
Abstract
In the last few decades, a substantial body of evidence underlined the pivotal role of bradykinin in certain types of angioedema. The formation and breakdown of bradykinin has been studied thoroughly; however, numerous questions remained open regarding the triggering, course, and termination of angioedema attacks. Recently, it became clear that vascular endothelial cells have an integrative role in the regulation of vessel permeability. Apart from bradykinin, a great number of factors of different origin, structure, and mechanism of action are capable of modifying the integrity of vascular endothelium, and thus, may participate in the regulation of angioedema formation. Our aim in this review is to describe the most important permeability factors and the molecular mechanisms how they act on endothelial cells. Based on endothelial cell function, we also attempt to explain some of the challenging findings regarding bradykinin-mediated angioedema, where the function of bradykinin itself cannot account for the pathophysiology. By deciphering the complex scenario of vascular permeability regulation and edema formation, we may gain better scientific tools to be able to predict and treat not only bradykinin-mediated but other types of angioedema as well.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
At the site of microcirculation, convection together with active, directed transcellular transport, and diffusion are responsible for the exchange of metabolites and gases. These processes are majorly controlled by the permeability/barrier (P/B) state of the endothelial cells (ECs). The P/B state depends on the anatomy of capillaries, as adapted to the function of distinct organs. This, however, can also quickly change according to the actual, local requirements. Therefore, increased (as a response) or high (as local tissue characteristics) vascular permeability (IVP/HVP) are not always considered pathological conditions. In other words, IVP/HVP and edema are not synonymous terms. Edema means “a swelling” in ancient Greek (oidēma). It occurs when excessive amount of fluid accumulates in the tissues, which may vary in its location, onset, and duration (Fig. 1). According to the Starling equation, edema has two requirements: a driving force of transvascular fluid flux (i.e., a non-zero sum of hydrostatic and osmotic pressure gradient) and the opening of the endothelial barriers (IVP/HVP). A good example is the liver (an HVP organ), where the net water flux is minimal and edema does not normally occur, despite the large intercellular gaps between the sinusoidal ECs. Such an equilibrium exists when the osmotic and hydrostatic pressures are equal between the interstitial and intracapillary space [1, 2]. Moreover, several built-in negative feedback loops exist to avoid the accumulation of fluid in the tissues [3]. Although there are some exceptions, the pathological background of most edemas is well-described. However, through certain vicious circles, multiple factors collaborate in the pathomechanism of almost every edematous disease. This is the case in chronic venous insufficiency, where the venous valvular incompetence (i.e., elevated hydrostatic pressure) induces turbulent blood flow, inadequate perfusion, inflammation, and consequently, increased endothelial permeability, which together lead to limb edema [4].

Main features of tissue edema. Pathophysiology, location, and temporal relations are the main features of edema. A few examples are also included under “pathophysiology.” Color shading highlights the main topic of the current review—the vascular endothelium
Although several pathophysiological processes can result in edema formation (Fig. 1), we will focus in this review on types of edema where dysregulation of ECs is the leading pathogenetic factor in the impairment of the P/B state. This is the case in bradykinin (BK)- or histamine-mediated angioedema or in capillary leak syndrome (CLS) [5]. The active participation of ECs in edema can be observed in inflammatory and infectious diseases (i.e., sepsis, and even in COVID-19). In these conditions, regardless of the active or passive role of endothelium, IVP is transiently induced. By eliminating the underlying pathologic factors, the edema also resolves. However, several distinct diseases cause recurrent elevation of EC permeability leading to angioedema attacks without any sign of hypoxia/reoxygenation (H/R) injury, severe inflammation, or injury to the endothelium. Wu et al. proposed to name these transient conditions as paroxysmal permeability disorders (PPDs), characterized by the return of the ECs to normal function after resolution of the angioedema attack [6, 7].
Although the term “angioedema” should be applied to every edematous disease where the primary cause is the damage or dysregulation of vascular endothelium, it is restrictively reserved to mast cell (MC)-mediated and BK-mediated PPDs. In MC-mediated angioedema, where histamine is the main mediator, a great number of other MC-derived factors and secondary factors produced by other cell types also participate in the pathomechanism. Typically, MC-mediated angioedema has a rather instant onset, beginning within minutes after allergen challenge [8].
As a potent vasoactive oligopeptide, BK has been brought into the focus of research during the exploration of the pathophysiology of angioedema [9]. During an episode of BK-mediated angioedema, BK is overproduced by the uncontrolled activation of the kallikrein-kinin system (KKS) due to C1-inhibitor (C1-INH) deficiency [10], or is elevated due to reduced breakdown caused by angiotensin converting enzyme inhibitors (ACEIs) [11, 12].
The picture of pathophysiology of angioedema is more complicated than we thought earlier. Research in the recent years has shown that BK also has a role in certain types of angioedemas that were not classified as BK-mediated before. For instance, heparin-initiated BK production plays a fundamental role in MC-mediated diseases [13]. It was recently proposed that BK also has a critical role in the lung inflammation of COVID-19. Consequently, targeted therapies originally developed for the management of BK-mediated angioedemas might also be effective for the treatment of COVID-19 [14].
BK-mediated angioedema involves a variety of plasma enzyme cascades (KKS, coagulation, fibrinolysis, and complement) [15, 16], vascular endothelium [7, 17, 18], lymphatic endothelium [19], and cells of the immune system (e.g., neutrophil granulocytes) [20]. Various signaling pathways (such as ANGPT1-Tie2 signaling in HAE patients with ANGPT1 mutation) [21], hormonal effects [22], the autonomic nervous system [23], and other mechanisms yet to be discovered [24] may also contribute to the pathogenesis.
Maintaining the hemodynamic balance of an organism—including the P/B state of the endothelium—is a complex process, which requires the precise cooperation of many organ systems. The pathophysiology of ECs in C1-INH deficiency provides a good example for such complexity. In this review, we aim to provide an insight into the molecular mechanisms of permeability-increasing and permeability-decreasing agents. Furthermore, we expose some of the current controversies in angioedema from the perspective of ECs, and finally, attempt to outline an alternative, complex hypothesis on the role of ECs in the pathogenesis of BK-mediated angioedema.
Molecular Mechanisms Behind Regulation of Permeability
When investigating the pathomechanism of edematous conditions, it is of key importance to reveal the P/B state of ECs and identify the relevant permeability modulating agents. To accomplish this, one needs to understand how permeability is regulated at the cellular/molecular level. At physiologic conditions, transcellular transport is controlled by chemical potential-driven diffusion and active vesicle transport, while paracellular transport is controlled by opening gaps between ECs. Transcellular and paracellular transport contribute to the exchange of materials between the blood and the tissues. The proportion of these transport mechanisms are substantially organ dependent. For example, in the brain, transcellular transport is almost exclusive, whereas in sinusoid endothelia (in liver, bone marrow, and spleen) paracellular transport is the dominant mechanism.
This review will focus on the mechanism of paracellular transport, since it is the predominant route of fluid transport during pathological conditions. To function as a barrier, ECs must have well-organized tight junctions (TJs) involving several adhesion molecules, such as occludin, junctional adhesion molecules (JAMs), claudins, angulins, and tricellulin. Through intracellular adaptor proteins from the zonula occludens protein family (ZOs), their intracellular domains are connected to actin cytoskeleton. TJs are physically supported by belt-like adherent junctions (AJs) and patchy desmosomes (DSs). Vascular endothelial cadherin (VE-cadherin), the predominant homotypic adhesion molecule of AJs in ECs, is anchored to actin fibers and behaves as a force transducer between ECs [25]. DSs and hemidesmosomes (HDs) are also important structures participating in cell-cell and cell-basal membrane connections [26]. Besides TJs (as complete sealers even for water molecules) and AJs (which retain blood cells and macromolecules with the largest size), glycocalyx also contributes to the barrier function by repelling negatively charged molecules, even where TJs are not intact or fully closed [27]. A well-structured cortical actin cytoskeleton is also required for the normal barrier function. Both the members of the cellular adhesion complexes described above and the acto-myosin network are regulated by several mechanisms including phosphorylation/dephosphorylation, internalization of receptors, and adaptor assembly/disassembly controlled by Ca-ions. Normally, these effects are completely reversible in order to effectively restore the original tissue functions. Agonist-induced disruption of the endothelial barrier occurs through the rearrangement of actin cytoskeleton, accompanied by the activation of the contractile apparatus and the disassembly of cellular junctions (especially VE-cadherin containing AJs). As a result, paracellular gaps form, causing endothelial hyperpermeability. The above mechanism is summarized in Fig. 2.

Common intercellular signaling events of endothelial permeability regulation. The most important signaling events leading to barrier protection (tight junction and adherent junction stabilization, formation of cortical actin network) are emphasized by green color, whereas those leading to barrier disruption and hyperpermeability (phosphorylation and dissociation of cell junction components, phosphorylation of myosin light chain and formation of actin stress fibers) by red color
After receptor activation, intracellular Ca2+ level rises, which in turn activates myosin light chain kinase (MLCK), RhoA/Rho kinase (ROCK), and protein kinase C (PKC) signaling. These pathways lead to the phosphorylation and activation of myosin light chains (MLC), actin stress fiber formation, increased contractility, and phosphorylation (and thereby dissociation) of intracellular AJ components [28]. This primary process of hyperpermeability takes place rather quickly, within minutes. Then why do some types of edema caused by hyperpermeability develop much slower? This can be the case if agonist receptors and other factors of permeability signaling are induced transcriptionally or if triggering factors act via other cell types, which in turn locally produce hyperpermeability agonists.
It is also important to comprehend how normal barrier function is maintained. The signaling of barrier protective mediators converge predominantly at the level of Rac1, a small GTPase with a crucial role in controlling RhoA activity and the cytoskeletal rearrangement. A well-known pathway leading to Rac1 activation is cyclic adenosine monophosphate (cAMP) signaling. cAMP interferes with RhoA as a result of a very intricate signaling network: either through activation of the PKA/Rac1 axis or Epac signaling (the latter also leads to the stabilization of microtubules) [29]. The signaling of other barrier enhancing mediators can promote Rac1 activation through protein kinase B (PKB/Akt) or Ras GTPase-activating-like protein (IQGAP1) activation, without increasing cAMP production. The importance of the above mentioned barrier stabilizing mechanisms is well illustrated during inflammation or sepsis, where an impaired cAMP/Rac1 axis contributes to the barrier disrupting effects of hyperpermeability mediators [30].
Taken together, theoretically, every mediator influencing the expression or the function of (1) permeability inducing or decreasing factors, (2) their receptors, (3) Ca2+ channels, (4) compounds of the Rho/ROCK/MLCK/Rac1 system, (5) cAMP metabolism, or (6) adaptors or adhesion molecules of TJs, AJs, DSs, and HDs can potentially modify endothelial permeability.
Permeability Modifying Mediators
Molecules known to affect endothelial permeability represent great diversity in their mechanisms of action. For the purpose of this review, we arbitrarily defined permeability modifying factors as “C1-INH sensitive” or “C1-INH non-sensitive.” C1-INH sensitive factors interact with C1-INH (i.e., they can be inhibited by C1-INH, can cleave C1-INH or their metabolism is influenced by C1-INH). The highly effective therapeutic C1-INH concentrates may also act at the level of these factors.
C1-INH non-sensitive factors (i.e., those without any obvious connection with C1-INH) may also attract attention, since they can modify the frequency, severity, localization, and resolution of angioedema. We present permeability modifying mediators in groups based on their structure, receptor usage or origin (Table 1).
Vasoactive Peptides and Peptidases
The group of vasoactive peptides comprise small oligopeptides that are usually produced as pro-peptides. They gain activity after cleavage of the pro-peptide, remain active for a relatively short period, bind to cell surface receptors, and can be inactivated by soluble or cell surface peptidases.
BK and Kallidin
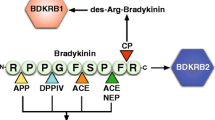
BK is a 9-amino acid-long oligopeptide, cleaved by plasma kallikrein (PKa) from high molecular weight kininogen (HK) mostly on the surface of ECs. Its predominant role in angioedemas have previously been discussed thoroughly [9, 31,32,33]. The plasma level of BK was found to be elevated during angioedema attacks [32]. BK represents a C1-INH-sensitive permeability factor, since its production is inhibited by C1-INH. Moreover, complement MASP-1, another serine protease that can be inhibited by C1-INH, is also able to cleave HK and form BK, although with less efficiency than PKa [34]. Kallidin (or Lys-BK) is a 10-amino acid-long oligopeptide, cleaved by tissue kallikrein 1 (KLK1) from low molecular weight kininogen. In contrast to BK, kallidin is C1-INH non-sensitive, as KLK1 is not inhibited by C1-INH [35]. Both BK and kallidin act on BK receptor B2 (BKRB2), and, with a lower affinity, on BKRB1 (both expressed on ECs). The first breakdown products of BK and kallidin, desArg-BK, and desArg-kallidin, respectively, are also regarded as permeability-increasing factors, acting solely on BKRB1. Both BK receptors belong to the G-protein coupled receptor (GPCR) family and induce Ca-signaling, PLA2/C/D, PKCδ, and NFκB activation [36]. They are regulated by GPCR kinase 2 (GRK2), which was recently found to be an important determinant of attack severity in C1-INH-HAE patients [37].
The first step of BK and kallidin breakdown is catalyzed by carboxypeptidase N, which cleaves BK and kallidin into their still vasoactive desArg forms, whereas other peptidases carry on further cleavage, resulting in completely inactive peptide fragments. This metalloproteinase group comprises, amongst others, carboxypeptidase M, neutral endopeptidase, angiotensin-converting enzymes (ACE, ACE2), and dipeptidyl-peptidase (DPP-IV). The latter peptidases can be blocked by ACE inhibitors (ACEI) and SARS-CoV2 or DPP-IV inhibitors (gliptins), respectively, which may all cause drug-induced angioedema by the inhibition of BK/kallidin breakdown [38,39,40]. Interestingly, BK is much more active as a permeability-increasing factor in vivo than in vitro [41]. A plausible explanation of this discrepancy is that BK is an extremely potent stimulator of the sensory nerves [42], which, in response to BK, produce several permeability increasing or potentiating factors [43, 44].
Angiotensin II
Angiotensin II, as the most active component of the renin-angiotensin system, is a very potent blood-pressure elevating vasoactive peptide and also influences permeability. It increased blood–brain barrier (BBB) permeability in murine models via its AT1 receptor, without having direct effects on TJ components or actin cytoskeleton [45]; however, it involves reactive oxygen species [46]. Angiotensin II antagonized the increased microvascular permeability induced by ATP in a rat model [47]. It is worth noting that the effects of angiotensin II on endothelial permeability are, at least partially, mediated by vascular endothelial growth factor (VEGF) [48, 49]. Although angiotensin II can influence vascular permeability, its converting enzymes (ACE and ACE2) are more important permeability regulators as they take part in the breakdown of BK.
ET-1
Endothelin-1 (ET-1) is a 21-amino acid peptide, synthesized mostly in ECs. Besides being the most potent vasopressor agent yet identified acting on vascular smooth muscle cells, ET-1 also has receptors on ECs. ET-1 was shown to decrease ATP- or BK-induced permeability as well as hydraulic permeability [50]; however, in other models, it increased or had no effect on vascular permeability in vivo [51,52,53]. This can partially be explained by the secondary effects of ET-1 as a strong inducer of both permeability-increasing thromboxane A2 (TXA2) and permeability decreasing adrenomedullin (ADM) [51, 54]. It is yet unclear whether the elevated level of ET-1 during C1-INH-HAE attacks [55] indicates a pathogenetic or a moderating role for ET-1 in edema formation.
AVP
Arginine-vasopressin (AVP) is an antidiuretic vasoactive nonapeptide of neuronal, cardiac, and adrenal gland origin. However, it also has a barrier stabilizing role, which may result from similar receptor signaling as its antidiuretic effect. AVP alone had no effect on the permeability of the BBB in a rat model [53], but in hamsters, it potently inhibited the permeability-increasing effects of histamine [56]. We previously found that AVP concentration was elevated during attacks in C1-INH-HAE patients, which also suggests its regulatory role in permeability [55].
NP Family
Natriuretic peptide (NP) family consists of three structurally similar peptides: the 28-amino acid ANP, the 32-amino acid BNP, and the 22-amino acid CNP. ANP and BNP are predominantly produced by the atrial muscle cells, whereas CNP is synthesized mainly by the vascular ECs. ANP has a well-described permeability decreasing effect. It suppresses both the basal permeability [57], and permeability induced by thrombin [58], tumor necrosis factor alpha (TNFα) [59], oxidants [60], VEGF [61], and histamine [62]. BNP has similar effects to those of ANP, it could also block thrombin-induced permeability [63]; however, the role of CNP is more controversial. Compared to ANP and BNP, CNP had no effect on permeability [63]; however, in another study, it increased BBB permeability by reducing ZO-1 expression [64]. ANP acts predominantly by inducing Rac1/PAK1 activation, stabilization of microtubules, and blocking RhoA pathway [65]. We found that ANP levels were lower during attack-free periods of C1-INH-HAE patients than in controls, suggesting that it is indeed a hyperpermeability factor [66].
