
Abstract
Mast cells (MCs) are physiologically activated by binding of stem cell factor (SCF) to the extracellular domains of the Kit receptor. This binding increases the proliferation and prolongs the survival of normal mature MCs, as well as intensifies the release of mediators. In mastocytosis, somatic mutations of the coding Kit gene cause autocrine dysregulation and lead to constitutive KIT activation even in the absence of its ligand SCF. Clinical symptoms are caused by MC-mediator release and/or infiltration of MCs into tissues. Aberrant KIT activation may result in increased production of MCs in the skin and extracutaneous organs. Depending on the affected organ(s), the disease can be divided into cutaneous mastocytosis (CM), systemic mastocytosis (SM), and localized MC tumors. The updated classification of WHO discriminates between several distinct subvariants of CM and SM. While the prognosis in CM and indolent SM (ISM) is excellent with (almost) normal life expectancy, the prognosis in aggressive SM (ASM) and MC leukemia (MCL) is dismal. The symptoms may comprise urticaria, angioedema, flush, pruritus, abdominal pain, diarrhea, hypotension, syncope, and musculoskeletal pain and are the results of MC infiltration and mediator release into target organs, i.e., the skin, gastrointestinal tract, liver, spleen, lymph nodes, and bone marrow. Mastocytosis differs from a lot of other hematological disorders because its pathology is not only based on the lack of normal function of a specific pathway or of a specific cell type but additionally is a proliferative disease. Currently available treatments of mastocytosis include symptomatic, antimediator and cytoreductive targeted therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mast cells (MCs) are normal residents of mucosal tissues, but their numbers and anatomical location change markedly during immune responses, infections, and other disorders [1]. In most settings, MCs have become infamous for their detrimental actions, i.e., anaphylaxis, allergy, arthritis, atherosclerosis, and cancer while in some settings, notably host defense against bacteria, parasites, and envenomation, their biologic function is in favor of maintaining health [2]. MC progenitor cells express the tyrosine kinase receptor KIT (CD117). Normally, the interaction between this oncogenic receptor and its ligand, stem cell factor (SCF), induces MC development in uncommitted and MC-committed hematopoietic precursor cells [3]. Mastocytosis is a heterogeneous group of disorders involving MCs and their CD34+/CD117+ progenitors. It is a group of rare clonal disorders of bone marrow origin characterized by abnormal growth and/or accumulation of clonal MCs primarily in the skin and bone marrow. Neoplastic MCs expressing CD25 and/or CD2 were described in systemic mastocytosis especially in aggressive systemic mastocytosis (ASM) and mast cell leukemia (MCL) [4, 5]. The symptoms of mast cell activation include sudden onset of flush, urticaria, angioedema, pruritus, abdominal pain, headache, diarrhea, hypotension, syncope, and musculoskeletal pain which are the results of MC mediator release and infiltration into target organs [4]. The heterogeneity of clinical presentation in mastocytosis is a result of MC burden and MC activity [6], the type of skin lesions, the patient’s age at the onset, and the associated hematological disorders. The typical mastocytosis of childhood is usually cutaneous and transient whereas in adulthood the systemic form is more common [7]. MCs respond to surrounding stimuli through the expression of a variety of receptors including FcεRI and KIT (CD117). MCs require stem cell factor (SCF) binding to their surface receptor KIT for homeostasis [8]. In mastocytosis, the presence of mutations within different regions of KIT in the extracellular, transmembrane, juxtamembrane domains, or activating loop interrupts the normal signaling cascade characterized by constitutive receptor activation independent from SCF [9].
Mast Cell Origin and Development
MCs are innate immune cells known for their role in allergic and anaphylactic reactions. They functionally can be considered a double-edged knife with both good and bad sides. The bad consists in type I allergic immune responses through crosslinking FcεR1 via allergen-bound IgE. On the good side the MC cell plays a protective key role in the battle against some environmental threats such as the venoms of reptiles and insects [10, 11]. In this regard, MC-derived carboxypeptidase plays a key role in degradation of the snake venom toxin safarotoxin [12]. Owing to expression of a wide range of receptors and release of a broad spectrum of mediators, they play a key role in acquired and innate immunity [13]. MCs arise from hematopoietic progenitor cells and mature MCs ordinarily do not circulate in the blood but migrate into peripheral tissues where they acquire their mature phenotype [14]. CD34+/CD117+ pluripotent progenitor cells from bone marrow origin circulate in the blood as committed precursors and under influence of SCF develop into mature FcεR1+ and CD117+ MCs in peripheral tissues [15,16,17]. SCF, which is produced by a variety of cells including fibroblasts and endothelial cells, promotes the recruitment of MC progenitors into tissues, as well as their local maturation and activation. Its receptor, c-Kit (CD117), is a type III tyrosine kinase broadly expressed on mature MCs and eosinophils [18]. In addition to SCF, MC growth and survival modulators include nerve growth factor (NGF) [15], IL-9 [19], CXCL12, IL-3, IL-4, IL-10, IL-33, and TGF-β [1, 14].
