG (2016) Monitoring allergic patients on omalizumab with free and total serum IgE measurements. J Allergy Clin Immunol Pract 4(2):366–368. https://doi.org/10.1016/j.jaip.2015.12.002
Probst M, Gogolka A, Krüll M, Noga O (2019) In search of clinically relevant parameters to monitor successful omalizumab therapy in allergic asthma. Allergol Select 2(1):49–55. https://doi.org/10.5414/ALX01377E
Acknowledgements
The authors would like to acknowledge the contributions of Cecile T.J. Holweg during the development of this manuscript.
Funding
This study was funded by Genentech, Inc., a member of the Roche Group, and Novartis Pharma AG. Third-party writing assistance was provided by Jack W. Pike, PhD, CMPP, of Envision Pharma Group, and funded by Genentech, Inc., a member of the Roche Group, and Novartis Pharmaceuticals Corporation.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conceptualization, writing, reviewing, and editing of the submitted review article.
Corresponding author
Ethics declarations
Conflict of Interest
Philippe Gevaert has received consulting fees, honoraria for lectures, and/or research funding from 3NT, Ablynx, ALK, Argenx, AstraZeneca, Bekaert Textiles, Genentech, Inc., Hal Allergy, Medtronic, Novartis, Regeneron, Roche, Sanofi Genzyme, Stallergenes Greer, Teva, and Thermo Fisher. Kit Wong is an employee of Genentech, Inc. Lauren A. Millette is an employee of Genentech, Inc. Tara F. Carr has received consulting fees from Aimmune Therapeutics, AstraZeneca, GlaxoSmithKline, Novartis, Regeneron, and Sanofi Genzyme.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gevaert, P., Wong, K., Millette, L.A. et al. The Role of IgE in Upper and Lower Airway Disease: More Than Just Allergy!. Clinic Rev Allerg Immunol 62, 200–215 (2022). https://doi.org/10.1007/s12016-021-08901-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-021-08901-1