ADM and the CGRP Family
Adrenomedullin (ADM) is a 52-amino acid peptide of the calcitonin gene-related-peptide (CGRP) family produced by a wide range of tissues including ECs themselves. ADM induces vasodilation, natriuresis, and cell growth. Moreover, it enhances the barrier function of ECs by cAMP induction, adhesion, and adaptor protein recruitment into the TJs and AJs, and Rap1-dependent cytoskeleton stabilization. Using several in vitro as well as in vivo models, ADM effectively decreased both basal and pre-induced permeability [67,68,69,70]. We found that ADM concentration was higher during attacks of C1-INH-HAE, than in symptom-free periods [55]. Intermedin (ADM2), another member of the CGRP family, also has barrier protecting activities via cAMP/PKA and cAMP/Epac signaling pathways [71]. Although CGRP, the denominator of the family, released mainly from sensory neurons, has less pronounced direct effects on vascular permeability. Early studies demonstrate a synergistic permeability-increasing role for CGRP [43, 72].
SP and NKs
Substance P (SP), NKA, and NKB belong to the family of tachykinins. These three closely related small peptides (11, 10, and 10 amino acids, respectively) mainly produced by sensory neurons are known as permeability-increasing factors.
Of the GPCR family neurokinin (NK) receptors, NK1 mediates the most pronounced permeability-increasing effect on ECs [73]. SP possesses the highest affinity to NK1 and is the strongest permeability-increasing factor in the tachykinin family. Although most studies concentrate on the SP/BBB interaction, SP acts on lung and skin vasculatures as well [44]. The neurogenic inflammation mediated by SP also has indirect effects on vascular permeability, as it is a potent histamine releasing factor [74]. The tachykinin family members are also prone to cleavage by ACE, ACE2, neutral endopeptidase and other cell surface peptidases.
Relaxin
Relaxin is a 52-amino acid polypeptide, a member of the insulin family, produced predominantly by the reproductive systems and the heart. Although it can induce VEGF expression [75, 76] leading to a late-onset increase in permeability, in short-term, relaxin is a potent barrier protecting factor. It prevents the permeability-increasing effects of H/R injury, hydrogen peroxide (H2O2) and cytokines [77].
PAR Agonists
Most BK-mediated angioedemas are closely related to plasma enzyme cascade systems involving more than 20 enzymes. The majority of these enzymes belong to the serine protease family and many of them are inhibited by C1-INH. Therefore, except when specifically the BK breakdown is impaired, disturbances of these cascade systems are observed in BK-mediated angioedemas [78, 79].
The participating enzymes, as well as several other plasma and tissue enzymes, act on ECs via protease activated receptors (PARs) and in that way they can cause increased vascular permeability (Table 2). PARs, belonging to the GPCR family, have a unique activation mechanism [80]. They are activated by a proteolytic cleavage on their N-terminal domain and the newly acquired N-terminal sequence becomes a tethered ligand, which leans back to the extracellular part of the 7TM domain and activates the receptor. PARs (PAR1-4) are expressed ubiquitously in the human body, but predominantly on vascular, epithelial, intestinal, and immune cells, as well as the nervous system. PARs can be cleaved at canonical or non-canonical cleavage sites, resulting in diverse signaling and cellular responses. The non-canonical cleavage causes the so-called biased signaling (incomplete or different signaling compared to the ones that have been described originally) [80], which has been observed in the case of all PARs [81,82,83,84,85,86]. In the following discussion, we describe the permeability modulating effects of a few important serine proteases of the PAR agonist group, as well as the non-enzymatic PAR agonist C4a, and how PAR-cleaving enzymes and other agonists act on vascular permeability.
Thrombin
Thrombin is a multifunctional serine protease being cleaved and activated from prothrombin in the blood. It participates in blood coagulation by forming fibrin from fibrinogen, and by converting coagulation factors FXI, FVIII, FV, and FXIII to their active forms. It also has major pro-inflammatory effects on ECs [87]. Thrombin cleaves PAR1 and PAR4, but it cannot activate PAR2 [88], while its PAR3 activation capability is still controversial. Thrombin directly increases endothelial permeability [89] via PAR-1 activation [90, 91] by disrupting the VE-cadherin/catenin complexes [92] in the AJs.
MASP-1
Mannan-binding lectin-associated serine protease 1 (MASP-1) is best known as being a key component in the activation of complement lectin pathway [93]. Our group showed that, similarly to thrombin, it directly activates ECs via PARs. The difference is their enzymatic activities and receptor preferences, i.e., MASP-1 can cleave PAR2 in addition to PAR1 and PAR4 [94]. The receptor activation manifests in pro-inflammatory activation of ECs [87, 94,95,96] and increased endothelial permeability by rearranging the actin-cytoskeleton, disrupting TJ/AJ components and altering transcription of several permeability related proteins [97].
MASP-2
Mannan-binding lectin-associated serine protease 2 (MASP-2) is activated by MASP-1, and by cleaving C2 and C4 it has a predominant role in complement lectin pathway activation. Using a human umbilical vein EC (HUVEC) model, we showed that MASP-2 is also a potent permeability-increasing enzyme. Although both the mechanism and the kinetics are very similar to those of thrombin and MASP-1 [98], the involvement of PARs in its activity is yet to be proven. We found a significant increase in MASP-2 levels in the 1/3 of patients during angioedema attacks [99], which supports the notion that MASP-2 may contribute to edema formation.
PKa
Plasma kallikrein (PKa) is formed from prekallikrein (PK) by FXIIa. It releases BK from HK, cleaves FXII and plasminogen, and is able to activate ECs via PAR1 and PAR2 [100, 101]. Increased leukocyte trafficking and enhanced ICAM-1 and VCAM-1 expression was demonstrated in the presence of PKa [101], which may modulate permeability indirectly through pro-inflammatory cytokines. PKa can cleave endothelial VE-cadherin in AJs [102]; moreover, as we have shown, it can activate ECs (possibly via PAR cleavage) and thereby directly increases endothelial permeability [98].
Plasmin
Plasmin is released from the inactive precursor plasminogen and after binding to clots or cell surfaces, can be activated by a variety of enzymes (e.g., tPA, uPA, PKa, FXII) [103]. Plasmin is best known as a fibrinolytic enzyme, but it can also activate several types of collagenases and cleave HK to BK [104]; furthermore, it cleaves all four types of PARs [88]. Although various permeability-increasing effects of plasmin were demonstrated (e.g., proteolytic damage of ECs, activation of matrix metalloproteinase MMP-3/9), the exact molecular mechanism is still controversial [105, 106]. Tranexamic acid, an antifibrinolytic drug, inhibits the formation of plasmin and has long been used in the prophylaxis of BK-mediated angioedema. Interestingly, it is also capable of protecting EC glycocalyx integrity independent of plasmin action, and thus protects barrier function [107].
Complement Fragment C4a
C4a, a complement activation product formed by C1s or MASP-2 (both are C1-INH sensitive serine proteases), turned out to increase endothelial permeability as an untethered agonist for PAR1 and PAR4 [108].
Signaling of PAR Agonists
PARs can signal through G-protein subunits Gβγ and almost all the Gα-s, which are major regulators of endothelial permeability. In general, the activation of the Gαq and Gαo subunits activates PLC-β, the Gαi subunit is responsible for adenylate cyclase inhibition, while Gα12/13 activation induces GTPases from the Rho signaling pathway and this way regulates actin cytoskeleton.
In ECs, PAR1 is coupled with Gαq [109, 110], Gαi [111], and Gα12/13 [110, 112], while PAR2 is associated with Gαq [113] subunits. We and others showed that PAR1 agonists are very potent permeability-increasing factors of human ECs in vitro [97, 114] as well as ex vivo in mice [90] and in vivo rats [115]. In human ECs, PAR1 agonists induce strong RhoA activation [114], Ca2+ influx [97], formation of actin stress fibers [114], and ring-like actin filaments [116], as well as inhibition of Rac [117]. PAR2 agonists also induce RhoA activation [114], Ca2+ influx [97], and actin stress fiber formation [114]; however, they can also stabilize the barrier function of human ECs via Rac activation [117]. Interestingly, although PAR2 agonists induce edema when injected into rat paws [118], this effect is obtained indirectly, by the release of vasoactive peptides (i.e., SP and CGRP), which originate from the sensory nerves activated by the PAR2 agonist [119]. This effect of PAR2 activation nicely demonstrates that hyperpermeability can be effectively induced indirectly, by utilizing other cell types (just as in the case of BK). PAR4 agonists induce actin stress fiber formation [116] and a weak Ca2+ influx [97] in human ECs. Jong-Sup et. al showed that PAR4 also plays a direct role in increasing endothelial permeability. When they blocked PAR1 with an antagonist peptide, thrombin could still induce permeability change in the endothelial layer [120], and we showed that PAR4 agonist directly increased endothelial permeability in HUVECs [97]. The role of PAR3 in the regulation of permeability is still unexplored. The role of PARs in edema formation is further complicated by simultaneous signaling and inhibition by protease catalyzed disarming [121, 122].
Amino Acid Derivatives
Histamine
Histamine is a well-known permeability-increasing agonist, synthesized by the decarboxylation of the amino acid histidine. Being produced and stored in mast cells and basophil granulocytes, it is the main mediator of acute allergic reaction. Its permeability-increasing effect causes edema and anaphylactic shock [123, 124]. Histamine has four receptors on ECs (H1R to H4R), and although all of them are GPCRs, they have opposing effects on endothelial permeability. H1R is responsible for the majority of the histamine-induced endothelial hyperpermeability. H1R is coupled to Gαq and its activation leads to intracellular Ca2+ mobilization, and activates MLCK, RhoA/ROCK, and p38 MAP kinase signaling, resulting in MLC phosphorylation, actin stress fiber formation, and cell contraction. This causes a rapid and fast-expiring increase in endothelial permeability (maximal response in 3 min) [125, 126]. The Gαs-coupled H2R has the opposite effect, it stabilizes the endothelial barrier as it stimulates the production of cAMP [127]. The mechanism of action of H3R and H4R is less understood, but these receptors are thought to contribute to the permeability-increasing effect of histamine in certain tissues, such as the skin [125].
Serotonin
Serotonin (or 5-hydroxytryptamine, 5-HT) is produced from tryptophan by enterochromaffin cells and MCs. It is released into the circulation, from where platelets take it up and store it in dense granules. Upon inflammation, platelets release serotonin back into the circulation, which leads to increased endothelial permeability, as demonstrated in endotoxemia [128, 129].
Catecholamines
Catecholamines (i.e., dopamine, adrenaline, noradrenaline) are the derivatives of phenylalanine/tyrosine and comprise several neurotransmitters and hormones. The permeability modulation by catecholamines is rather complex and sometimes controversial. Although the effect of adrenaline on β-adrenergic receptors induce cAMP synthesis, leading to decreased permeability [130], it may also induce α-adrenergic receptors and the breakdown of EC glycocalyx, both resulting in diminished barrier function [107, 131]. Noradrenaline may also act as a permeability-increasing or decreasing factor depending on the actual state of the ECs and their surroundings [132, 133]. Interestingly, no effects of catecholamines on lung microvascular permeability was found in ex vivo experiments [134]. Nevertheless, the direct effects of catecholamines may be exceeded by their secondary impact on the production and accessibility of other permeability modulating factors, such as ET-1 [135] and VEGF [136].
Cytokines and Growth Factors
Several cytokines affect endothelial/epithelial barrier function to either increase or decrease monolayer permeability. It is no surprise that pro-inflammatory cytokines, such as tumor necrosis factor-α (TNFα), interferon-γ (IFNγ), the majority of the interleukin-1 (IL-1) family, IL-4, IL-6, IL-8, and IL-13 act mostly as permeability-increasing agonists, while anti-inflammatory cytokines (IL-1Ra, IL-10) tend to protect the endothelial barrier. Although cytokines can modify vascular permeability directly by initiating intracellular signaling pathways in ECs, they also have pleiotropic secondary permeability-regulating effects via activation or suppression of leukocytes and other non-endothelial cell types. The vascular effects of cytokines are observed in the case of capillary-leak syndromes, referred to as “cytokine-storm,” but rarely connected to BK-mediated angioedema. Growth factors (GFs) regulate the proliferation and differentiation of various cell types and will be discussed here together with the cytokines because their signaling events and effects have many common characteristics.
TNFα
Tumor necrosis factor alpha (TNFα) is a potent pro-inflammatory cytokine that can increase endothelial permeability via the receptor TNFR1 or the combined signaling of TNFR1/TNFR2, although the underlying mechanisms are still quite elusive [41, 137]. The delayed but prolonged hyperpermeability effect of TNFα (8–24 h) was suggested to be caused by the removal/redistribution of TJ proteins without early RhoA activation and MLC phosphorylation [138], but in a more recent study, ROCK and MLCK inhibitors prevented the TNFα-induced hyperpermeability in vitro [139]. Moreover, TNFα stimulation upregulates the expression of TRPC1, the predominant isoform of store-operated cation channel in ECs, which results in an augmented Ca2+ influx, and, consequently, an enhanced permeability increase in response to other GPCR-utilizing agonists [140].
IFNγ
Interferon gamma (IFNγ) is predominantly produced by TH1, TC1, and NK cells during viral infections. The permeability-increasing effect of IFNγ is long-known [141]. There is one known receptor for IFNγ, IFNGR, that is comprised of two ligand-binding IFNGR1 and two associated signal transducing IFNGR2 chains [142]. Results of in vitro experiments on endothelial/epithelial monolayers showed that IFNγ induces the activation of the p38 MAPK and the RhoA/ROCK axis, resulting in MLC phosphorylation, acto-myosin contraction, cell-shape changes, and internalization of TJ-associated adhesion molecules [143, 144]. Nevertheless, IFNγ cannot induce a rapid permeability change but has a remote effect on endothelial permeability, which may be achieved by transcriptomic regulation (e.g., upregulation of Rho/ROCK axis proteins [145]). In vivo significance of IFNγ-induced permeability seems well-established by the observation that EC-specific IFNGR2-knockout mice showed significantly reduced vascular permeability in an experimental colitis model [146]. Although IFNγ itself belongs to the C1-INH non-sensitive permeability modifying factors, according to multiple studies and our unpublished data, it seems to be the strongest inducer of C1-INH production in ECs and hepatocytes [147,148,149]. Based on these observations, IFNγ may have a regulatory role in the termination of HAE attacks by elevating C1-INH concentrations.
IL-1 Family
Interleukin-1 (IL-1) has two isoforms, IL-1α and IL-1β, both being very potent pro-inflammatory cytokines with pleiotropic effects and key functions in the acute phase reaction, as well as in angiogenesis and permeability regulation. To exert their biological functions, both IL-1 isoforms use the same IL-1 receptor complex (IL-1R1) and both can increase endothelial permeability, although the effect of IL-1β is more pronounced [150]. In vivo and in vitro results suggest that the increased endothelial permeability observed in the IL-1β-treated lung is due to the combination of its direct and indirect effects. IL-1β induces RhoA/ROCK activation, stress fiber formation, and junctional disassembly. Moreover, it activates transforming growth factor β (TGFβ) production in the adjacent epithelial cells, which in turn increases endothelial permeability [151]. IL-1Ra inhibits the permeability-increasing effects of IL-1α and IL-1β as a natural IL-1R antagonist. IL-18 has an ambiguous role in permeability regulation. Although it stimulates the expression of permeability-increasing factors, IL-18 was also shown to have permeability-decreasing and anti-edema effects in the BBB and the retina by upregulating the actin-binding proteins dystrophin and claudin-5, leading to TJ stabilization [152, 153].
TGFβ
The anti-inflammatory cytokine Transforming growth factor beta (TGFβ) can also have both permeability-increasing and permeability-decreasing effects. TGFβ, besides its role in angiogenesis, development, and immune-regulation, can mediate endothelial/epithelial barrier protection through a canonical SMAD-dependent stabilization of TJ-associated proteins ZO-1, occludin, and claudin-2 [154, 155], whereas it is also able to induce barrier disruption by non-canonical TGFBR signaling and the activation of the RhoA/ROCK axis [156, 157].
IL-10
Interleukin 10 (IL-10), a pleiotropic anti-inflammatory cytokine produced by TREGs, B cells, and several other cell types, can decrease EC permeability by inhibiting the production of most pro-inflammatory cytokines. In an in vivo murine model, IL-10 inhibited the increase of microvascular permeability in response to endotoxin [158]. We showed that IL-10 strongly downregulated the expression of VEGF [159], which may also result in the protection of EC barrier function (see below).
VEGFs
The most well-characterized interaction of the vascular endothelial growth factor (VEGF) family and their receptors (VEGFR1-3) is between VEGF-A and VEGFR-2. Due to its role in tumor vascularization, it is the target of anti-angiogenic agents. The binding of VEGF-A to EC surface VEGFR-2 induces tyrosine phosphorylation of the receptor and initiates the PLC-γ, PI3K, and p38 MAPK signaling cascades to induce cell proliferation, survival, vasodilation, hyperpermeability, and cell migration [160]. The permeability-increasing effect of VEGF-A—which peaks approximately 15 min after treatment—was found to be dependent mainly on the PI3K-Akt mediated activation of endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) synthesis. Increased NO levels contribute to RhoA activation and cytoskeletal rearrangements, as well as the Src-dependent phosphorylation and destabilization of VE-cadherin in AJs, causing endothelial hyperpermeability [161]. VEGF-A and VEGF-C plasma concentration was found higher in C1-INH-HAE patients in remission than in controls, and the level of both VEGFs further increased during attacks, which supports their major permeability regulating role in BK-mediated angioedemas [162].