Mast Cell Tissue Homing
Tissue homing is a multifaceted process controlled by chemokines and expression of integrin and adhesion molecules both on the surface of progenitors and cells of residing tissues [15, 20]. For instance, trafficking of MC progenitors into the lung tissues requires expression of α4β7 and α4β1 integrins by progenitors and vascular cell adhesion molecule-1 (VCAM-1) by endothelium [8]. α4β7 integrin, expressed on MCs, interacts with (MAdCAM-1) or VCAM-1 on the endothelium and contributes to maintenance of MC number in the small intestine. Moreover, CXCR2, expressed on progenitors, has a role in their directed migration to the small intestine [21] (Fig. 1)

MCs develop from CD34+/CD117+ progenitors of bone marrow origin. Mast cell progenitors (MCP) are released from bone marrow into circulation. There they follow a controlled trafficking pattern with the help of interaction between integrins and their receptors. Finally, they reach the target tissues where under influence of growth factors they mature into mast cells
Mast Cell Mediators and Receptors
MCs are specialized secretory cells of the innate immune system that play an important role in host defense by producing and releasing proinflammatory mediators, chemotactic factors, and immunoregulatory cytokines. Upon stimulation of their surface Fc receptors with IgE, they immediately release a large number of secretory granules containing histamine, serotonin, and other inflammatory mediators. These mediators play a central role in both the immediate and late-phase inflammatory reaction [22]. MCs produce three categories of effector molecules: (1) pre-formed mediators stored in granules such as serotonin, histamine, heparin, tryptase, and chymase; (2) mediators synthesized de novo upon cell stimulation, among them mainly the lipid mediators PAF, PDG2, and LTB4 and LTD4; and (3) cytokines including IL-1, IL-3, IL-5, IL-8, IL-10, GM-CSF, TNF-α, TGF-β, and VEGF [23]. Human MCs can be categorized into MCT (express high levels of the MC-specific tryptase) and MCTC (express tryptase and chymase) [1, 24]. Morphologically, MCs are characterized by numerous, electron dense cytoplasmic granules containing biogenic amines, enzymes, cytokines, and proteoglycans [25, 26]. They express a wide range of receptors such as FcεRI, Fcγ receptors, complement, cytokine, chemokine, hormone receptors, and toll-like receptors (TLRs). This enables them to secrete a diverse and wide range of biologically active products that enhance as well as suppress immune responses [27] (Fig. 2). Signaling through the high-affinity receptor for IgE immunoglobulins (FcεRI) after binding of type I-allergens is the major pathway for the activation of MCs [28]. However, immunoglobulin free light chains, anaphylatoxins (C3a and C5a) [29], hormones (including corticotropin-releasing hormone (CRH)) [30], and neuropeptides (substance P (SP), hemokinin, neurotensin (NT) and NGF) are alternative ways to activate MCs [13]. After antigen sensitization and specific IgE production by plasma cell, the IgE molecules bind to FcεRI receptors on the surface of tissue MCs and circulating basophils. Re-exposure to the original antigen (or a crossreactive bivalent or multivalent antigen) results in the crosslinking of adjacent FcεRI-bound IgE and the consequent aggregation of surface FcεRI [31]. FcεRI exists in two forms. It can be expressed as a trimeric variant expressed on a variety of immune cells such as monocytes, eosinophils, Langerhans cells, or as a tetrameric variant primarily on MCs and basophils. The tetrameric variant is composed of an IgE-binding α chain, a membrane-tetraspanning β chain that is absent in the trimeric receptor, and a disulfide-linked homodimer of γ chains [32]. The trimeric FcεRI consists of an α-subunit and two γ-subunits. In this form, the α-subunit is a transmembrane protein which binds IgE only insufficiently. The two domains of its extracellular portion adopt the shape of an inverted “v,” the second of which binds one dimeric IgE-Fc molecule asymmetrically through interactions at two sites. IgE binding to FcεRIα results in adopting a unique bent conformation of IgE, and this conformational change contributes to the remarkably slow dissociation rate from FcεRI. The γ-subunit of FcεRI is a transmembrane protein that acts as a common adaptor molecule for various Fc receptors including FcγRI (CD64). The γ-subunit associates as a homodimer formed via a disulfide bond linked between N-terminal cysteine amino acids [33].

MCs express a variety of chemokine and cytokine receptors on their surface. They release mediators including cytokines, de novo synthesized and stored mediators. MCs rapidly respond to environmental stimuli such as IgE-bound allergen and interact with other immune/non-immune cells
Stem Cell Factor
The knowledge about the molecular structure of the KIT receptor and the further intracellular signaling cascade upon binding to SCF is a prerequisite for a better understanding of the pathogenesis of mastocytosis and is also the basis for treatments targeting this receptor. The discovery of the dimeric molecule SCF (also known as Kit Ligand, Steel Factor, or Mast Cell Growth Factor) and KIT was based on experimental single gene-induced anemias in mice which led to the identification of the W and Steel (Sl) loci. Mutations at either of these loci were shown to cause alterations of fur color, anemia and lack of tissue MCs [34]. Genomic investigations by Geissler et al. revealed that SCF in humans maps on chromosome 12, between 12q14.3 and 12qter [35]. SCF exists both as a membrane-bound and a soluble form expressed by fibroblasts and endothelial cells throughout the whole body [36, 37]. It is synthesized from two alternatively spliced messenger RNAs (mRNAs) as transmembrane proteins which are enzymatically cleaved to produce soluble forms or act as cell associated molecules [38]. Both SCF variants have distinct roles in the survival and proliferation of hematopoietic cells. Using the Sl/Sld mouse model in which mutants were generated that express only soluble SCF or membrane restricted SCF, it was reported that soluble SCF is responsible for proliferation of myeloid progenitors (in concert with other cytokines), while only the membrane bound form is able partially to correct the runting and the bone marrow hypocellularity that are seen in these mice [39].
SCF-KIT Interaction and Mediated Signaling
The c-Kit receptor, encoded by the oncogene c-kit [36], is a type III receptor tyrosine kinase with five extracellular immunoglobulin-like domains followed by a single transmembrane-spanning region. The first three Ig-like domains possess complementary shape and charge and are capable of binding to SCF, while domains 4 and 5 are involved in the KIT receptor dimerization. A juxtamembrane region is located nearly 30 amino acids between the plasma membrane and the kinase domain and forms the first part of the intracellular section contributing to the regulation of c-Kit kinase activity. The kinase domain consists of two subdomains, tyrosine kinase domain 1 and 2, and is interrupted by a kinase insert sequence [40]. SCF-mediated c-Kit signaling plays important roles in mediating angiogenesis, migration, cell survival, and proliferation of MCs [41]. Binding of SCF to KIT leads to homodimerization of c-Kit by interactions between the Ig-like domains 4/5 of two monomeric KIT receptors. Such interactions pave the way for the consecutive transphosphorylation in the regions “juxtamembrane,” “kinase insert,” “kinase domain,” and finally, “COOH-terminal tail” [40]. Phosphorylated residues act as docking sites for signaling molecules such as Src and Shc kinase, phosphoinositide 3-kinase (PI3K), and phospholipase Cγ (PLCγ). GTP exchanger Sos, PI3K, PLCγ, and JAK2 activate the Ras-Raf-Map kinase (MAPK) cascade which results in Ca2+-influx and activation of transcription factors required for MC activation [42] (Fig. 3). SCF-induced activation of JAK2 in human MCs results in STAT5 and STAT6 activation. STAT5 activation promotes MC development, survival, and proliferation. Interleukin-3 (IL-3) is crucial for MC expansion and activates JAK2, STAT3, and STAT5. Interestingly IL-3 and SCF share overlapping or synergistic functions in MCs because of their concomitant STAT5 activation [43].