ANGs
Angiopoietins (ANGs) have a predominant role in the regulation of EC growth, survival, and barrier function. Despite using the same receptor complex (Tie-2 and Tie-1), the effects of ANG-1 and ANG-2 on the vasculature are rather opposite. ANG-1 is produced by mesenchymal cells and is a strong agonist for Tie-2, supporting EC survival, vessel stability, and barrier functions [163]. ANG-1/Tie-2 signaling was shown to further reduce basal permeability and prevent VEGF- and thrombin-induced endothelial hyperpermeability by reducing VE-cadherin, occludin, and PECAM-1 phosphorylation, while increasing β-catenin/VE-cadherin and occludin/ZO-1 association, leading to AJ/TJ stabilization [164, 165]. Interestingly, the ANG-1-induced barrier protection against VEGF was found to be mediated by RhoA; however, this did not activate ROCK but another RhoA target mDia, which bound and sequestered Src, depriving VEGFR2 of its essential downstream signaling molecule [166]. Moreover, ANG-1 was shown to be the predominant effector of mesenchymal stem cell (MSC) microvesicles, which have the potential to restore normal barrier function in lung ECs treated with pro-inflammatory cytokines [167].
Recently, ANG-1 has also gained attention among HAE experts as a missense mutation of the ANGPT1 gene, leading to a reduced ability of the ANG-1 protein to bind Tie-2, was found to be associated with a new type of hereditary angioedema, named ANGPT1-HAE [21]. The underlying mechanism was suggested to be haplo-insufficiency, where the loss-of-function mutant of ANG-1 fails to stabilize VE-cadherin in AJs and could not effectively reduce VEGF- or BK- induced stress fiber formation, leading to increased vascular leakage [168]. ANG-2, on the other hand, can be a weak agonist or an antagonist of Tie-2 in a context-dependent manner. ECs can produce ANG-2 and store it in their Weibel-Palade bodies. Under basal conditions, autocrine ANG-2 may act as a weak Tie-2 agonist in a Tie-1 dependent manner, leading to barrier stabilization. However, during inflammation, ANG-2 expression/release increases, and Tie-1 exodomain cleavage and shedding eliminates Tie-1 from the receptor complex, acting as a molecular switch turning ANG-2 into a Tie-2 antagonist, which causes barrier disruption [163]. In line with the differential permeability modulating effects of ANG-1 and ANG-2, only ANG-2 was found to be elevated during angioedema attacks in C1-INH-HAE patients [162].
bFGF
Basic fibroblast growth factor (bFGF) has an important role in angiogenesis and EC proliferation [169]. Recent findings of in vivo experiments imply that BBB protection is achieved by the FGF-receptor mediated activation of the PI3K-Akt-Rac1 signaling pathway that leads to reduced activity of the RhoA/ROCK axis and stabilization of junctional catenins [170]. Additionally, in vitro results suggest the involvement of the ERK signaling in endothelial barrier protection against oxygen–glucose deprivation/reperfusion [171].
EGF
Epidermal growth factor (EGF) is an essential growth factor not only for epidermal cells but for almost every epithelial/endothelial/mesothelial cell type. EGF was recently found to protect the BBB after ischemic injury by preventing ischemia-induced decrease of ZO-1 and claudin-5 [172]. Since media of in vitro EC culture usually contain EGF, bFGF and VEGF, it warns us to treat in vitro permeability results with special care, and that the different GF content of these EC media, having opposing effect on EC permeability, may explain several contradictions found in the literature.
Lipid Mediators
PAF
Platelet-activating factor (PAF) is a phospholipid that acts on multiple cell types including ECs. PAF exerts its endothelial effects (i.e., cell migration, angiogenesis, and hyperpermeability) by binding to its G-protein coupled receptor (PAF-R), which signals through Gαq and cSrc, inducing actin cytoskeletal rearrangement and interfering with actin polymerization in a Rac1-dependent but RhoA-independent manner. Very interestingly, PAF-R signaling leads to the disassembly of inter-endothelial junctions without MLC phosphorylation and cell contraction [173].
AA Metabolites
The membrane phospholipid derivative arachidonic acid (AA) is the source of many types of eicosanoids important for the regulation of the inflammatory response. These AA metabolites include prostanoids (prostaglandins and thromboxanes), leukotrienes, lipoxins, hydroxyeicosatetraenoic acids (HETEs), and epoxyeicosatrienoic acids. All are potent modulators of vascular function, including endothelial permeability in several cases. Thromboxane A2 (TXA2), as well as prostaglandins PGD2, PGE2, PGF2, and PGI2 exert their strong regulatory function through five basic types of GPCRs, and their effect on vascular permeability depends on the receptor subtype used and the tissue affected [174]. TXA2 for example affects the endothelial barrier acting on the TP prostanoid receptor subtype, activating the Rho/ROCK pathway and disrupting AJs, leading to increased vascular permeability within 15–20 min. This TXA2/TP signaling was found to contribute to tissue edema formation in an acute lung injury model [175], as well as to exacerbate microvascular dysfunction after ischemia/reperfusion [176]. Prostacyclin (PGI2), on the other hand, is a potent barrier protecting prostanoid. It can reduce the LPS- or thrombin-elicited permeability increase by inhibiting RhoA activation through cAMP/Epac or cAMP/Rac1 pathways [177,178,179].
Ceramide and S1P
Ceramide, a sphingolipid, is an important second messenger and mediator of apoptosis in several cell types, including ECs. Ceramide was found to increase endothelial permeability independently of apoptosis and without inducing actin cytoskeleton rearrangement [180]. In contrast to ceramide, its derivative, sphingosine-1-phosphate (S1P), is one of the most potent endothelial barrier protecting agents [181]. Extracellular sphingosine is effectively taken up by red blood cells (RBCs), in which S1P is generated by phosphorylation. RBCs, as the major source of S1P in the plasma, can store S1P in their plasma membranes in large quantities, from where extracellular serum albumin and high-density lipoprotein (HDL) can extract it. HDL and albumin bound S1P is then recognized by its S1P1 receptor (a GPCR) on ECs, and maintains the barrier functions under physiological conditions [182]. Barrier stabilization is achieved via two mechanisms: S1P1 receptor activation leads to a Rac1-dependent stabilization of endothelial junctions [183], and it also inhibits matrix metalloprotease (MMP) activity, thereby protecting EC surface glycocalyx, an important part of the endothelial barrier [184, 185].
Complement Components
Historically, the complement system was described as the first system involved in BK-mediated angioedema. We previously discussed the role of MASP-1, MASP-2, and C4a that use PARs to regulate endothelial permeability. Here, we focus on two additional molecules of the complement system leaving PAR signaling untouched.
C5a
C5a is a potent anaphylatoxin produced by the cleavage of C5 during complement activation. It mediates pro-inflammatory effects via C5aR, a GPCR family member receptor expressed on ECs [186]. In vitro experiments on various types of ECs showed that both C3a and C5a reorganize the actin cytoskeleton, but only C5a is capable of increasing monolayer permeability in a histamine-independent manner [187].
C1r
C1r is a serine protease indispensable for the complement classical pathway. We showed that C1r can also increase endothelial permeability, but this effect is independent of its enzymatic activity, therefore, it may not be mediated by PARs [98]. The exact mechanism awaits explanation.
Purinergic Mediators
Adenosine and ATP
Extracellular nucleotides and nucleosides—purinergic mediators—can be derived from every type of cells either by membrane transport or by cellular damage. ECs express numerous types of purinergic receptors, out of which nucleotide signals (e.g., ATP, ADP) are mediated by cation-channel type or GPCR type P2 receptors, while the most important extracellular nucleosides (e.g., adenosine) signal through G-protein-coupled P1 adenosine receptors [188]. Adenosine was shown to enhance the endothelial barrier function in the vasa vasorum through stabilization of cortical actin [189] and prevent the detrimental effects of TH1 cytokines on the BBB [190], whereas selective activation of the same receptors (A1R and A2AR) was found to be disruptive to the BBB [191]. ATP was also known to either increase or decrease endothelial permeability, but interestingly, newer results suggest that ATP itself induces barrier stabilization through P2Y receptors, while its metabolite adenosine is responsible for the barrier disrupting effect [192].
Steroid Compounds
Estrogens
Besides having an indispensable role in reproduction, estrogens, a group of sex steroids, regulate numerous metabolic and cardiovascular functions. Estrogens decrease the expression of claudin-5 in uterine ECs at the transcription level [193]; however, they restore barrier function in BBB insulted by oxygen–glucose deprivation/reperfusion via classical ERα and ERβ receptors [194]. In most endothelia, estrogens induce HIF1α-dependent VEGF expression [195], which increases permeability as a remote effect. Finally, estrogens were shown to have significant regulatory role in the synthesis of plasma enzyme cascade components relevant to mechanisms of angioedema (i.e., PK and FXII) [22, 196].
HC and Dexamethasone
Hydrocortisone (HC) is an anti-inflammatory glucocorticoid frequently used as a supplement to strengthen the barrier function of in vitro cultured ECs. HC decreases the basal vascular permeability by the cAMP signaling pathway as well as diminishes TNFα-induced hyperpermeability in BBB [197]. HC can directly upregulate the expression of occludin in cerebral ECs [198]. Its synthetic derivative, dexamethasone, is frequently used in diseases accompanied by edema, including COVID-19. Besides its anti-inflammatory effects (decreasing pro-inflammatory cytokines), dexamethasone downregulates the expression of VEGF, thus preserving barrier function [199].
Danazol
The attenuated androgen danazol has been used as prophylactic agent in C1-INH-HAE for decades. In addition to acting as inducer of C1-INH production in the liver, it has a direct effect on endothelial permeability. In nanomolar doses, it stabilizes the cortical actin ring, thereby protecting barrier function, however, in micromolar concentrations, danazol induces actin stress fiber formation and increases permeability [200]. Its biphasic dose-curve may explain why the effectiveness of danazol has not been straightforward.
Free Radicals
Free radicals, either generated as byproducts of respiration or as “on purpose” molecules, have numerous biological effects, which occasionally result in completely different outcomes at low and at high concentrations.
ROS
Reactive oxygen species (ROS) are produced mainly by different NADPH oxidases (Nox1–5 and Duox1,2) [201]. Superoxide anions (O2−) generated by Nox1,2,3 and 5 are very potent pro-inflammatory agents via induction of oxidative stress. O2− can activate oxidant-sensitive transient receptor potential (TRP) channels that are permeable for Ca2+, thereby increased intracellular [Ca2+] initiates signaling pathways causing endothelial hyperpermeability [202]. Interestingly, Nox4, Duox1, and Duox2 produce predominantly H2O2 instead of O2−, which has (in low concentration) a substantial role in redox signaling and preservation of endothelial barrier function [203].
Reactive Nitrogen Species
Nitric oxide (NO) is probably the most pleiotropic effector molecule produced by ECs. Therefore, it is not surprising that NO also has different effects on barrier function, depending on its concentration and the redox state of ECs. When ECs are in a reductive state (i.e., reduced glutathione is present), and produce NO in low nanomolar concentration by eNOS enzyme, RhoA activity is inhibited by its S-nitrosylation, which effectively protects the barrier function [204, 205]. During oxidative stress, however, NO and O2− are converted into peroxynitrite (ONOO−), which activates RhoA by Tyr-nitration, leading to hyperpermeability [206]. Moreover, NO can mediate the permeability-increasing effects of several factors, including shear stress, thrombin, PAF, ADP, serotonin, VEGF, and BK, when produced in high nanomolar concentration by eNOS [207, 208] or TNFα by inducible NOS (iNOS) [209]. NO may activate cGMP production and MAP kinases, and it can directly bind to catenins entailing their S-nitrosylation and dissociation from VE-cadherin, leading to barrier disruption [208].
Gasotransmitters and pH
H2S and CO
Endogenous hydrogen sulfide (H2S) and carbon monoxide (CO) are important gasotransmitters in the cardiovascular system with potent anti-inflammatory and barrier protecting effects when produced in a near-physiological concentration [210, 211].
Acidic Conditions
Changes in the pH of extracellular fluids may reflect several physiological and pathological processes. ECs highly express proton-sensing receptor GPR4, a GPCR, which elevates permeability in response to acidification through Gα12/13/Rho GTPase signaling pathway [212].
Viral, Fungal, and Bacterial Compounds
Besides endogenous permeability-modifying mediators, microbial factors can also affect the barrier function of the endothelium.
Viral Compounds
Viruses predominantly modify the vascular permeability by cytotoxicity, reprogramming the cellular metabolism and inducing cytokine production from the infected cells or from the surrounding leukocytes; however, direct effects were also described. Enterovirus A71 capsid protein VP1 reduced claudin-5 and increased vimentin expression in a murine BBB model, both of which may help the virus to enter the brain [213]. SARS-CoV2 can also modify endothelial permeability either directly or indirectly. The spike glycoprotein of SARS-CoV2 was found to directly induce a rapid decrease in the barrier electrical resistance of endothelial monolayers (with the effect starting immediately and peaking between 12 and 14 h) [214]. The indirect effect of SARS-CoV2 on endothelial permeability involves the depletion of ACE2, a cell surface enzyme serving as a receptor for SARS-CoV2 entry to ECs. As ACE2 is necessary for BK metabolism, its depletion results in the accumulation of des-Arg-BK. It was recently proposed that this BK metabolite binds to BK1R, which is upregulated by pro-inflammatory cytokines in response to the viral activity, thereby enhancing local pulmonary angioedema in a BK-dependent manner [39]. Additionally, many viruses can induce IFNγ production, which may increase C1-INH expression and modulate BK-mediated angioedema.
Fungal Compounds
Fungal compounds are also able to modulate vascular permeability. The mycotoxin cytochalasin D induces actin depolymerization leading to junctional destabilization and increased permeability [215].
LPS
Bacterial lipopolysaccharide (LPS) is a well-known bacterial permeability-increasing compound, a major component of the Gram-negative outer-membrane. LPS is recognized by a “pattern-recognition receptor,” toll-like receptor 4, expressed by ECs [216, 217]. LPS induces ROCK activation, MLC phosphorylation, stress fiber formation, and disruption of cell junctions, thereby increasing endothelial permeability. Its effect is sustained for up to 24 h and mediated by p38 MAPK, JNK, and NFκB activation, as well as the suppression of barrier stabilizing AMP-activated protein kinase (AMPK) [218, 219]. Moreover, LPS is able to upregulate the receptor expression of several permeability modifying factors in ECs (e.g., BK, histamine, thrombin, and MASP-1) as well as hyperpermeability-inducing cytokines at the site of inflammation (e.g., IL-1β, TNFα) [220,221,222,223,224].
Other Bacterial Toxins
Clostridium difficile toxin A and B can also strongly disrupt the epithelial barrier independent of cellular damage, mediated by the rearrangement of the actin cytoskeleton and the disintegration of TJs [225], as well as by the upregulation of intestinal VEGF-A production that increases endothelial permeability [226]. Clostridium botulinum toxin C2 is also an extremely potent inducer of endothelial hyperpermeability [227] acting via G-actin capping and causing consequent actin depolymerization [228]. Pertussis toxin from Bordetella pertussis also increases permeability in low concentrations without modifying actin cytoskeleton or junctional molecules; however, in higher concentrations, it protects barrier function by stimulating cAMP production [229]. Cholera toxin from Vibrio cholerae, on the other hand, is known to decrease endothelial permeability by the stimulation of cAMP production, which leads to the inhibition of MLC phosphorylation and acto-myosin contraction and thereby enhances the endothelial barrier and counteracts the effects of barrier destabilizing agents [230, 231]. Another toxin produced by Vibrio cholerae is zonula occludens toxin (Zot), which mimics the effects of an endogenous protein, zonulin (pre-haptoglobin 2, a serine-protease homolog). Both Zot and zonulin contain a PAR2 activating motif and increase permeability through PAR2 activated, PKCα-dependent phosphorylation and displacement of TJ component ZO-1, as well as cytoskeletal rearrangement [232, 233]. Although Zot and zonulin were thought to be involved in the regulation of gastrointestinal tract epithelial cell permeability, similar effects were shown in a lung model, involving both epithelial cells and ECs [234].
Although there is no compelling evidence whether microbial compounds indeed directly trigger angioedema attacks, we previously described a decreased frequency of abdominal attacks in C1-INH-HAE patients following eradication of the stomach mucosal-resident pathogen Helicobacter pylori [235], which suggests that bacterial macromolecules may indeed be permeability modifying trigger-factors either directly, or via the activation of immune and/or neural system.
Current Controversies in BK-Mediated Angioedema
During the more than 130-year-long period that has elapsed since hereditary angioedema was first described, many details of its pathomechanism have been elucidated. However, the “pieces of the puzzle” have not yet come together. Many questions and contradictions remain unanswered, and there are phenomena that appear controversial due to the lack of sufficient information.
The Mystery of Angioedema Localization
Why do angioedema attacks manifest only in certain body regions, whereas others are spared? Upper airway edema involves the mucosa of the mesopharynx, the larynx and the hypopharynx. Why does it spare the nasal or the bronchial mucosa? Why are the skin and the gastrointestinal tract frequently affected, but others like the lungs, heart, liver or kidneys are not? These questions puzzle many experts in the angioedema field and we possess some plausible answers. Hofman et al. speculated how a systemic activation due to lack of inhibitory effect of C1-INH leads to local manifestations [236]. We would like to add some novel hints to complement the aforementioned explanation.