Main signaling pathways in mast cell biology include Kit and FcεRI signaling. While the first pathway (in green) is associated with the cell survival and proliferation, the second pathway (in blue) has a key role in the production of the delayed de novo mediators and degranulation of the immediately released preformed mediators. SCF binding to the first three Ig-like domains of extracellular region of KIT induces structural changes resulting in dimerization of two KIT receptors following phosphorylation of intracellular regions and activation of signaling pathways. Crosslinking of IgE-bound allergen triggers the FcεRI signaling pathway (blue) initiates and results in mast cell degranulation
Molecular Mechanisms
Nagata et al. identified a point mutation consisting of a substitution of valine for aspartic acid in the catalytic domain of c-KIT (ASP816VAL or D816V) in the peripheral blood of patients with mastocytosis. A year later, this discovery was followed by identification of the same mutation in CM and aggressive systemic mastocytosis (SM) [42]. Indeed, more than 90% of patients with SM are found to have a somatic gain-of-function mutation in the KIT receptor tyrosine kinase, primarily an aspartic acid to valine substitution (D816V) in the second catalytic domain. This point mutation at molecular level results in enhanced survival and cell autonomous growth of neoplastic MCs [44]. Zappulla et al. generated transgenic mice (by pronuclear injection of the linear Bchm/Asp816Val Kit transgene into fertilized (C57BL/6 DBA2) F2 zygotes) expressing the human D816VKit transgene in MCs to provide a line of evidence for a contribution of D816V mutation in development of mastocytosis. Transgenic mice were harboring a fusion transgene consisting of the 571 bp primate chymase gene (571-bchm) promoter fragment (required for specific expression of the transgene to MCs) in addition to human Kit protooncogene cDNA with the codon 816 Asp → Val substitution. Transgenic and non-transgenic mice were killed at different ages to evaluate the histopathological changes associated with Asp816Val Kit expression. An abnormal accumulation of MCs was accompanied by organ involvement observed in four lines of 12- to 18-month transgenic mice. The spleen as a primary site of MC disease in SM was found to be frequently populated by large MC aggregates in particular within the subcapsular area. Moreover, lymph node and heart involvement were also reported in some transgenic mice. Consistent with the lack of transgene expression in bone marrow, there were no signs of bone marrow involvement. Apparently, this observation was due to use of the chymase promoter, which specifically targets differentiated MCs with expression of mouse MC protease 5. This group of researchers sought to analyze the impact of the Asp816Val Kit mutation on MC proliferation and prepared bone marrow-derived cultured mast cells (BMMCs) from both transgenic and non-transgenic mice. Not surprisingly, only BMMCs from transgenic mice could express the transgene. Transgenic BMMCs could successfully be maintained through continuous cultures for over 24 months. After several months, they became independent of growth factors such as SCF or IL-3. However, exogenous IL-3 was necessary in the initial phases suggesting that, while Asp816Val Kit is necessary for growth factor independent proliferation, other factors are required to confer such ability [45] (Fig. 4). There are other non-specific oncogenic mutations identified recently in patients with mastocytosis including TET2 (TET oncogene family member 2), a putative tumor suppressor gene and N-RAS [5]. Tefferi et al. reported that loss-of-function mutations in TET2 occur at a high frequency in systemic mastocytosis (SM) associated with KITD816V mutations [46]. Although KIT mutations play a key role in the pathogenesis of mastocytosis, they are also the most common additional genetic abnormalities in t(8;21) AML with a reported incidence ranging from 26 to 47% [47]. It must be kept in mind that dysregulation of MC apoptosis plays a key role in the pathogenesis of mastocytosis. Interestingly, upregulation of the antiapoptotic protein Bcl-2 in aggressive mastocytosis and also upregulation of another antiapoptotic protein Bcl-X have been reported in bone marrow of patients with ISM [7]. Most recently, the role of programmed cell death protein-1 (PD-1) has been investigated in mastocytosis. The PD-1 receptor is expressed on T and B lymphocytes, while its ligand (PD-L1) is expressed on tumor cells. In addition to various other tumor cells, PD-L1 has been found to be expressed physiologically on MCs and dendritic cells. It had been shown, previously, that tumor cells evade the immune response by abrogating this signaling [48, 49]. Kuklinski et al. investigated skin biopsies from patients with mastocytosis by immunohistochemistry and reported increased expression of PDL1 in MC proliferations [49]. Interestingly, Kataoka et al. reported the expression of PD-1 receptor in clinical samples of human cutaneous mastocytosis and also a human mastocytosis cell line LAD2. PD-1 is categorized as an inhibitory receptor that contains immunoreceptor tyrosine-based inhibitory (ITIM) motifs within its cytosolic domains. It is thought that activation of such inhibitory receptors recruits the non-receptor protein phosphatases, such as SHP-1 or SHP-2, to the ITIMs. The activation of ITIMs followed by reversing the action of tyrosine kinase cascades results in downregulation of AKT [48]. Recently, signaling molecules involved in KIT signaling have been gaining attention as possible therapeutic targets for mastocytosis. For instance, due to its involvement in KIT signaling, AKT has been described as phosphorylated in patients with KIT D816V+ SM. Similarly, phosphorylated AKT has been reported in the HMC-1 cell line (human KIT D816V+ leukemia MC line) which suggests the involvement of AKT activation in the pathogenesis of mastocytosis [50]. Alterations in KIT mRNA processing have been shown to play a role in SM, as novel KIT transcripts have been detected in aggressive mast cell malignancies [51].

Transgenic mice expressing the human D816VKit transgene in MCs were assessed at different ages for developing SM symptoms and compared with normal non-transgenic mice. They were found to have organ involvement in the spleen, heart, lymph nodes, and stomach but not in the bone marrow. Comparing BMMCs of these two groups of mice revealed that BMMCs from the transgenic ones became independent to SCF for proliferation and activation
Symptoms of Mediator Release
The symptoms exhibited in mastocytosis are the consequences of increased MC numbers in tissues, and the results of increased mediator release which cause both local and distant effects, when distributed via the circulation. Histamine is the most important mediator acting through four receptors, H1–H4, that mediate vasopermeability, vasodilation, and constriction of bronchial and gastrointestinal smooth muscles, enhancing gastric acid production by parietal cells (via H2 receptors), and pruritus [4]. Generally, H1 receptors control the tone and permeability of the vascular bed, the tone of the intestinal and bronchial smooth muscle, mucus production, heart rate, and flushing responses [52]. Histamine-induced itch is triggered by the excitation of a subset of unmyelinated C-fibers. Neuronal H1 receptors participate in the sensation of itch through activation of phospholipase C. Thus, not surprisingly, H1R blockers (antihistamines) are widely used to manage and alleviate itch symptoms [53]. MC-derived histamine by acting through H1 receptors stimulates fibroblast proliferation and collagen synthesis [54]. Elevated serum tryptase and histamine are general findings in patients with mastocytosis. Upon spontaneous activation of tissue infiltrated MCs, released mediators induce different effects on both tissue residing cells and immune cells [52]. Clinically, these pathophysiologic effects can lead to anaphylaxis. H2 receptors control the vascular permeability, gastric acid secretion, and airway mucus production [55]. H4 receptor is involved in mediating pruritus in mice. The intradermal injection of H4 receptor agonist 4-methylhistamine could induce itch in mice. H4 receptors are expressed in the dorsal root ganglion (DRG) neurons of humans and rats, and their mRNAs have been found in the sensory neurons [56]. Patients with mastocytosis show higher incidence of severe anaphylaxis following hymenoptera stings than in the normal population and baseline serum tryptase should be determined in these patients. A value above 11.4 mg/l is often a clinical clue unmasking an underlying mastocytosis and indicates a high risk of very severe anaphylaxis following re-sting [57]. Not only histamine but also other vasoactive substances such as serotonin (5-HT) and substance P provoke flushing [58]. Chymase is potentiating fibrogenesis by activating TGF-β-induced Smad-dependent pathways. Fibrotic changes in the BM, liver, spleen, and lymph nodes in patients with SM could result from MC released IL-13 and TGF-β [52, 59]. A small proportion of patients with ISM suffer from increased IL-6 levels leading to dysgammaglobulinemia with elevated IgG and IgM levels and monoclonal IgG κ in serum electrophoresis. Tissue eosinophilia may be the result of IL-5 release from MCs. Osteoporosis, osteosclerosis, or osteolysis can be observed in patients with mastocytosis and may be mediated by MC-mass itself as well as IL-1β and IL-6 secreted by MCs [52]. Moreover, heparin and MC-proteases play a role in osteoporosis in patients with mastocytosis [52]. MC cytokines, mainly, TGF-β, FGF, and VEGF, may be associated with tissue remodeling by interacting with endothelium, epithelium, fibroblasts, and macrophages. MCs are a source of IL-31 which is known as a potent mediator of itch. IL-31 levels have been reported to be correlated with disease severity, tryptase levels, and percentages of BM infiltration [60]. Histamine, leukotrienes, endothelin, and PAF cause hypotension and swelling through affecting the endothelial walls of vessels. TGF-β, another MC mediator, induces fibrosis in surrounding tissues [4] (Fig. 5).

Features associated with release of MC mediators during mastocytosis at two levels of organ involvement (a) and cell-cytokine interactions of MCs and tissue cells (b)
MCAS
The umbrella term mast cell activation disease (MCAD) comprises the full spectrum of primary systemic MC disease, i.e., systemic mastocytosis (SM) which is further divided into the subtypes primary MC activation syndrome (MCAS) and MC leukemia (MCL) [61]. Aberrant release of mast cell mediators is responsible for related symptoms in mast cell activation disorders. Histamine is responsible for the immediate symptoms including headache, hypotension, pruritus, urticaria, angioedema, diarrhea, and anaphylaxis. Symptoms such as cardiac arrhythmias, myocardial infarction, and hypotension may be attributed to aberrant chymase release. Abdominal cramping, pulmonary edema, urticaria, bronchoconstriction, hypotension, arrhythmia, and anaphylaxis are associated with the release of platelet activating factor (PAF). Prostaglandin D2 is responsible for symptoms such as flushing, mucus secretion, bronchoconstriction, vascular instability, headache, nausea, and abdominal pain [62]. Mast cell activation syndromes (MCAS) are a group of disorders that typically present with symptoms of MC mediator release including itching, flushing, whealing, flaring, angioedema, tachycardia, headache, and gastrointestinal manifestations such as abdominal pain and diarrhea [63]. The proposed diagnostic criteria for MCAS include episodic recurrent symptoms consistent with MC activation in more than one organ; decrease in frequency/severity of symptoms in response to MC mediator therapy such as H1 and H2 antihistamines, leukotrienes, cromolyn, and glucocorticoids; and increased MC activation products mainly tryptase above baseline in at least two symptomatic episodes [64]. Other MC products that can be found above normal during MCAS include heparin and chromogranin A in the blood or histamine and its metabolites (e.g., N-methylhistamine) in the urine [61]. Lack of distinguishing cardinal signs and symptoms makes it hard to clearly distinguish mastocytosis and MCAS. Widespread distribution of MCs and various pattern of aberrant mediator expression result in a great diversity in the clinical presentation of MCAS. Generally, MCAS does not include the entire body, but may involve a specific organ, such as the bladder or GI tract [65]. Unlike SM, urticaria and angioedema are often present in MCAS. MC shape in bone marrow differs in both diseases in which spindled MCs could be seen in mastocytosis while MCs are round and fully granulated in bone marrow specimens obtained from patients with MCAS [64]. In patients with MCAS, hymenoptera stings, alcohol, and heat are the most common triggers of symptoms [62].