ECs show significant functional and morphological heterogeneity [237], which causes differences in basal and inducible permeability state across the vascular system (Fig. 3). As an example, expression pattern of junctional molecules claudin-5, VE-cadherin, occludin, ZO-1, and JAM-C is different in human dermal microvascular EC (HDMEC), lung MEC, HUVEC, and aortic EC [238]. Although the resting pattern of leukocyte homing receptors (ICAM-1, VCAM-1, and E-selectin) is similar in HUVECs and human intestinal MECs, the kinetics of induction as well as inducibility by pro-inflammatory factors are fairly different [239]. Another example is the case of four histamine receptors, which are present on HUVECs, HDMECs, and human cerebral MECs, but their distribution is distinct, and induced trans-endothelial resistance showed different kinetics and recovery time [125]. It appears that although PAR1,2 and 4 are widely expressed in the human body, their distribution in ECs from various anatomical sites differs significantly. PAR1 is usually the most abundant, followed by PAR2 then PAR4, however, the pattern of their relative expression is different amongst pulmonary, dermal, and umbilical cord ECs [240,241,242,243,244]. Also, iNOS inhibitors increase binding of leukocytes to cytokine- and LPS-activated human intestinal MECs but not to similarly activated HUVECs [245]. The differences in induction of permeability are also observed at the functional level. Basal albumin leak was found to be significantly lower in human pulmonary MECs (HPMEC) than in HUVECs, but septicemia increases the leak across both types. Moreover, the presence of neutrophils enhances sepsis-increased albumin leak in HPMEC but not in HUVECs [246].

Characteristics of endothelial permeability regulation in different tissues. The brain, skin and liver are shown as examples. The green area indicates normal activity, whereas the red area indicates pathological intensity of paracellular transport. Gray areas on the dials indicate zone of permeability that is rarely reached even in pathological conditions (never or very few times during a lifetime). Note that skin microvasculature becomes frequently hyperpermeable (indicated by red line without grey area) during a normal lifespan (e.g., in response to minor traumas, mosquito bites, allergic reactions, etc.)
Taken together, the very same permeability-increasing stimuli may provoke significantly different response in various parts of the vascular tree, which adds an “EC perspective” to the enigma of edema localization in angioedema.
The Secret of Unpredictable Occurrence and Severity of Angioedema Attacks
Why do HAE patients not experience continuous attacks, while their C1-INH level is permanently low? Why there is no correlation between C1-INH level and severity of the disease? Why does mechanical trauma (as a triggering factor) provoke an attack on one occasion but not on another? These questions may indicate that BK-mediated angioedema comprises a heterogeneous group of multifactorial diseases, even if their pathomechanism can be linked to mutations of a single gene (SERPING1, FXII, ANGPT1, etc.) or to an extrinsic factor (e.g., ACE- and DPP-IV-inhibitors).
Trigger-factors, acting either alone or in combination, are also able to modify vascular permeability. Therefore, together with the preexisting genetic and environmental background, they contribute to defining the frequency, severity, and localization of the attacks. Figure 4 summarizes the permeability-increasing effects of trigger-factors identified or suspected in C1-INH-HAE [247].

Proposed mechanisms of common trigger-factors of angioedema attacks. The figure shows the most important mechanisms by which trigger factors of hereditary angioedema exert their vascular permeability effects on endothelial cells
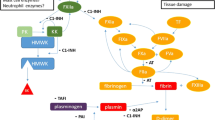
Even minor trauma has multiple effects on permeability. Activation of coagulation and fibrinolytic pathways forms thrombin and other PAR agonist enzymes from their zymogens, which have direct effects on ECs. Negatively charged surfaces activate the KKS and generate BK, but activated enzymes of KKS concurrently act directly on ECs (via PARs) and stimulate sensory neurons, which, in turn, enhance permeability by the secretion of SP and other neurotransmitters [119]. Finally, even “sterile” trauma may induce local inflammation, which involves the complement system and leukocytes, thereby increasing permeability through MASP-1, anaphylatoxins, and pro-inflammatory cytokines. Pathomechanism of non-sterile trauma also involves microbial compounds with permeability-enhancing potentials.
Physical exertion was also shown to induce pro-inflammatory cytokines including those that increase permeability (IL-1, TNFα, IFNγ) [248], and to cause micro-injuries to the skeletal muscles (sterile trauma). Moreover, local acidification of the muscles by accumulated lactic acid increases permeability through the pH-sensing GPR4 [212].
Menstruation is one of the most frequent and reliably identified trigger-factors of angioedema attacks in females. However, its effect on permeability is very complex. Besides being a sort of moderate trauma, steroid hormone pattern changes dramatically during menstruation. The first peak of estrogen precedes the beginning of menstruation by 1–2 days, which coincides with the long-onset down-regulation of claudin-5 in endometrial ECs [193]. VEGF, PK, and FXII inducing capability of estrogens may explain the remote permeability-increasing effects. Finally, menstruation may evidently cause mental stress in a great proportion of individuals (see below).
Pregnancy may alleviate or aggravate the angioedema attacks. Interestingly, C1-INH-HAE patients being pregnant with fetus afflicted by C1-INH-HAE register significantly more attacks than those with healthy fetus [249]. Since C1-INH is supposed to be unable to cross fetal-maternal interface, other, not yet identified trigger-factors should disturb the maternal P/B state in these cases.
Weather changes comprise a drift in air-pressure, temperature, and/or atmospheric electrical charges. Although their exact mechanism on human physiology is still the objects of extended studies, alteration in electrical charges is long known to interfere with the circadian-rhythm by the release of serotonin [250], or by the changes of serotonin responsiveness [251]. Since serotonin can modulate permeability [252], weather changes may directly trigger angioedema attacks by serotonin release.
Impact of mental stress on angioedema formation is still largely unexplored. Even if solid evidence is missing, there are some putative explanations. Mental stress causes hemoconcentration in otherwise healthy people, which entail subclinical systemic increase in endothelial permeability. In patients with angioedema, at some predisposed locations, together with elevated BK level or decreased activity of ANG-1, mental stress may trigger angioedema attacks. Although noradrenaline was shown to be elevated in a tilt-induced stress model of C1-INH-HAE patients [23], the pathogenetic role of noradrenaline in increasing permeability is controversial [132,133,134]. Glucocorticoids are also increased during stress; however, their effects are questionable similarly to catecholamines, as glucocorticoids decrease permeability directly [197] as well as indirectly by reducing pro-inflammatory cytokine production of T cells and other leukocytes. Nevertheless, IL-1β and TNFα levels are significantly elevated during stress [253], which may contribute to the increased permeability, but the exact mechanism leading to the elevation of these pro-inflammatory cytokines in the presence of the high cortisol conditions is still not clear. A more promising explanation is stress-induced MC activation [254], in which numerous compounds that increase endothelial permeability are released, including histamine, serotonin, lipid mediators, cytokines, growth factors, and proteases [255].
Infection can induce angioedema by several related mechanisms. Microbial compounds themselves may trigger the loss of barrier function, and they also modulate permeability via activating the complement cascade, leukocytes, and cells of the infected tissue locally, which results the release of pro-inflammatory cytokines, purinergic mediators, and PAR agonist enzymes.
Certain drugs hindering BK catabolism, such as ACE inhibitors and DPP-IV inhibitors, can cause angioedema that is usually regarded as an acquired form. These drugs may also be potential trigger-factors in HAE.
The Riddle of Response to Therapy
Why does the administration of C1-INH concentrate prove effective in BK-mediated angioedema despite normal plasma levels of C1-INH? In contrast, why do patients with BK-mediated angioedema have any remaining active C1-INH before or during angioedema attacks? Why is the concentration of C1-INH complexes with plasma enzymes similar in healthy people and in patients with angioedema [256, 257]?
C1-INH concentrate can effectively improve or terminate attacks even in patients with normal C1-INH levels, such as in ACEI-AAE patients [38], which underlines the permeability signal integrating role of ECs. It can be hypothesized that in patients with BK-mediated angioedema, excess of active serine proteases cause a low and constant, but still controllable permeability stimulation to ECs; however, edema formation requires an additional trigger-factor. We and others demonstrated that circulating C1-INH is complexed with several plasma serine proteases both in healthy individuals and C1-INH-HAE patients [256, 257], which supports the hypothesis that a small fraction of these enzymes is activated spontaneously and continuously (tick-over mechanism). Administration of C1-INH inhibits serine proteases, lowers the basal permeability, and thus prevents the effect of the trigger-factor to reach the threshold for edema formation.
Another paradox is that C1-INH is not completely depleted during attacks. This can be resolved by the fact that angioedema does not depend on a single serine protease. The extra permeability-increasing effect of the trigger-factors, with or without minor elevation in PKa activity [256] is sufficient to initiate an attack when ECs are pre-sensitized by BK, PKa, or other serine proteases. The P/B state of ECs relies on a fine-tuned delicate balance. The above observations indicate that the P/B state can be impaired not only by a drastic alteration in one particular compound, but by tiny changes in many factors impacting the ECs.
Conclusions and Future Perspectives
ECs have a great variety of physiological functions. At the level of microcirculation, one of their most important functions is the regulation of a proper P/B state. ECs act as signal integrators with a vast repertoire of cell surface receptors, adhesion molecules and a highly developed cytoskeleton system, by which they respond to the requirements of their microenvironment. ECs also have several built-in control mechanisms to shut down exaggerated modifications of the P/B state. One of the most well-studied negative feed-back mechanisms is receptor desensitization, achieved either by irreversible impairment of the receptor (PARs) or by endocytosis (e.g., BK receptors, histamine receptors). Thus, in the majority of cases, including angioedema, multiple impacts on ECs are needed to form a sustained hyperpermeability state leading to edema. This would explain the great number of controversial findings about BK-mediated angioedemas described in the previous chapter.
Taken together, we attempt to outline how angioedema attacks evolve (Fig. 5). First, the genetic background determines how susceptible the ECs are to different permeability inducing agents in general. This is modulated by the person’s lifestyle and other environmental effects, which may explain why there is such big a difference in attack frequency, severity, and localization even in the same type of angioedema. Secondly, a strong permeability increasing impact is required, which elevates either the permeability or the inducibility of ECs in response to permeability increasing agents. A mutation in any of the permeability-related genes (e.g., SERPING1, FXII, ANGPT1) or a permeability modifying drug (i.e., ACE or DPP-IV inhibitors) can contribute to such instability. Such an effect may differ amongst organs because of their diverse endothelial lining. The third component is the actual trigger-factor that pushes the permeability over the limit of controllable range.

Role of endothelial signal integration in the permeability response. Predisposing mutations and genetic background together with ever-changing factors (i.e., drugs, lifestyle, and trigger-factors) act on endothelial cells, which integrate these signals. The output of this signal integration (i.e., controlled hyperpermeability or uncontrolled edema) could only be estimated if each of these signals were fully known
The edema resolves without medical intervention, when (1) the sum concentration of the permeability increasing factors drops below a certain level, (2) the sum concentration of the barrier supporting factors elevates as a built-in barrier protecting mechanism, and (3) the receptors of permeability increasing factors are desensitized at protein or transcriptional level, or because of the natural combination of the previous mechanisms.
To understand the complex pathomechanism of angioedema, research strategy should be directed toward simultaneous investigation of several stimuli that act on ECs. Also, utilization of genome-, transcriptome-, proteome-, and metabolome-based systems-biological approaches would enable to study the simultaneous, multiple effects on ECs. International collaborations resulting in high-number, standardized, good quality samples (bio-banking supported with fully registered clinical data) would allow us to recognize weak-interactions and network effects. Improved analytics would be also required to measure crucial factors (e.g., BK itself) in a quantitative, reproducible, and affordable manner. Interstitial fluid samples taken from the very site of edema would help comprehend the local driving forces of endothelial based, BK-mediated angioedema. These methodological advancements in angioedema research together will be necessary to understand and reverse the deleterious effects of permeability-increasing factors, the molecular dambusters.
Abbreviations
- AA:
-
Arachidonic acid
- ACE:
-
Angiotensin-converting enzyme
- ACEI:
-
Angiotensin-converting enzyme inhibitor
- ADM:
-
Adrenomedullin
- AJ:
-
Adherent junction
- AMPK:
-
AMP activated protein kinase
- ANGs:
-
Angiopoietins
- AVP:
-
Arginine-vasopressin
- BAEC:
-
Bovine aortic endothelial cell
- BBB:
-
Blood–brain barrier
- bFGF:
-
Basic fibroblast growth factor
- BK:
-
Bradykinin
- BKRB1:
-
Bradykinin receptor B1
- BKRB2:
-
Bradykinin receptor B2
- C1-INH:
-
C1-inhibitor
- cAMP:
-
Cyclic adenosine monophosphate
- CGRP:
-
Calcitonin gene-related-peptide
- CLS:
-
Capillary leak syndrome
- CNS:
-
Central nervous system
- CO:
-
Carbon monoxide
- DPP-IV:
-
Dipeptidyl peptidase 4
- DS:
-
Desmosome
- ECs:
-
Endothelial cells
- EGF:
-
Epidermal growth factor
- eNOS:
-
Endothelial nitric oxide synthase
- ET-1:
-
Endothelin-1
- GF:
-
Growth factor
- GPCR:
-
G-protein coupled receptor
- H2O2 :
-
Hydrogen peroxide
- HAE:
-
Hereditary angioedema
- HAEC:
-
Human aortic endothelial cell
- HC:
-
Hydrocortisone
- HD:
-
Hemidesmosome
- HDL:
-
High-density lipoprotein
- HDMEC:
-
Human dermal microvascular endothelial cell
- HETE:
-
Hydroxyeicosatetraenoic acid
- HK:
-
High molecular weight kininogen
- HPMEC:
-
Human pulmonary microvascular endothelial cell
- H/R:
-
Hypoxia/reoxygenation
- HUVEC:
-
Human umbilical vein endothelial cell
- HVP:
-
High vascular permeability
- IFNγ:
-
Interferon γ
- iNOS:
-
Inducible nitric oxide synthase
- IL:
-
Interleukin
- IVP:
-
Increased vascular permeability
- JAM:
-
Junctional adhesion molecule
- KKS:
-
Kallikrein kinin system
- KLK-1:
-
Tissue kallikrein 1
- LPS:
-
Lipopolysaccharide
- MASP-1:
-
Mannan-binding lectin-associated serine protease 1
- MASP-2:
-
Mannan-binding lectin-associated serine protease 2
- MC:
-
Mast cell
- MLC:
-
Myosin light chain
- MLCK:
-
Myosin light chain kinase
- MMP:
-
Matrix metalloprotease
- MSC:
-
Mesenchymal stem cell
- NK:
-
Neurokinin
- NO:
-
Nitric oxide
- NP:
-
Natriuretic peptide
- PACAP:
-
Pituitary adenylate cyclase-activating polypeptide
- PAR:
-
Protease activated receptor
- P/B state:
-
Permeability/barrier state
- PAF:
-
Platelet-activating factor
- PGI2:
-
Prostacyclin
- PK:
-
Prekallikrein
- PKa:
-
Plasma kallikrein
- PKB:
-
Protein kinase B
- PKC:
-
Protein kinase C
- PPD:
-
Paroxysmal permeability disorder
- RBC:
-
Red blood cell
- ROS:
-
Reactive oxygen species
- SP:
-
Substance P
- S1P:
-
Sphingosine-1-phosphate
- TGFβ:
-
Transforming growth factor β
- TJ:
-
Tight junction
- TNFα:
-
Tumor necrosis factor α
- TRP:
-
Transient receptor potential
- TXA2:
-
Thromboxane A2
- VEGF:
-
Vascular growth factor
- VIP:
-
Vasoactive intestinal peptide
- ZO:
-
Zonula occludens
- Zot:
-
Zonula occludens toxin
- 5-HT:
-
Serotonin/5-hydroxytryptamine
References
Stokum JA, Gerzanich V, Simard JM (2016) Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab 36(3):513–538. https://doi.org/10.1177/0271678X15617172
Ballet F (1990) Hepatic circulation: potential for therapeutic intervention. Pharmacol Ther 47(2):281–328. https://doi.org/10.1016/0163-7258(90)90091-f
Scallan J, Huxley VH, Korthuis RJ (2010). In: Capillary fluid exchange: Regulation, functions, and pathology. Integrated systems physiology: from molecule to function to disease. San Rafael (CA)
McDonagh PF (1993) The microvascular pathophysiology of chronic venous insufficiency. Yale J Biol Med 66(1):27–36
Siddall E, Khatri M, Radhakrishnan J (2017) Capillary leak syndrome: etiologies, pathophysiology, and management. Kidney Int 92(1):37–46. https://doi.org/10.1016/j.kint.2016.11.029
Wu MA, Tsvirkun D, Bureau L, Boccon-Gibod I, Inglebert M, Duperray A, Bouillet L, Misbah C, Cicardi M (2019) Paroxysmal permeability disorders: development of a microfluidic device to assess endothelial barrier function. Front Med (Lausanne) 6:89. https://doi.org/10.3389/fmed.2019.00089
Wu MA, Bova M, Berra S, Senter R, Parolin D, Caccia S, Cicardi M (2020) The central role of endothelium in hereditary angioedema due to C1 inhibitor deficiency. Int Immunopharmacol 82:106304. https://doi.org/10.1016/j.intimp.2020.106304
Busse PJ, Smith T (2017) Histaminergic angioedema. Immunol Allergy Clin North Am 37(3):467–481. https://doi.org/10.1016/j.iac.2017.03.001
Nussberger J, Cugno M, Cicardi M, Agostoni A (1999) Local bradykinin generation in hereditary angioedema. J Allergy Clin Immunol 104(6):1321–1322. https://doi.org/10.1016/s0091-6749(99)70030-8
Zeerleder S, Levi M (2016) Hereditary and acquired C1-inhibitor-dependent angioedema: from pathophysiology to treatment. Ann Med 48(4):256–267. https://doi.org/10.3109/07853890.2016.1162909
Byrd JB, Adam A, Brown NJ (2006) Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 26(4):725–737. https://doi.org/10.1016/j.iac.2006.08.001
Montinaro V, Cicardi M (2020) ACE inhibitor-mediated angioedema. Int Immunopharmacol 78:106081. https://doi.org/10.1016/j.intimp.2019.106081
Oschatz C, Maas C, Lecher B, Jansen T, Bjorkqvist J, Tradler T, Sedlmeier R, Burfeind P, Cichon S, Hammerschmidt S, Muller-Esterl W, Wuillemin WA, Nilsson G, Renne T (2011) Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 34(2):258–268. https://doi.org/10.1016/j.immuni.2011.02.008
Meini S, Zanichelli A, Sbrojavacca R, Iuri F, Roberts AT, Suffritti C, Tascini C (2020) Understanding the pathophysiology of COVID-19: could the contact system be the key? Front Immunol 11:2014. https://doi.org/10.3389/fimmu.2020.02014
Csuka D, Veszeli N, Varga L, Prohaszka Z, Farkas H (2017) The role of the complement system in hereditary angioedema. Mol Immunol 89:59–68. https://doi.org/10.1016/j.molimm.2017.05.020
van Geffen M, Cugno M, Lap P, Loof A, Cicardi M, van Heerde W (2012) Alterations of coagulation and fibrinolysis in patients with angioedema due to C1-inhibitor deficiency. Clin Exp Immunol 167(3):472–478. https://doi.org/10.1111/j.1365-2249.2011.04541.x
Kajdacsi E, Jani PK, Csuka D, Varga LA, Prohaszka Z, Farkas H, Cervenak L (2014) Endothelial cell activation during edematous attacks of hereditary angioedema types I and II. J Allergy Clin Immunol 133(6):1686–1691. https://doi.org/10.1016/j.jaci.2013.12.1072
Bouillet L, Vilgrain I (2014) VE-cadherin, a potential marker for endothelial cell activation during hereditary angioedema attacks. J Allergy Clin Immunol 134(1):241. https://doi.org/10.1016/j.jaci.2014.04.016
Kempe S, Fois G, Brunner C, Hoffmann TK, Hahn J, Greve J (2020) Bradykinin signaling regulates solute permeability and cellular junction organization in lymphatic endothelial cells. Microcirculation 27(2):e12592. https://doi.org/10.1111/micc.12592
Veszeli N, Csuka D, Zotter Z, Imreh E, Jozsi M, Benedek S, Varga L, Farkas H (2015) Neutrophil activation during attacks in patients with hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis 10:156. https://doi.org/10.1186/s13023-015-0374-y
Bafunno V, Firinu D, D’Apolito M, Cordisco G, Loffredo S, Leccese A, Bova M, Barca MP, Santacroce R, Cicardi M, Del Giacco S, Margaglione M (2018) Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol 141(3):1009–1017. https://doi.org/10.1016/j.jaci.2017.05.020
Gompel A, Fain O, Boccon-Gibod I, Gobert D, Bouillet L (2020) Exogenous hormones and hereditary angioedema. Int Immunopharmacol 78:106080. https://doi.org/10.1016/j.intimp.2019.106080
Wu MA, Casella F, Perego F, Suffritti C, Afifi Afifi N, Tobaldini E, Zanichelli A, Cogliati C, Montano N, Cicardi M (2017) Hereditary angioedema: assessing the hypothesis for underlying autonomic dysfunction. PLoS One 12(11):e0187110. https://doi.org/10.1371/journal.pone.0187110
Germenis AE, Speletas M (2016) Genetics of hereditary angioedema revisited. Clin Rev Allergy Immunol 51(2):170–182. https://doi.org/10.1007/s12016-016-8543-x
Barry AK, Wang N, Leckband DE (2015) Local VE-cadherin mechanotransduction triggers long-ranged remodeling of endothelial monolayers. J Cell Sci 128(7):1341–1351. https://doi.org/10.1242/jcs.159954
Bazzoni G, Dejana E (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84(3):869–901. https://doi.org/10.1152/physrev.00035.2003
Alphonsus CS, Rodseth RN (2014) The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia 69(7):777–784. https://doi.org/10.1111/anae.12661
Sukriti S, Tauseef M, Yazbeck P, Mehta D (2014) Mechanisms regulating endothelial permeability Pulm Circ 4(4):535–551. https://doi.org/10.1086/677356
Roberts OL, Dart C (2014) cAMP signalling in the vasculature: the role of Epac (exchange protein directly activated by cAMP). Biochem Soc Trans 42(1):89–97. https://doi.org/10.1042/BST20130253
Schlegel N, Waschke J (2014) cAMP with other signaling cues converges on Rac1 to stabilize the endothelial barrier- a signaling pathway compromised in inflammation. Cell Tissue Res 355(3):587–596. https://doi.org/10.1007/s00441-013-1755-y
Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE 3rd (2002) Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest 109(8):1057–1063. https://doi.org/10.1172/JCI14211
Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A (1998) Plasma bradykinin in angio-oedema. Lancet 351(9117):1693–1697. https://doi.org/10.1016/S0140-6736(97)09137-X
Kaplan AP, Joseph K (2017) Pathogenesis of hereditary angioedema: the role of the bradykinin-forming cascade. Immunol Allergy Clin North Am 37(3):513–525. https://doi.org/10.1016/j.iac.2017.04.001
Dobo J, Major B, Kekesi KA, Szabo I, Megyeri M, Hajela K, Juhasz G, Zavodszky P, Gal P (2011) Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS One 6(5):e20036. https://doi.org/10.1371/journal.pone.0020036
Jean M, Raghavan A, Charles ML, Robbins MS, Wagner E, Rivard GE, Charest-Morin X, Marceau F (2016) The isolated human umbilical vein as a bioassay for kinin-generating proteases: An in vitro model for therapeutic angioedema agents. Life Sci 155:180–188. https://doi.org/10.1016/j.lfs.2016.05.010
Prado GN, Taylor L, Zhou X, Ricupero D, Mierke DF, Polgar P (2002) Mechanisms regulating the expression, self-maintenance, and signaling-function of the bradykinin B2 and B1 receptors. J Cell Physiol 193(3):275–286. https://doi.org/10.1002/jcp.10175
Gambardella J, Sorriento D, Bova M, Rusciano M, Loffredo S, Wang X, Petraroli A, Carucci L, Mormile I, Oliveti M, Bruno Morelli M, Fiordelisi A, Spadaro G, Campiglia P, Sala M, Trimarco B, Iaccarino G, Santulli G, Ciccarelli M (2020) Role of endothelial G protein-coupled receptor kinase 2 in angioedema. Hypertension 76(5):1625–1636. https://doi.org/10.1161/HYPERTENSIONAHA.120.15130
Greve J, Bas M, Hoffmann TK, Schuler PJ, Weller P, Kojda G, Strassen U (2015) Effect of C1-Esterase-inhibitor in angiotensin-converting enzyme inhibitor-induced angioedema. Laryngoscope 125(6):E198-202. https://doi.org/10.1002/lary.25113
van de Veerdonk FL, Netea MG, van Deuren M, van der Meer JW, de Mast Q, Bruggemann RJ, van der Hoeven H (2020) Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome Elife 9. https://doi.org/10.7554/eLife.57555
Scott SI, Andersen MF, Aagaard L, Buchwald CV, Rasmussen ER (2018) Dipeptidyl peptidase-4 inhibitor induced angioedema - an overlooked adverse drug reaction? Curr Diabetes Rev 14(4):327–333. https://doi.org/10.2174/1573399813666170214113856
Uhlig S, Yang Y, Waade J, Wittenberg C, Babendreyer A, Kuebler WM (2014) Differential regulation of lung endothelial permeability in vitro and in situ. Cell Physiol Biochem 34(1):1–19. https://doi.org/10.1159/000362980
Walker K, Perkins M, Dray A (1995) Kinins and kinin receptors in the nervous system. Neurochem Int 26 (1):1–16; discussion 17–26. https://doi.org/10.1016/0197-0186(94)00114-a
Brain SD, Williams TJ (1985) Inflammatory oedema induced by synergism between calcitonin gene-related peptide (CGRP) and mediators of increased vascular permeability. Br J Pharmacol 86(4):855–860. https://doi.org/10.1111/j.1476-5381.1985.tb11107.x
Baluk P (1997) Neurogenic inflammation in skin and airways. J Investig Dermatol Symp Proc 2(1):76–81. https://doi.org/10.1038/jidsymp.1997.15
Fleegal-DeMotta MA, Doghu S, Banks WA (2009) Angiotensin II modulates BBB permeability via activation of the AT(1) receptor in brain endothelial cells. J Cereb Blood Flow Metab 29(3):640–647. https://doi.org/10.1038/jcbfm.2008.158
Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T (2010) Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience 171(3):852–858. https://doi.org/10.1016/j.neuroscience.2010.09.029
Victorino GP, Newton CR, Curran B (2002) Effect of angiotensin II on microvascular permeability. J Surg Res 104(2):77–81. https://doi.org/10.1006/jsre.2002.6412
Pupilli C, Lasagni L, Romagnani P, Bellini F, Mannelli M, Misciglia N, Mavilia C, Vellei U, Villari D, Serio M (1999) Angiotensin II stimulates the synthesis and secretion of vascular permeability factor/vascular endothelial growth factor in human mesangial cells. J Am Soc Nephrol 10(2):245–255
Williams B, Baker AQ, Gallacher B, Lodwick D (1995) Angiotensin II increases vascular permeability factor gene expression by human vascular smooth muscle cells. Hypertension 25(5):913–917. https://doi.org/10.1161/01.hyp.25.5.913
Victorino GP, Newton CR, Curran B (2004) Endothelin-1 decreases microvessel permeability after endothelial activation. J Trauma 56(4):832–836. https://doi.org/10.1097/01.ta.0000057228.45839.3c
Sirois MG, Filep JG, Rousseau A, Fournier A, Plante GE, Sirois P (1992) Endothelin-1 enhances vascular permeability in conscious rats: role of thromboxane A2. Eur J Pharmacol 214(2–3):119–125. https://doi.org/10.1016/0014-2999(92)90108-g
Eibl G, Hotz HG, Faulhaber J, Kirchengast M, Buhr HJ, Foitzik T (2000) Effect of endothelin and endothelin receptor blockade on capillary permeability in experimental pancreatitis. Gut 46(3):390–394. https://doi.org/10.1136/gut.46.3.390
Faraci FM (1989) Effects of endothelin and vasopressin on cerebral blood vessels. Am J Physiol 257(3 Pt 2):H799-803. https://doi.org/10.1152/ajpheart.1989.257.3.H799
Eto T, Kato J, Kitamura K (2003) Regulation of production and secretion of adrenomedullin in the cardiovascular system. Regul Pept 112(1–3):61–69. https://doi.org/10.1016/s0167-0115(03)00023-5
Kajdacsi E, Jani PK, Csuka D, Varga L, Prohaszka Z, Farkas H, Cervenak L (2016) Novel vasoregulatory aspects of hereditary angioedema: the role of arginine vasopressin, adrenomedullin and endothelin-1. J Clin Immunol 36(2):160–170. https://doi.org/10.1007/s10875-016-0239-8
Adamski SW, Svensjo E, Grega GJ (1985) Effects of AVP and DDAVP on histamine-induced increases in macromolecular permeability in the hamster cheek pouch. Microcirc Endothelium Lymphatics 2(1):41–53
Hempel A, Noll T, Bach C, Piper HM, Willenbrock R, Hohnel K, Haller H, Luft FC (1998) Atrial natriuretic peptide clearance receptor participates in modulating endothelial permeability. Am J Physiol 275(5):H1818-1825. https://doi.org/10.1152/ajpheart.1998.275.5.H1818
Lofton CE, Newman WH, Currie MG (1990) Atrial natriuretic peptide regulation of endothelial permeability is mediated by cGMP. Biochem Biophys Res Commun 172(2):793–799. https://doi.org/10.1016/0006-291x(90)90744-8
Kiemer AK, Weber NC, Furst R, Bildner N, Kulhanek-Heinze S, Vollmar AM (2002) Inhibition of p38 MAPK activation via induction of MKP-1: atrial natriuretic peptide reduces TNF-alpha-induced actin polymerization and endothelial permeability. Circ Res 90(8):874–881. https://doi.org/10.1161/01.res.0000017068.58856.f3
Lofton CE, Baron DA, Heffner JE, Currie MG, Newman WH (1991) Atrial natriuretic peptide inhibits oxidant-induced increases in endothelial permeability. J Mol Cell Cardiol 23(8):919–927. https://doi.org/10.1016/0022-2828(91)90134-8
Lara-Castillo N, Zandi S, Nakao S, Ito Y, Noda K, She H, Ahmed M, Frimmel S, Ablonczy Z, Hafezi-Moghadam A (2009) Atrial natriuretic peptide reduces vascular leakage and choroidal neovascularization. Am J Pathol 175(6):2343–2350. https://doi.org/10.2353/ajpath.2009.090439
Furst R, Bubik MF, Bihari P, Mayer BA, Khandoga AG, Hoffmann F, Rehberg M, Krombach F, Zahler S, Vollmar AM (2008) Atrial natriuretic peptide protects against histamine-induced endothelial barrier dysfunction in vivo. Mol Pharmacol 74(1):1–8. https://doi.org/10.1124/mol.108.045773
Klinger JR, Warburton R, Carino GP, Murray J, Murphy C, Napier M, Harrington EO (2006) Natriuretic peptides differentially attenuate thrombin-induced barrier dysfunction in pulmonary microvascular endothelial cells. Exp Cell Res 312(4):401–410. https://doi.org/10.1016/j.yexcr.2005.11.001
Bohara M, Kambe Y, Nagayama T, Tokimura H, Arita K, Miyata A (2014) C-type natriuretic peptide modulates permeability of the blood-brain barrier. J Cereb Blood Flow Metab 34(4):589–596. https://doi.org/10.1038/jcbfm.2013.234
Tian X, Tian Y, Gawlak G, Sarich N, Wu T, Birukova AA (2014) Control of vascular permeability by atrial natriuretic peptide via a GEF-H1-dependent mechanism. J Biol Chem 289(8):5168–5183. https://doi.org/10.1074/jbc.M113.493924
Kajdacsi E, Varga L, Prohaszka Z, Farkas H, Cervenak L (2016) Atrial natriuretic peptide as a novel biomarker of hereditary angioedema. Clin Immunol 165:45–46. https://doi.org/10.1016/j.clim.2016.03.007
Dunworth WP, Fritz-Six KL, Caron KM (2008) Adrenomedullin stabilizes the lymphatic endothelial barrier in vitro and in vivo. Peptides 29(12):2243–2249. https://doi.org/10.1016/j.peptides.2008.09.009