Clinical Classification
Categories of mastocytosis include the following:
-
1.
Cutaneous mastocytosis (CM) which is the most frequent form with favorable prognosis and no organ involvement besides the skin.
-
2.
Systemic mastocytosis (SM) characterized by infiltration of MCs in extracutaneous organs such as the spleen, liver [66], and bone marrow [67]. (Fig. 6)

Comparison between CM and SM due to clinical manifestations
According to the 2008 World Health Organization (WHO) classification system, mastocytosis can be classified into several subtypes: (1) cutaneous mastocytosis, (2) extracutaneous mastocytosis, (3) mast cell sarcoma, and (4) systemic mastocytosis (SM). SM can be further subdivided into the following subcategories: (1) indolent systemic mastocytosis (ISM); (2) SM associated with another clonal hematological non-mast cell lineage disease (SM-AHNMD), most commonly chronic myelomonocytic leukemia (CMML); (3) aggressive SM (ASM); and (4) mast cell leukemia [68]. (Table 1) The clinical course of patients with SM is highly variable ranging from frequent indolent to rare highly aggressive variants, affecting multiorgan involvement and overall survival. According to WHO, SM is diagnosed when at least one major and one minor or at least three minor SM criteria are fulfilled [7, 69]. (Table 2) Childhood-onset mastocytosis is usually accompanied by a self-limited course, anaphylaxis frequency rates below 10% and a basal serum tryptase level (BST) of <20 μg/l [70]. Children with typical cutaneous lesions usually do not require bone marrow biopsy. However, this procedure may be considered if hepatosplenomegaly, lymphadenopathy, or peripheral-blood abnormalities are observed [71]. BST levels >20 μg/l in pediatric CM have been shown to reflect extensive skin involvement and the possibility of systemic disease [72]. Increased total BST in the absence of acute MC mediator release has long been documented in ISM [73]. Only the small proportion of children who do not remit spontaneously before puberty will experience transformation into SM. According to the most recent classification of CM, proposed by an international task force involving experts from different organizations, adulthood-onset mastocytosis is characterized by a chronic course with 50% prevalence of anaphylaxis [70]. The presence of small monomorphic maculopapular lesions distributed on thigh and trunk is a key feature of this type of CM. In this classification, the typical tryptase level reported is over 20 μg/l and the location of KIT mutation is within exon 17 (most frequently KIT D816V). In case of the presence of a KIT mutation, it is most frequently localized in exon 8, 9, 11, or 17. Large polymorphic maculopapular cutaneous lesions distributed on trunk, head, and extremities are the dominant clinical feature [70]. One of the most important results of this classification is bridging the size of lesions and the age of development with the persistence as a prognostic marker. In this regard, it is suggested that if a monomorphic variant develops in children, it often persists into adulthood, while the polymorphic variant may resolve around puberty [70]. Familial transmission of mastocytosis has been rarely reported since it usually occurs as a result of spontaneous mutation in the C-kit gene [74]. As one of the exceptions to this rule, Wöhrl et al. demonstrated a familial transmission of a mutation in exon 18 at position 849 (S849I) [75].
Diagnosis
The diagnosis of mastocytosis can be based on the histological examination of a skin biopsy for CM (if necessary and in patients who are suspected clinically in accordance with the recommendation of WHO), and the BM biopsy for the systemic forms according to the recommendation of WHO [76]. The co-expression of KIT and tryptase in MCs in BM makes them easy to detect in histological sections by immunostaining [77]. MCs in SM are characterized by the expression of CD25 and CD2 and an abnormal spindle-shaped hypogranular morphology tending to form clusters around blood vessels and paratrabecular and interstitial areas in the BM [9]. Indolent systemic mastocytosis (ISM) is the least severe systemic variant which is not a life threatening disease [78]. ISM without skin lesions has been frequently reported in those with systemic allergic reactions to hymenoptera venom and raised basal serum tryptase [79]. Gastrointestinal symptoms including abdominal pain, diarrhea, nausea, vomiting, and bloating are found in SM [80]. Up to 28% of patients with SM have peripheral eosinophilia (>650 cells/mm3), and this frequency increases in advanced forms [81]. Neoplastic MCs in ASM and MCL, but not ISM, preferentially express CD30 which correlates with a poor overall prognosis [5]. Additionally, myeloproliferative or myelodysplastic syndromes can be observed in patients with SM [71]. Several different staging investigations need to be performed from the BM in patients with SM including microscopic investigations on smears stained with Wright-Giemsa, histology and immunohistochemistry (IHC), cytogenetics, flow cytometry for documenting the expression of CD2 and/or CD25 on neoplastic MCs, and PCR to detect KIT D816V [3, 82]. Generally, the life expectancy in patients with SM depends on the diagnosed variant in which indolent forms do not shorten life expectancy whereas advanced SM variants, including MCL, SM-AHNMD, and ASM have survival rates ranging from months to a few years despite cytoreductive therapy [83].
Approximately 40% suffer from SM. SM may be associated with other hematologic neoplasms not confined to the MC lineage [84]. For example, Tschandl et al. described in a case report a 61-year-old patient with three diseases occurring synchronously: CMML, xanthogranulomas, and systemic mastocytosis [85]. Coincidence of SM with other diseases such as pulmonary interstitial disease [86], refractory pruritus and cirrhosis [87], and Kounis syndrome [88] has been documented in case reports. New potential biomarkers for predicting episodes of mediator release and monitoring the treatment were described recently. For example, Ehara et al. found the Melanoma inhibitory activity (MIA), an 11-kD protein used as a serum marker for malignant melanoma, to be elevated in children with CM [89]. Not only serum biomarkers but also MC surface markers may be of importance in the diagnosis and classification of the disease. Although co-expression of CD2 and CD25 has been known as a typical feature of neoplastic MCs for several decades, recently also other markers such as CD63, CD69, CD58, CD33, and several complement-associated molecules such as CD11c and CD35 have been found to be overexpressed in these cells. Immunophenotyping of MCs using these markers through flow cytometry revealed that MCs in ASM are of the CD25+CD2−CD63+CD69+ whereas MCs in MCL of the CD25+CD2−CD63−CD69− phenotype [68].
Treatment
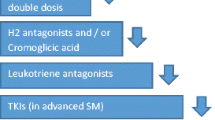
Control of the immediate, possibly severe symptoms is a common component in disease management of mastocytosis regardless of the subtype. For instance, H1-antihistamines are commonly used for the reduction of pruritus and flushing, H2-antihistamines to treat gastrointestinal (GI) symptoms, and corticosteroids and/or analgesics for mitigating bone pain and other symptoms [90]. Cromoglicinic acid is only weakly effective but may act as adjuvant MC stabilizer through reducing the calcium influx for MC degranulation following FcεR1 crosslinking especially for GI symptoms. Furthermore, ketotifen acts in the same way as other H1-antihistamines because of its stabilizing effects. Omalizumab (Xolair®, Novartis), a humanized murine monoclonal antibody with ability to conjugate in vivo with free serum IgE, is able to reduce binding to FCεRI on MCs and basophils and can be used as an additional measure for controlling the immediate symptoms of MC activation [91]. Oral glucocorticoids, through decreasing the number of connective tissue MCs in a dose-dependent fashion, inhibit SCF production and decrease FcεR1 expression and chemokine receptors including CCR3. Acetylic salicylic acid (Aspirin®, Bayer), a non-steroidal antiinflammatory drug (NSAID), has positive effects in patients with mastocytosis by normalizing the levels of PGD2 metabolites through irreversible inhibition of the cyclooxygenase (COX) isoenzymes. Interferon-α (IFN-α) and cladribine (2-CdA) are used in patients with aggressive SM. IFN-α acts through several mechanisms including decreasing MC mediator release, organ infiltration and normalization