Kis B, Deli MA, Kobayashi H, Abraham CS, Yanagita T, Kaiya H, Isse T, Nishi R, Gotoh S, Kangawa K, Wada A, Greenwood J, Niwa M, Yamashita H, Ueta Y (2001) Adrenomedullin regulates blood-brain barrier functions in vitro. NeuroReport 12(18):4139–4142. https://doi.org/10.1097/00001756-200112210-00055
Temmesfeld-Wollbruck B, Brell B, David I, Dorenberg M, Adolphs J, Schmeck B, Suttorp N, Hippenstiel S (2007) Adrenomedullin reduces vascular hyperpermeability and improves survival in rat septic shock. Intensive Care Med 33(4):703–710. https://doi.org/10.1007/s00134-007-0561-y
Garcia Ponce A, Citalan Madrid AF, Vargas Robles H, Chanez Paredes S, Nava P, Betanzos A, Zarbock A, Rottner K, Vestweber D, Schnoor M (2016) Loss of cortactin causes endothelial barrier dysfunction via disturbed adrenomedullin secretion and actomyosin contractility. Sci Rep 6:29003. https://doi.org/10.1038/srep29003
Aslam M, Pfeil U, Gunduz D, Rafiq A, Kummer W, Piper HM, Noll T (2012) Intermedin (adrenomedullin2) stabilizes the endothelial barrier and antagonizes thrombin-induced barrier failure in endothelial cell monolayers. Br J Pharmacol 165(1):208–222. https://doi.org/10.1111/j.1476-5381.2011.01540.x
Inoue H, Nagata N, Koshihara Y (1993) Profile of capsaicin-induced mouse ear oedema as neurogenic inflammatory model: comparison with arachidonic acid-induced ear oedema. Br J Pharmacol 110(4):1614–1620. https://doi.org/10.1111/j.1476-5381.1993.tb14009.x
Holzer P (1998) Neurogenic vasodilatation and plasma leakage in the skin. Gen Pharmacol 30(1):5–11. https://doi.org/10.1016/s0306-3623(97)00078-5
Vena GA, Cassano N, Di Leo E, Calogiuri GF, Nettis E (2018) Focus on the role of substance P in chronic urticaria. Clin Mol Allergy 16:24. https://doi.org/10.1186/s12948-018-0101-z
Unemori EN, Erikson ME, Rocco SE, Sutherland KM, Parsell DA, Mak J, Grove BH (1999) Relaxin stimulates expression of vascular endothelial growth factor in normal human endometrial cells in vitro and is associated with menometrorrhagia in women. Hum Reprod 14(3):800–806. https://doi.org/10.1093/humrep/14.3.800
Kaczmarek MM, Blitek A, Kaminska K, Bodek G, Zygmunt M, Schams D, Ziecik AJ (2008) Assessment of VEGF-receptor system expression in the porcine endometrial stromal cells in response to insulin-like growth factor-I, relaxin, oxytocin and prostaglandin E2. Mol Cell Endocrinol 291(1–2):33–41. https://doi.org/10.1016/j.mce.2008.04.020
Gao XM, Su Y, Moore S, Han LP, Kiriazis H, Lu Q, Zhao WB, Ruze A, Fang BB, Duan MJ, Du XJ (2019) Relaxin mitigates microvascular damage and inflammation following cardiac ischemia-reperfusion. Basic Res Cardiol 114(4):30. https://doi.org/10.1007/s00395-019-0739-9
Hofman Z, de Maat S, Hack CE, Maas C (2016) Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol 51(2):152–161. https://doi.org/10.1007/s12016-016-8540-0
Kaplan AP, Maas C (2017) The search for biomarkers in hereditary angioedema. Front Med (Lausanne) 4:206. https://doi.org/10.3389/fmed.2017.00206
Zhao P, Metcalf M, Bunnett NW (2014) Biased signaling of protease-activated receptors. Front Endocrinol (Lausanne) 5:67. https://doi.org/10.3389/fendo.2014.00067
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120(3):303–313. https://doi.org/10.1016/j.cell.2004.12.018
Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W (2002) Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 296(5574):1880–1882. https://doi.org/10.1126/science.1071699
Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Blaber M, Blaber SI, Scarisbrick I, Diamandis EP, Hollenberg MD (2006) Kallikrein-mediated cell signalling: targeting proteinase-activated receptors (PARs). Biol Chem 387(6):817–824. https://doi.org/10.1515/BC.2006.104
Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, Hoxie JA, Schechter N, Woolkalis M, Brass LF (1997) Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem 272(7):4043–4049. https://doi.org/10.1074/jbc.272.7.4043
Stavenuiter F, Mosnier LO (2014) Noncanonical PAR3 activation by factor Xa identifies a novel pathway for Tie2 activation and stabilization of vascular integrity. Blood 124(23):3480–3489. https://doi.org/10.1182/blood-2014-06-582775
Thibeault PE, LeSarge JC, Arends D, Fernandes M, Chidiac P, Stathopulos PB, Luyt LG, Ramachandran R (2020) Molecular basis for activation and biased signaling at the thrombin-activated GPCR proteinase activated receptor-4 (PAR4). J Biol Chem 295(8):2520–2540. https://doi.org/10.1074/jbc.RA119.011461
Jani PK, Schwaner E, Kajdacsi E, Debreczeni ML, Ungai-Salanki R, Dobo J, Doleschall Z, Rigo J Jr, Geiszt M, Szabo B, Gal P, Cervenak L (2016) Complement MASP-1 enhances adhesion between endothelial cells and neutrophils by up-regulating E-selectin expression. Mol Immunol 75:38–47. https://doi.org/10.1016/j.molimm.2016.05.007
Altrogge LM, Monard D (2000) An assay for high-sensitivity detection of thrombin activity and determination of proteases activating or inactivating protease-activated receptors. Anal Biochem 277(1):33–45. https://doi.org/10.1006/abio.1999.4356
Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW 2nd, Malik AB (1986) Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol 128(1):96–104. https://doi.org/10.1002/jcp.1041280115
Vogel SM, Gao X, Mehta D, Ye RD, John TA, Andrade-Gordon P, Tiruppathi C, Malik AB (2000) Abrogation of thrombin-induced increase in pulmonary microvascular permeability in PAR-1 knockout mice. Physiol Genomics 4(2):137–145. https://doi.org/10.1152/physiolgenomics.2000.4.2.137
Troyanovsky B, Alvarez DF, King JA, Schaphorst KL (2008) Thrombin enhances the barrier function of rat microvascular endothelium in a PAR-1-dependent manner. Am J Physiol Lung Cell Mol Physiol 294(2):L266-275. https://doi.org/10.1152/ajplung.00107.2007
Rabiet MJ, Plantier JL, Rival Y, Genoux Y, Lampugnani MG, Dejana E (1996) Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol 16(3):488–496. https://doi.org/10.1161/01.atv.16.3.488
Heja D, Harmat V, Fodor K, Wilmanns M, Dobo J, Kekesi KA, Zavodszky P, Gal P, Pal G (2012) Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and -2 Are essential for lectin pathway activation and reveal structural plasticity of MASP-2. J Biol Chem 287(24):20290–20300. https://doi.org/10.1074/jbc.M112.354332
Megyeri M, Mako V, Beinrohr L, Doleschall Z, Prohaszka Z, Cervenak L, Zavodszky P, Gal P (2009) Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J Immunol 183(5):3409–3416. https://doi.org/10.4049/jimmunol.0900879
Jani PK, Kajdacsi E, Megyeri M, Dobo J, Doleschall Z, Futosi K, Timar CI, Mocsai A, Mako V, Gal P, Cervenak L (2014) MASP-1 induces a unique cytokine pattern in endothelial cells: a novel link between complement system and neutrophil granulocytes. PLoS One 9(1):e87104. https://doi.org/10.1371/journal.pone.0087104
Schwaner E, Nemeth Z, Jani PK, Kajdacsi E, Debreczeni ML, Doleschall Z, Dobo J, Gal P, Rigo J, Andras K, Hegedus T, Cervenak L (2017) Transcriptome analysis of inflammation-related gene expression in endothelial cells activated by complement MASP-1. Sci Rep 7(1):10462. https://doi.org/10.1038/s41598-017-09058-8
Debreczeni ML, Nemeth Z, Kajdacsi E, Schwaner E, Mako V, Masszi A, Doleschall Z, Rigo J, Walter FR, Deli MA, Pal G, Dobo J, Gal P, Cervenak L (2019) MASP-1 increases endothelial permeability. Front Immunol 10:991. https://doi.org/10.3389/fimmu.2019.00991
Debreczeni ML, Szekacs I, Kovacs B, Saftics A, Kurunczi S, Gal P, Dobo J, Cervenak L, Horvath R (2020) Human primary endothelial label-free biochip assay reveals unpredicted functions of plasma serine proteases. Sci Rep 10(1):3303. https://doi.org/10.1038/s41598-020-60158-4
Csuka D, Munthe-Fog L, Hein E, Zotter Z, Prohaszka Z, Farkas H, Varga L, Garred P (2014) Activation of the ficolin-lectin pathway during attacks of hereditary angioedema. J Allergy Clin Immunol 134 (6):1388–1393 e1381. https://doi.org/10.1016/j.jaci.2014.05.030
Abdallah RT, Keum JS, El-Shewy HM, Lee MH, Wang B, Gooz M, Luttrell DK, Luttrell LM, Jaffa AA (2010) Plasma kallikrein promotes epidermal growth factor receptor transactivation and signaling in vascular smooth muscle through direct activation of protease-activated receptors. J Biol Chem 285(45):35206–35215. https://doi.org/10.1074/jbc.M110.171769
Gobel K, Asaridou CM, Merker M, Eichler S, Herrmann AM, Geuss E, Ruck T, Schungel L, Groeneweg L, Narayanan V, Schneider-Hohendorf T, Gross CC, Wiendl H, Kehrel BE, Kleinschnitz C, Meuth SG (2019) Plasma kallikrein modulates immune cell trafficking during neuroinflammation via PAR2 and bradykinin release. Proc Natl Acad Sci U S A 116(1):271–276. https://doi.org/10.1073/pnas.1810020116
Bouillet L, Mannic T, Arboleas M, Subileau M, Massot C, Drouet C, Huber P, Vilgrain I (2011) Hereditary angioedema: key role for kallikrein and bradykinin in vascular endothelial-cadherin cleavage and edema formation. J Allergy Clin Immunol 128(1):232–234. https://doi.org/10.1016/j.jaci.2011.02.017
Maas C (2019) Plasminflammation-an emerging pathway to bradykinin production. Front Immunol 10:2046. https://doi.org/10.3389/fimmu.2019.02046
Marcos-Contreras OA, Martinez de Lizarrondo S, Bardou I, Orset C, Pruvost M, Anfray A, Frigout Y, Hommet Y, Lebouvier L, Montaner J, Vivien D, Gauberti M (2016) Hyperfibrinolysis increases blood-brain barrier permeability by a plasmin- and bradykinin-dependent mechanism. Blood 128(20):2423–2434. https://doi.org/10.1182/blood-2016-03-705384
Okajima K, Abe H, Binder BR (1995) Endothelial cell injury induced by plasmin in vitro. J Lab Clin Med 126(4):377–384
Niego B, Medcalf RL (2014) Plasmin-dependent modulation of the blood-brain barrier: a major consideration during tPA-induced thrombolysis? J Cereb Blood Flow Metab 34(8):1283–1296. https://doi.org/10.1038/jcbfm.2014.99
Diebel ME, Martin JV, Liberati DM, Diebel LN (2018) The temporal response and mechanism of action of tranexamic acid in endothelial glycocalyx degradation. J Trauma Acute Care Surg 84(1):75–80. https://doi.org/10.1097/TA.0000000000001726
Wang H, Ricklin D, Lambris JD (2017) Complement-activation fragment C4a mediates effector functions by binding as untethered agonist to protease-activated receptors 1 and 4. Proc Natl Acad Sci U S A 114(41):10948–10953. https://doi.org/10.1073/pnas.1707364114
Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB (2002) Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ Res 91(5):398–405. https://doi.org/10.1161/01.res.0000033520.95242.a2
McLaughlin JN, Shen L, Holinstat M, Brooks JD, Dibenedetto E, Hamm HE (2005) Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem 280(26):25048–25059. https://doi.org/10.1074/jbc.M414090200
Ellis CA, Malik AB, Gilchrist A, Hamm H, Sandoval R, Voyno-Yasenetskaya T, Tiruppathi C (1999) Thrombin induces proteinase-activated receptor-1 gene expression in endothelial cells via activation of Gi-linked Ras/mitogen-activated protein kinase pathway. J Biol Chem 274(19):13718–13727. https://doi.org/10.1074/jbc.274.19.13718
Gavard J, Gutkind JS (2008) Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Galpha12/13 and Galpha11/q. J Biol Chem 283(44):29888–29896. https://doi.org/10.1074/jbc.M803880200
Suzuki H, Motley ED, Eguchi K, Hinoki A, Shirai H, Watts V, Stemmle LN, Fields TA, Eguchi S (2009) Distinct roles of protease-activated receptors in signal transduction regulation of endothelial nitric oxide synthase. Hypertension 53(2):182–188. https://doi.org/10.1161/HYPERTENSIONAHA.108.125229
Klarenbach SW, Chipiuk A, Nelson RC, Hollenberg MD, Murray AG (2003) Differential actions of PAR2 and PAR1 in stimulating human endothelial cell exocytosis and permeability: the role of Rho-GTPases. Circ Res 92(3):272–278. https://doi.org/10.1161/01.res.0000057386.15390.a3
Kawabata A, Kuroda R, Nishikawa H, Asai T, Kataoka K, Taneda M (1999) Enhancement of vascular permeability by specific activation of protease-activated receptor-1 in rat hindpaw: a protective role of endogenous and exogenous nitric oxide. Br J Pharmacol 126(8):1856–1862. https://doi.org/10.1038/sj.bjp.0702513
Fujiwara M, Jin E, Ghazizadeh M, Kawanami O (2005) Activation of PAR4 induces a distinct actin fiber formation via p38 MAPK in human lung endothelial cells. J Histochem Cytochem 53(9):1121–1129. https://doi.org/10.1369/jhc.4A6592.2005
Vouret-Craviari V, Grall D, Van Obberghen-Schilling E (2003) Modulation of Rho GTPase activity in endothelial cells by selective proteinase-activated receptor (PAR) agonists. J Thromb Haemost 1(5):1103–1111. https://doi.org/10.1046/j.1538-7836.2003.00238.x
Vergnolle N, Hollenberg MD, Sharkey KA, Wallace JL (1999) Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR2)-activating peptides in the rat paw. Br J Pharmacol 127(5):1083–1090. https://doi.org/10.1038/sj.bjp.0702634
Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW (2000) Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med 6(2):151–158. https://doi.org/10.1038/72247
Bae JS, Yang L, Manithody C, Rezaie AR (2007) The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood 110(12):3909–3916. https://doi.org/10.1182/blood-2007-06-096651
Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D (2003) Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol 28(3):339–346. https://doi.org/10.1165/rcmb.4908
Loew D, Perrault C, Morales M, Moog S, Ravanat C, Schuhler S, Arcone R, Pietropaolo C, Cazenave JP, van Dorsselaer A, Lanza F (2000) Proteolysis of the exodomain of recombinant protease-activated receptors: prediction of receptor activation or inactivation by MALDI mass spectrometry. Biochemistry 39(35):10812–10822. https://doi.org/10.1021/bi0003341
Leach L, Eaton BM, Westcott ED, Firth JA (1995) Effect of histamine on endothelial permeability and structure and adhesion molecules of the paracellular junctions of perfused human placental microvessels. Microvasc Res 50(3):323–337. https://doi.org/10.1006/mvre.1995.1062
Ashina K, Tsubosaka Y, Nakamura T, Omori K, Kobayashi K, Hori M, Ozaki H, Murata T (2015) Histamine induces vascular hyperpermeability by increasing blood flow and endothelial barrier disruption in vivo. PLoS One 10(7):e0132367. https://doi.org/10.1371/journal.pone.0132367
Adderley SP, Zhang XE, Breslin JW (2015) Involvement of the H1 histamine receptor, p38 MAP kinase, myosin light chains kinase, and Rho/ROCK in histamine-induced endothelial barrier dysfunction. Microcirculation 22(4):237–248. https://doi.org/10.1111/micc.12189
Kugelmann D, Rotkopf LT, Radeva MY, Garcia-Ponce A, Walter E, Waschke J (2018) Histamine causes endothelial barrier disruption via Ca(2+)-mediated RhoA activation and tension at adherens junctions. Sci Rep 8(1):13229. https://doi.org/10.1038/s41598-018-31408-3
Takeda T, Yamashita Y, Shimazaki S, Mitsui Y (1992) Histamine decreases the permeability of an endothelial cell monolayer by stimulating cyclic AMP production through the H2-receptor. J Cell Sci 101(Pt 4):745–750
Lee HZ, Wu CH (1999) Serotonin-stimulated increase in cytosolic Ca(2+) in cultured rat heart endothelial cells. Eur J Pharmacol 384(1):53–60. https://doi.org/10.1016/s0014-2999(99)00672-x
Walther A, Petri E, Peter C, Czabanka M, Martin E (2007) Selective serotonin-receptor antagonism and microcirculatory alterations during experimental endotoxemia. J Surg Res 143(2):216–223. https://doi.org/10.1016/j.jss.2006.08.021
Spindler V, Waschke J (2011) Beta-adrenergic stimulation contributes to maintenance of endothelial barrier functions under baseline conditions. Microcirculation 18(2):118–127. https://doi.org/10.1111/j.1549-8719.2010.00072.x
Borges N, Shi F, Azevedo I, Audus KL (1994) Changes in brain microvessel endothelial cell monolayer permeability induced by adrenergic drugs. Eur J Pharmacol 269(2):243–248. https://doi.org/10.1016/0922-4106(94)90092-2
Sarmento A, Borges N, Azevedo I (1991) Adrenergic influences on the control of blood-brain barrier permeability. Naunyn Schmiedebergs Arch Pharmacol 343(6):633–637. https://doi.org/10.1007/BF00184295
Langeler EG, van Hinsbergh VW (1991) Norepinephrine and iloprost improve barrier function of human endothelial cell monolayers: role of cAMP. Am J Physiol 260(5 Pt 1):C1052-1059. https://doi.org/10.1152/ajpcell.1991.260.5.C1052
Shibamoto T, Wang HG, Tanaka S, Koyama S (1995) No effects of large doses of catecholamines on vascular permeability in isolated blood-perfused dog lungs. Acta Physiol Scand 155(2):127–135. https://doi.org/10.1111/j.1748-1716.1995.tb09957.x
Nickel T, Deutschmann A, Hanssen H, Summo C, Wilbert-Lampen U (2009) Modification of endothelial biology by acute and chronic stress hormones. Microvasc Res 78(3):364–369. https://doi.org/10.1016/j.mvr.2009.07.008
Zhou J, Liu Z, Zhang L, Hu X, Wang Z, Ni H, Wang Y, Qin J (2020) Activation of beta2-adrenergic receptor promotes growth and angiogenesis in breast cancer by down-regulating PPARgamma. Cancer Res Treat 52(3):830–847. https://doi.org/10.4143/crt.2019.510
Royall JA, Berkow RL, Beckman JS, Cunningham MK, Matalon S, Freeman BA (1989) Tumor necrosis factor and interleukin 1 alpha increase vascular endothelial permeability. Am J Physiol 257(6 Pt 1):L399-410. https://doi.org/10.1152/ajplung.1989.257.6.L399
McKenzie JA, Ridley AJ (2007) Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J Cell Physiol 213(1):221–228. https://doi.org/10.1002/jcp.21114
Xu C, Wu X, Hack BK, Bao L, Cunningham PN (2015) TNF causes changes in glomerular endothelial permeability and morphology through a Rho and myosin light chain kinase-dependent mechanism. Physiol Rep 3 (12). https://doi.org/10.14814/phy2.12636
Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB (2006) Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13(8):693–708. https://doi.org/10.1080/10739680600930347
Martin S, Maruta K, Burkart V, Gillis S, Kolb H (1988) IL-1 and IFN-gamma increase vascular permeability. Immunology 64(2):301–305
Kak G, Raza M, Tiwari BK (2018) Interferon-gamma (IFN-gamma): exploring its implications in infectious diseases. Biomol Concepts 9(1):64–79. https://doi.org/10.1515/bmc-2018-0007
Ng CT, Fong LY, Sulaiman MR, Moklas MA, Yong YK, Hakim MN, Ahmad Z (2015) Interferon-gamma increases endothelial permeability by causing activation of p38 MAP kinase and actin cytoskeleton alteration. J Interferon Cytokine Res 35(7):513–522. https://doi.org/10.1089/jir.2014.0188
Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A (2005) Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell 16(10):5040–5052. https://doi.org/10.1091/mbc.e05-03-0193
Bonney S, Seitz S, Ryan CA, Jones KL, Clarke P, Tyler KL, Siegenthaler JA (2019) Gamma Interferon alters junctional integrity via rho kinase, resulting in blood-brain barrier leakage in experimental viral encephalitis. mBio 10 (4). https://doi.org/10.1128/mBio.01675-19
Langer V, Vivi E, Regensburger D, Winkler TH, Waldner MJ, Rath T, Schmid B, Skottke L, Lee S, Jeon NL, Wohlfahrt T, Kramer V, Tripal P, Schumann M, Kersting S, Handtrack C, Geppert CI, Suchowski K, Adams RH, Becker C, Ramming A, Naschberger E, Britzen-Laurent N, Sturzl M (2019) IFN-gamma drives inflammatory bowel disease pathogenesis through VE-cadherin-directed vascular barrier disruption. J Clin Invest 129(11):4691–4707. https://doi.org/10.1172/JCI124884
Vastag M, Skopal J, Kramer J, Kolev K, Voko Z, Csonka E, Machovich R, Nagy Z (1998) Endothelial cells cultured from human brain microvessels produce complement proteins factor H, factor B, C1 inhibitor, and C4. Immunobiology 199(1):5–13. https://doi.org/10.1016/S0171-2985(98)80059-4
Gulati P, Lemercier C, Guc D, Lappin D, Whaley K (1993) Regulation of the synthesis of C1 subcomponents and C1-inhibitor. Behring Inst Mitt 93:196–203
Lappin DF, Guc D, Hill A, McShane T, Whaley K (1992) Effect of interferon-gamma on complement gene expression in different cell types. Biochem J 281(Pt 2):437–442. https://doi.org/10.1042/bj2810437
Fahey E, Doyle SL (2019) IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front Immunol 10:1426. https://doi.org/10.3389/fimmu.2019.01426
Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, Sheppard D, Violette SM, Weinreb PH, Horan GS, Matthay MA, Pittet JF (2008) Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res 102(7):804–812. https://doi.org/10.1161/CIRCRESAHA.107.161067
Jung HK, Ryu HJ, Kim MJ, Kim WI, Choi HK, Choi HC, Song HK, Jo SM, Kang TC (2012) Interleukin-18 attenuates disruption of brain-blood barrier induced by status epilepticus within the rat piriform cortex in interferon-gamma independent pathway. Brain Res 1447:126–134. https://doi.org/10.1016/j.brainres.2012.01.057
Shen J, Choy DF, Yoshida T, Iwase T, Hafiz G, Xie B, Hackett SF, Arron JR, Campochiaro PA (2014) Interleukin-18 has antipermeablity and antiangiogenic activities in the eye: reciprocal suppression with VEGF. J Cell Physiol 229(8):974–983. https://doi.org/10.1002/jcp.24575
Walshe TE, Saint-Geniez M, Maharaj AS, Sekiyama E, Maldonado AE, D’Amore PA (2009) TGF-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS One 4(4):e5149. https://doi.org/10.1371/journal.pone.0005149
Howe KL, Reardon C, Wang A, Nazli A, McKay DM (2005) Transforming growth factor-beta regulation of epithelial tight junction proteins enhances barrier function and blocks enterohemorrhagic Escherichia coli O157:H7-induced increased permeability. Am J Pathol 167(6):1587–1597. https://doi.org/10.1016/s0002-9440(10)61243-6
Clements RT, Minnear FL, Singer HA, Keller RS, Vincent PA (2005) RhoA and Rho-kinase dependent and independent signals mediate TGF-beta-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am J Physiol Lung Cell Mol Physiol 288(2):L294-306. https://doi.org/10.1152/ajplung.00213.2004
Antonov AS, Antonova GN, Fujii M, ten Dijke P, Handa V, Catravas JD, Verin AD (2012) Regulation of endothelial barrier function by TGF-beta type I receptor ALK5: potential role of contractile mechanisms and heat shock protein 90. J Cell Physiol 227(2):759–771. https://doi.org/10.1002/jcp.22785
Hickey MJ, Issekutz AC, Reinhardt PH, Fedorak RN, Kubes P (1998) Endogenous interleukin-10 regulates hemodynamic parameters, leukocyte-endothelial cell interactions, and microvascular permeability during endotoxemia. Circ Res 83(11):1124–1131. https://doi.org/10.1161/01.res.83.11.1124
Cervenak L, Morbidelli L, Donati D, Donnini S, Kambayashi T, Wilson JL, Axelson H, Castanos-Velez E, Ljunggren HG, Malefyt RD, Granger HJ, Ziche M, Bejarano MT (2000) Abolished angiogenicity and tumorigenicity of Burkitt lymphoma by interleukin-10. Blood 96(7):2568–2573
Melincovici CS, Bosca AB, Susman S, Marginean M, Mihu C, Istrate M, Moldovan IM, Roman AL, Mihu CM (2018) Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol 59(2):455–467
Di Lorenzo A, Lin MI, Murata T, Landskroner-Eiger S, Schleicher M, Kothiya M, Iwakiri Y, Yu J, Huang PL, Sessa WC (2013) eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J Cell Sci 126(Pt 24):5541–5552. https://doi.org/10.1242/jcs.115972
Loffredo S, Bova M, Suffritti C, Borriello F, Zanichelli A, Petraroli A, Varricchi G, Triggiani M, Cicardi M, Marone G (2016) Elevated plasma levels of vascular permeability factors in C1 inhibitor-deficient hereditary angioedema. Allergy 71(7):989–996. https://doi.org/10.1111/all.12862
Saharinen P, Eklund L, Alitalo K (2017) Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov 16(9):635–661. https://doi.org/10.1038/nrd.2016.278
Gamble JR, Drew J, Trezise L, Underwood A, Parsons M, Kasminkas L, Rudge J, Yancopoulos G, Vadas MA (2000) Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res 87(7):603–607. https://doi.org/10.1161/01.res.87.7.603
Siddiqui MR, Mayanil CS, Kim KS, Tomita T (2015) Angiopoietin-1 regulates brain endothelial permeability through PTPN-2 mediated tyrosine dephosphorylation of occludin. PLoS One 10(6):e0130857. https://doi.org/10.1371/journal.pone.0130857
Gavard J, Patel V, Gutkind JS (2008) Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell 14(1):25–36. https://doi.org/10.1016/j.devcel.2007.10.019
Hu S, Park J, Liu A, Lee J, Zhang X, Hao Q, Lee JW (2018) Mesenchymal stem cell microvesicles restore protein permeability across primary cultures of injured human lung microvascular endothelial cells. Stem Cells Transl Med 7(8):615–624. https://doi.org/10.1002/sctm.17-0278
d’Apolito M, Santacroce R, Colia AL, Cordisco G, Maffione AB, Margaglione M (2019) Angiopoietin-1 haploinsufficiency affects the endothelial barrier and causes hereditary angioedema. Clin Exp Allergy 49(5):626–635. https://doi.org/10.1111/cea.13349
Murakami M, Simons M (2008) Fibroblast growth factor regulation of neovascularization. Curr Opin Hematol 15(3):215–220. https://doi.org/10.1097/MOH.0b013e3282f97d98
Huang B, Krafft PR, Ma Q, Rolland WB, Caner B, Lekic T, Manaenko A, Le M, Tang J, Zhang JH (2012) Fibroblast growth factors preserve blood-brain barrier integrity through RhoA inhibition after intracerebral hemorrhage in mice. Neurobiol Dis 46(1):204–214. https://doi.org/10.1016/j.nbd.2012.01.008
Chen P, Zhang H, Zhang Q, Zhou W, Deng Y, Hu X, Zhang L (2019) Basic fibroblast growth factor reduces permeability and apoptosis of human brain microvascular endothelial cells in response to oxygen and glucose deprivation followed by reoxygenation via the fibroblast growth factor receptor 1 (FGFR1)/ERK pathway. Med Sci Monit 25:7191–7201. https://doi.org/10.12659/MSM.918626
Yang S, Jin H, Zhao ZG (2019) Epidermal growth factor treatment has protective effects on the integrity of the blood-brain barrier against cerebral ischemia injury in bEnd3 cells. Exp Ther Med 17(3):2397–2402. https://doi.org/10.3892/etm.2019.7186
Knezevic II, Predescu SA, Neamu RF, Gorovoy MS, Knezevic NM, Easington C, Malik AB, Predescu DN (2009) Tiam1 and Rac1 are required for platelet-activating factor-induced endothelial junctional disassembly and increase in vascular permeability. J Biol Chem 284(8):5381–5394. https://doi.org/10.1074/jbc.M808958200
Bogatcheva NV, Sergeeva MG, Dudek SM, Verin AD (2005) Arachidonic acid cascade in endothelial pathobiology. Microvasc Res 69(3):107–127. https://doi.org/10.1016/j.mvr.2005.01.007
Kobayashi K, Horikami D, Omori K, Nakamura T, Yamazaki A, Maeda S, Murata T (2016) Thromboxane A2 exacerbates acute lung injury via promoting edema formation. Sci Rep 6:32109. https://doi.org/10.1038/srep32109
Chiang CY, Chien CY, Qiou WY, Chang C, Yu IS, Chang PY, Chien CT (2018) Genetic depletion of thromboxane A2/thromboxane-prostanoid receptor signalling prevents microvascular dysfunction in ischaemia/reperfusion injury. Thromb Haemost 118(11):1982–1996. https://doi.org/10.1055/s-0038-1672206
Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N (2005) Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol 25(1):136–146. https://doi.org/10.1128/MCB.25.1.136-146.2005
Bundey RA, Insel PA (2006) Adenylyl cyclase 6 overexpression decreases the permeability of endothelial monolayers via preferential enhancement of prostacyclin receptor function. Mol Pharmacol 70(5):1700–1707. https://doi.org/10.1124/mol.106.028035
Birukova AA, Meng F, Tian Y, Meliton A, Sarich N, Quilliam LA (1852) Birukov KG (2015) Prostacyclin post-treatment improves LPS-induced acute lung injury and endothelial barrier recovery via Rap1. Biochim Biophys Acta 5:778–791. https://doi.org/10.1016/j.bbadis.2014.12.016
Lindner K, Uhlig U, Uhlig S (2005) Ceramide alters endothelial cell permeability by a nonapoptotic mechanism. Br J Pharmacol 145(1):132–140. https://doi.org/10.1038/sj.bjp.0706173
Winkler MS, Nierhaus A, Poppe A, Greiwe G, Graler MH, Daum G (2017) Sphingosine-1-phosphate: a potential biomarker and therapeutic target for endothelial dysfunction and sepsis? Shock 47(6):666–672. https://doi.org/10.1097/SHK.0000000000000814
Bode C, Sensken SC, Peest U, Beutel G, Thol F, Levkau B, Li Z, Bittman R, Huang T, Tolle M, van der Giet M, Graler MH (2010) Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem 109(6):1232–1243. https://doi.org/10.1002/jcb.22507
Adamson RH, Sarai RK, Altangerel A, Thirkill TL, Clark JF, Curry FR (2010) Sphingosine-1-phosphate modulation of basal permeability and acute inflammatory responses in rat venular microvessels. Cardiovasc Res 88(2):344–351. https://doi.org/10.1093/cvr/cvq184
Zeng Y, Adamson RH, Curry FR, Tarbell JM (2014) Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol 306(3):H363-372. https://doi.org/10.1152/ajpheart.00687.2013
Zhang L, Zeng M, Fan J, Tarbell JM, Curry FR, Fu BM (2016) Sphingosine-1-phosphate maintains normal vascular permeability by preserving endothelial surface glycocalyx in intact microvessels. Microcirculation 23(4):301–310. https://doi.org/10.1111/micc.12278
Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J (2009) The role of the anaphylatoxins in health and disease. Mol Immunol 46(14):2753–2766. https://doi.org/10.1016/j.molimm.2009.04.027
Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R (2002) Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J Immunol 169(4):2102–2110. https://doi.org/10.4049/jimmunol.169.4.2102
Ralevic V, Dunn WR (2015) Purinergic transmission in blood vessels. Auton Neurosci 191:48–66. https://doi.org/10.1016/j.autneu.2015.04.007
Umapathy SN, Kaczmarek E, Fatteh N, Burns N, Lucas R, Stenmark KR, Verin AD, Gerasimovskaya EV (2013) Adenosine A1 receptors promote vasa vasorum endothelial cell barrier integrity via Gi and Akt-dependent actin cytoskeleton remodeling. PLoS One 8(4):e59733. https://doi.org/10.1371/journal.pone.0059733
Liu Y, Alahiri M, Ulloa B, Xie B, Sadiq SA (2018) Adenosine A2A receptor agonist ameliorates EAE and correlates with Th1 cytokine-induced blood brain barrier dysfunction via suppression of MLCK signaling pathway. Immun Inflamm Dis 6(1):72–80. https://doi.org/10.1002/iid3.187
Carman AJ, Mills JH, Krenz A, Kim DG, Bynoe MS (2011) Adenosine receptor signaling modulates permeability of the blood-brain barrier. J Neurosci 31(37):13272–13280. https://doi.org/10.1523/JNEUROSCI.3337-11.2011
Gunduz D, Aslam M, Krieger U, Becker L, Grebe M, Arshad M, Sedding DG, Hartel FV, Abdallah Y, Piper HM, Voss RK, Noll T (2012) Opposing effects of ATP and adenosine on barrier function of rat coronary microvasculature. J Mol Cell Cardiol 52(5):962–970. https://doi.org/10.1016/j.yjmcc.2012.01.003
Hata M, Yamanegi K, Yamada N, Ohyama H, Yukitatsu Y, Nakasho K, Okamura H, Terada N (2014) Estrogen decreases the expression of claudin-5 in vascular endothelial cells in the murine uterus. Endocr J 61(7):705–715. https://doi.org/10.1507/endocrj.ej13-0442
Shin JA, Yoon JC, Kim M, Park EM (2016) Activation of classical estrogen receptor subtypes reduces tight junction disruption of brain endothelial cells under ischemia/reperfusion injury. Free Radic Biol Med 92:78–89. https://doi.org/10.1016/j.freeradbiomed.2016.01.010
Molitoris KH, Kazi AA, Koos RD (2009) Inhibition of oxygen-induced hypoxia-inducible factor-1alpha degradation unmasks estradiol induction of vascular endothelial growth factor expression in ECC-1 cancer cells in vitro. Endocrinology 150(12):5405–5414. https://doi.org/10.1210/en.2009-0884
Kobayashi H, Okada Y, Asahina T, Gotoh J, Terao T (1998) The kallikrein-kinin system, but not vascular endothelial growth factor, plays a role in the increased vascular permeability associated with ovarian hyperstimulation syndrome. J Mol Endocrinol 20(3):363–374. https://doi.org/10.1677/jme.0.0200363
Forster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D (2008) Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol 586(7):1937–1949. https://doi.org/10.1113/jphysiol.2007.146852
Forster C, Silwedel C, Golenhofen N, Burek M, Kietz S, Mankertz J, Drenckhahn D (2005) Occludin as direct target for glucocorticoid-induced improvement of blood-brain barrier properties in a murine in vitro system. J Physiol 565(Pt 2):475–486. https://doi.org/10.1113/jphysiol.2005.084038
Machein MR, Kullmer J, Ronicke V, Machein U, Krieg M, Damert A, Breier G, Risau W, Plate KH (1999) Differential downregulation of vascular endothelial growth factor by dexamethasone in normoxic and hypoxic rat glioma cells. Neuropathol Appl Neurobiol 25(2):104–112. https://doi.org/10.1046/j.1365-2990.1999.00166.x
Thomas GW, Rael LT, Bar-Or R, Mains CW, Slone DS, Boyd SR, Bar-Or D (2012) Biphasic effect of danazol on human vascular endothelial cell permeability and f-actin cytoskeleton dynamics. Biochem Biophys Res Commun 421(4):707–712. https://doi.org/10.1016/j.bbrc.2012.04.066
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W (2016) ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev 2016:4350965. https://doi.org/10.1155/2016/4350965
Di A, Mehta D, Malik AB (2016) ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium 60(3):163–171. https://doi.org/10.1016/j.ceca.2016.02.002
Burtenshaw D, Hakimjavadi R, Redmond EM, Cahill PA (2017) Nox, reactive oxygen species and regulation of vascular cell fate. Antioxidants (Basel) 6 (4). https://doi.org/10.3390/antiox6040090
Rafikov R, Dimitropoulou C, Aggarwal S, Kangath A, Gross C, Pardo D, Sharma S, Jezierska-Drutel A, Patel V, Snead C, Lucas R, Verin A, Fulton D, Catravas JD, Black SM (2014) Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J Biol Chem 289(8):4710–4722. https://doi.org/10.1074/jbc.M114.547596
Chen F, Wang Y, Rafikov R, Haigh S, Zhi WB, Kumar S, Doulias PT, Rafikova O, Pillich H, Chakraborty T, Lucas R, Verin AD, Catravas JD, She JX, Black SM, Fulton DJR (2017) RhoA S-nitrosylation as a regulatory mechanism influencing endothelial barrier function in response to G(+)-bacterial toxins. Biochem Pharmacol 127:34–45. https://doi.org/10.1016/j.bcp.2016.12.014
Choi S, Saxena N, Dhammu T, Khan M, Singh AK, Singh I, Won J (2019) Regulation of endothelial barrier integrity by redox-dependent nitric oxide signaling: implication in traumatic and inflammatory brain injuries. Nitric Oxide 83:51–64. https://doi.org/10.1016/j.niox.2018.12.007
Jin RC (2010) Loscalzo J (2010) Vascular nitric oxide: formation and function. J Blood Med 1:147–162. https://doi.org/10.2147/JBM.S7000
Duran WN, Beuve AV, Sanchez FA (2013) Nitric oxide, S-nitrosation, and endothelial permeability. IUBMB Life 65(10):819–826. https://doi.org/10.1002/iub.1204
Zhou Z, Gengaro P, Wang W, Wang XQ, Li C, Faubel S, Rivard C, Schrier RW (2008) Role of NF-kappaB and PI 3-kinase/Akt in TNF-alpha-induced cytotoxicity in microvascular endothelial cells. Am J Physiol Renal Physiol 295(4):F932-941. https://doi.org/10.1152/ajprenal.00066.2008
Beltowski J (2019) Synthesis, metabolism, and signaling mechanisms of hydrogen sulfide: an overview. Methods Mol Biol 2007:1–8. https://doi.org/10.1007/978-1-4939-9528-8_1
Basuroy S, Leffler CW, Parfenova H (2013) CORM-A1 prevents blood-brain barrier dysfunction caused by ionotropic glutamate receptor-mediated endothelial oxidative stress and apoptosis. Am J Physiol Cell Physiol 304(11):C1105-1115. https://doi.org/10.1152/ajpcell.00023.2013
Krewson EA, Sanderlin EJ, Marie MA, Akhtar SN, Velcicky J, Loetscher P, Yang LV (2020) The proton-sensing gpr4 receptor regulates paracellular gap formation and permeability of vascular endothelial cells. iScience 23 (2):100848. https://doi.org/10.1016/j.isci.2020.100848
Wang W, Sun J, Wang N, Sun Z, Ma Q, Li J, Zhang M, Xu J (2020) Enterovirus A71 capsid protein VP1 increases blood-brain barrier permeability and virus receptor vimentin on the brain endothelial cells. J Neurovirol 26(1):84–94. https://doi.org/10.1007/s13365-019-00800-8
Buzhdygan TP, DeOre BJ, Baldwin-Leclair A, McGary H, Razmpour R, Galie PA, Potula R, Andrews AM, Ramirez SH (2020) The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in vitro models of the human blood-brain barrier. bioRxiv. https://doi.org/10.1101/2020.06.15.150912
Waschke J, Curry FE, Adamson RH, Drenckhahn D (2005) Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol 288(3):H1296-1305. https://doi.org/10.1152/ajpheart.00687.2004
Dunzendorfer S, Lee HK, Soldau K, Tobias PS (2004) Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: roles of LBP and sCD14 in mediating LPS responses. FASEB J 18(10):1117–1119. https://doi.org/10.1096/fj.03-1263fje
Mako V, Czucz J, Weiszhar Z, Herczenik E, Matko J, Prohaszka Z, Cervenak L (2010) Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL-1beta, TNF-alpha, and LPS. Cytometry A 77(10):962–970. https://doi.org/10.1002/cyto.a.20952
Xing J, Birukova AA (2010) ANP attenuates inflammatory signaling and Rho pathway of lung endothelial permeability induced by LPS and TNFalpha. Microvasc Res 79(1):56–62. https://doi.org/10.1016/j.mvr.2009.11.006
Xing J, Wang Q, Coughlan K, Viollet B, Moriasi C, Zou MH (2013) Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am J Pathol 182(3):1021–1030. https://doi.org/10.1016/j.ajpath.2012.11.022
Raveendran VV, Tan X, Sweeney ME, Levant B, Slusser J, Stechschulte DJ, Dileepan KN (2011) Lipopolysaccharide induces H1 receptor expression and enhances histamine responsiveness in human coronary artery endothelial cells. Immunology 132(4):578–588. https://doi.org/10.1111/j.1365-2567.2010.03403.x
Chao HH, Chen PY, Hao WR, Chiang WP, Cheng TH, Loh SH, Leung YM, Liu JC, Chen JJ, Sung LC (2017) Lipopolysaccharide pretreatment increases protease-activated receptor-2 expression and monocyte chemoattractant protein-1 secretion in vascular endothelial cells. J Biomed Sci 24(1):85. https://doi.org/10.1186/s12929-017-0393-1
Murakami M, Ohta T, Ito S (2009) Lipopolysaccharides enhance the action of bradykinin in enteric neurons via secretion of interleukin-1beta from enteric glial cells. J Neurosci Res 87(9):2095–2104. https://doi.org/10.1002/jnr.22036
Madej MP, Topfer E, Boraschi D, Italiani P (2017) Different regulation of interleukin-1 production and activity in monocytes and macrophages: innate memory as an endogenous mechanism of IL-1 inhibition. Front Pharmacol 8:335. https://doi.org/10.3389/fphar.2017.00335
Remick DG, Strieter RM, Eskandari MK, Nguyen DT, Genord MA, Raiford CL, Kunkel SL (1990) Role of tumor necrosis factor-alpha in lipopolysaccharide-induced pathologic alterations. Am J Pathol 136(1):49–60
Hecht G, Pothoulakis C, LaMont JT, Madara JL (1988) Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest 82(5):1516–1524. https://doi.org/10.1172/JCI113760
Huang J, Kelly CP, Bakirtzi K, Villafuerte Galvez JA, Lyras D, Mileto SJ, Larcombe S, Xu H, Yang X, Shields KS, Zhu W, Zhang Y, Goldsmith JD, Patel IJ, Hansen J, Huang M, Yla-Herttuala S, Moss AC, Paredes-Sabja D, Pothoulakis C, Shah YM, Wang J, Chen X (2019) Clostridium difficile toxins induce VEGF-A and vascular permeability to promote disease pathogenesis. Nat Microbiol 4(2):269–279. https://doi.org/10.1038/s41564-018-0300-x
Ohishi I, Iwasaki M, Sakaguchi G (1981) Vascular permeability activity of botulinum C2 toxin elicited by cooperation of two dissimilar protein components. Infect Immun 31(3):890–895. https://doi.org/10.1128/IAI.31.3.890-895.1981
Barth H, Aktories K (2011) New insights into the mode of action of the actin ADP-ribosylating virulence factors Salmonella enterica SpvB and Clostridium botulinum C2 toxin. Eur J Cell Biol 90(11):944–950. https://doi.org/10.1016/j.ejcb.2010.11.007
Bruckener KE, el Baya A, Galla HJ, Schmidt MA (2003) Permeabilization in a cerebral endothelial barrier model by pertussis toxin involves the PKC effector pathway and is abolished by elevated levels of cAMP. J Cell Sci 116(Pt 9):1837–1846. https://doi.org/10.1242/jcs.00378
Stelzner TJ, Weil JV, O’Brien RF (1989) Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J Cell Physiol 139(1):157–166. https://doi.org/10.1002/jcp.1041390122
Patterson CE, Davis HW, Schaphorst KL, Garcia JG (1994) Mechanisms of cholera toxin prevention of thrombin- and PMA-induced endothelial cell barrier dysfunction. Microvasc Res 48(2):212–235. https://doi.org/10.1006/mvre.1994.1050
Goldblum SE, Rai U, Tripathi A, Thakar M, De Leo L, Di Toro N, Not T, Ramachandran R, Puche AC, Hollenberg MD, Fasano A (2011) The active Zot domain (aa 288–293) increases ZO-1 and myosin 1C serine/threonine phosphorylation, alters interaction between ZO-1 and its binding partners, and induces tight junction disassembly through proteinase activated receptor 2 activation. FASEB J 25(1):144–158. https://doi.org/10.1096/fj.10-158972
Sturgeon C, Fasano A (2016) Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers 4(4):e1251384. https://doi.org/10.1080/21688370.2016.1251384
Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang MS, Grailer JJ, Zetoune FS, Andjelkovic AV, Fasano A, Ward PA (2013) Zonulin as prehaptoglobin2 regulates lung permeability and activates the complement system. Am J Physiol Lung Cell Mol Physiol 304(12):L863-872. https://doi.org/10.1152/ajplung.00196.2012
Visy B, Fust G, Bygum A, Bork K, Longhurst H, Bucher C, Bouillet L, Cicardi M, Farkas H (2007) Helicobacter pylori infection as a triggering factor of attacks in patients with hereditary angioedema. Helicobacter 12(3):251–257. https://doi.org/10.1111/j.1523-5378.2007.00501.x
Hofman ZL, Relan A, Zeerleder S, Drouet C, Zuraw B, Hack CE (2016) Angioedema attacks in patients with hereditary angioedema: Local manifestations of a systemic activation process. J Allergy Clin Immunol 138(2):359–366. https://doi.org/10.1016/j.jaci.2016.02.041
Aird WC (2012) Endothelial cell heterogeneity. Cold Spring Harb Perspect Med 2(1):a006429. https://doi.org/10.1101/cshperspect.a006429
Kluger MS, Clark PR, Tellides G, Gerke V, Pober JS (2013) Claudin-5 controls intercellular barriers of human dermal microvascular but not human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol 33(3):489–500. https://doi.org/10.1161/ATVBAHA.112.300893
Haraldsen G, Kvale D, Lien B, Farstad IN, Brandtzaeg P (1996) Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells. J Immunol 156(7):2558–2565
Fujiwara M, Jin E, Ghazizadeh M, Kawanami O (2004) Differential expression of protease-activated receptors 1, 2, and 4 on human endothelial cells from different vascular sites. Pathobiology 71(1):52–58. https://doi.org/10.1159/000072962
Sacks RS, Firth AL, Remillard CV, Agange N, Yau J, Ko EA, Yuan JX (2008) Thrombin-mediated increases in cytosolic [Ca2+] involve different mechanisms in human pulmonary artery smooth muscle and endothelial cells. Am J Physiol Lung Cell Mol Physiol 295(6):L1048-1055. https://doi.org/10.1152/ajplung.90259.2008
Shpacovitch VM, Brzoska T, Buddenkotte J, Stroh C, Sommerhoff CP, Ansel JC, Schulze-Osthoff K, Bunnett NW, Luger TA, Steinhoff M (2002) Agonists of proteinase-activated receptor 2 induce cytokine release and activation of nuclear transcription factor kappaB in human dermal microvascular endothelial cells. J Invest Dermatol 118(2):380–385. https://doi.org/10.1046/j.0022-202x.2001.01658.x
Steinhoff M, Corvera CU, Thoma MS, Kong W, McAlpine BE, Caughey GH, Ansel JC, Bunnett NW (1999) Proteinase-activated receptor-2 in human skin: tissue distribution and activation of keratinocytes by mast cell tryptase. Exp Dermatol 8(4):282–294. https://doi.org/10.1111/j.1600-0625.1999.tb00383.x
Quan L, Jian Z, Ping Z, Weiming L (2009) Proteinase-activated receptor-1 mediates allogeneic CD8(+) T cell-induced apoptosis of vascular endothelial cells. Med Oncol 26(4):379–385. https://doi.org/10.1007/s12032-008-9132-5
Binion DG, Fu S, Ramanujam KS, Chai YC, Dweik RA, Drazba JA, Wade JG, Ziats NP, Erzurum SC, Wilson KT (1998) iNOS expression in human intestinal microvascular endothelial cells inhibits leukocyte adhesion. Am J Physiol 275(3):G592-603. https://doi.org/10.1152/ajpgi.1998.275.3.G592
Shelton JL, Wang L, Cepinskas G, Sandig M, Inculet R, McCormack DG, Mehta S (2006) Albumin leak across human pulmonary microvascular vs. umbilical vein endothelial cells under septic conditions. Microvasc Res 71 (1):40–47. https://doi.org/10.1016/j.mvr.2005.11.003
Zotter Z, Csuka D, Szabo E, Czaller I, Nebenfuhrer Z, Temesszentandrasi G, Fust G, Varga L, Farkas H (2014) The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis 9:44. https://doi.org/10.1186/1750-1172-9-44
Moldoveanu AI, Shephard RJ, Shek PN (2001) The cytokine response to physical activity and training. Sports Med 31(2):115–144. https://doi.org/10.2165/00007256-200131020-00004
Czaller I, Visy B, Csuka D, Fust G, Toth F, Farkas H (2010) The natural history of hereditary angioedema and the impact of treatment with human C1-inhibitor concentrate during pregnancy: a long-term survey. Eur J Obstet Gynecol Reprod Biol 152(1):44–49. https://doi.org/10.1016/j.ejogrb.2010.05.008
Sulman FG (1980) Migraine and headache due to weather and allied causes and its specific treatment. Ups J Med Sci Suppl 31:41–44
Dowdall M, De Montigny C (1985) Effect of atmospheric ions on hippocampal pyramidal neuron responsiveness to serotonin. Brain Res 342(1):103–109. https://doi.org/10.1016/0006-8993(85)91357-5
Qin L, Zhao D, Xu J, Ren X, Terwilliger EF, Parangi S, Lawler J, Dvorak HF, Zeng H (2013) The vascular permeabilizing factors histamine and serotonin induce angiogenesis through TR3/Nur77 and subsequently truncate it through thrombospondin-1. Blood 121(11):2154–2164. https://doi.org/10.1182/blood-2012-07-443903
Maarouf M, Maarouf CL, Yosipovitch G, Shi VY (2019) The impact of stress on epidermal barrier function: an evidence-based review. Br J Dermatol 181(6):1129–1137. https://doi.org/10.1111/bjd.17605
Huang M, Pang X, Letourneau R, Boucher W, Theoharides TC (2002) Acute stress induces cardiac mast cell activation and histamine release, effects that are increased in Apolipoprotein E knockout mice. Cardiovasc Res 55(1):150–160. https://doi.org/10.1016/s0008-6363(02)00336-x
Kelley JL, Chi DS, Abou-Auda W, Smith JK, Krishnaswamy G (2000) The molecular role of mast cells in atherosclerotic cardiovascular disease. Mol Med Today 6(8):304–308. https://doi.org/10.1016/s1357-4310(00)01747-0
Kajdacsi E, Jandrasics Z, Veszeli N, Mako V, Koncz A, Gulyas D, Kohalmi KV, Temesszentandrasi G, Cervenak L, Gal P, Dobo J, de Maat S, Maas C, Farkas H, Varga L (2020) Patterns of C1-inhibitor/plasma serine protease complexes in healthy humans and in hereditary angioedema patients. Front Immunol 11:794. https://doi.org/10.3389/fimmu.2020.00794
Konings J, Cugno M, Suffritti C, Ten Cate H, Cicardi M, Govers-Riemslag JW (2013) Ongoing contact activation in patients with hereditary angioedema. PLoS One 8(8):e74043. https://doi.org/10.1371/journal.pone.0074043
Acknowledgements
The study was supported by the grant of the National Research, Development and Innovation Office (NKFIH) K115623 (LC), and by the Higher Education Institutional Excellence Program {FIKP} of the Semmelweis University (MLD).
Funding
Open access funding provided by Semmelweis University.
Author information
Authors and Affiliations
Contributions
LC proposed the initial idea for the review. MLD, ZN, EK, and HF equally participated in the literature search and in writing parts of the review. LC merged the parts of the review and conducted the final revision. MLD produced all figures and ZN compiled the tables based on the idea of all authors.
Corresponding author
Ethics declarations
Conflict of Interest
Henriette Farkas has received research grants from CSL Behring, Shire/Takeda and Pharming and served as an advisor for these companies and Biocryst, and has participated in clinical trials/registries for BioCryst, CSL Behring, Pharming, Kalvista and Shire/Takeda. The other authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Debreczeni, M.L., Németh, Z., Kajdácsi, E. et al. Molecular Dambusters: What Is Behind Hyperpermeability in Bradykinin-Mediated Angioedema?. Clinic Rev Allerg Immunol 60, 318–347 (2021). https://doi.org/10.1007/s12016-021-08851-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-021-08851-8