of serum tryptase level [9]. Imatinib mesylate (Gleevec®, Novartis) is an ATP-competitive, orally bioavailable agent and the only FDA-approved inhibitor of various tyrosine kinases including ABL1, platelet-derived growth factor receptor (PDGFR), ARG, and KIT for use in patients with ASM without D816V KIT or wild type KIT or sporadic KIT mutant isoforms in SM, such as KIT F522C [66, 92]. Moreover, it has therapeutic applications in chronic myelogenous leukemia and gastrointestinal stromal tumors [93, 94]. Other tyrosine kinase inhibitors include Nilotinib (targets Breakpoint Cluster Region-Abelson (BCR-ABL), KIT, and PDGFR), Dasatinib (targets BCR-ABL, SRC, and KIT), Masitinib (multitargeted inhibitor of the KIT, PDGFR, fibroblast growth factor receptor 3, and Lyn tyrosine kinases), and Midostaurin also known as PKC412 (inhibitor of KIT, fms-related tyrosine kinase 3, vascular endothelial growth factor receptor 2, and PDGFR) [66]. Clinical trials are investigating treatment particularly with KIT D816V inhibitors to be approved officially by FDA [95]. Furthermore, narrowband ultraviolet (UVB) phototherapy is an alternative treatment option in patients with CM [96]. Ustun et al. investigated the effectiveness of allogeneic hematopoietic stem-cell transplantation (alloHCT) in patients with advanced SM, who had undergone either sibling or unrelated alloHCT. Three parameters were used to assess response before and after transplantation including the percentage of bone marrow MCs, serum tryptase levels, and organ involvement. The median bone marrow MC percentage in biopsies and the levels of serum tryptase showed a significant decrease after transplantation. They reported that alloHCT can confer long-term overall survival [83]. Baumgartner et al. showed that neoplastic MCs in patients with SM exhibit phosphorylated STAT5 (pSTAT5) in cytoplasm as an essential growth/survival factor, so targeting of pSTAT5 apparently could be an approach in treatment of SM [97]. Moreover, Sharma et al. investigated the role of SHP2/PTPN11 phosphatase in oncogenic KIT signaling using an aggressive SM mouse model. They reported that stable knockdown of SHP2 results in impaired growth, colony formation, and increased rates of apoptosis in mouse mastocytoma cell line P815 harboring KITD814Y mutation [98].
Conclusion
Mastocytosis is a group of rare clonal disorders characterized by abnormal expansion and accumulation of tissue MCs in one or multiple organs. Trafficking patterns of neoplastic MCs within the BM and also augmented angiogenesis in the BM during SM in particular are interesting aspects of this disease and should be addressed in further investigations. The clonal nature of the disease can be established through the demonstration of gain-of-function mutations involving the tyrosine kinase domain of KIT receptor in skin and/or BM cells. The heterogeneity of clinical presentations of mastocytosis relates to the tissue MC burden. There is much variation in the type of skin lesions, the patient’s age at the onset, and associated hematological disorders that—taken together—make the treatment of the disease challenging. The clinical symptoms are mediated by the release of MC mediators. Management of patients within all categories of mastocytosis includes avoidance of triggering factors such as allergen. Additionally, continuous training for the correct application of the rescue self-medication (including self-injectable intramuscular epinephrine and, as warranted, antihistamine and corticosteroids) for patients and children that are at increased risk of anaphylaxis is constantly required. In recent years, various tyrosine kinase inhibitors have also been employed in order to reduce the MC-load for SM. However, currently, there is no approved tyrosine kinase inhibitor to inhibit the D816V c-kit mutation for routine settings. Moreover, most drugs including KIT D816V blocking agents have not demonstrated a promising efficacy in achieving a long-lasting remission in patients with advanced SM.
Abbreviations
- MAdCAM-1:
-
Mucosal addressin cell adhesion molecule-1
- VCAM-1:
-
Vascular cell adhesion molecule-1
- NGF:
-
Nerve growth factor
- SM:
-
Systemic mastocytosis
- CM:
-
Cutaneous mastocytosis
- MC:
-
Mast cell
- SCF:
-
Stem cell factor
- ISM:
-
Indolent systemic mastocytosis
- BST:
-
Baseline serum tryptase
- alloHCT:
-
Allogeneic hematopoietic stem cell transplantation
- TK:
-
Tyrosine kinase
References
Reber LL, Sibilano R, Mukai K, Galli SJ (2015) Potential effector and immunoregulatory functions of mast cells in mucosal immunity. Mucosal Immunol 8(3):444–463. doi:10.1038/mi.2014.131
Ronnberg E, Melo FR, Pejler G (2012) Mast cell proteoglycans. J Histochem Cytochem Off J Histochem Soc 60(12):950–962. doi:10.1369/0022155412458927
Valent P (2013) Mastocytosis: a paradigmatic example of a rare disease with complex biology and pathology. Am J Cancer Res 3(2):159–172
Carter MC, Metcalfe DD, Komarow HD (2014) Mastocytosis. Immunol Allergy Clin N Am 34(1):181–196. doi:10.1016/j.iac.2013.09.001
Magliacane D, Parente R, Triggiani M (2014) Current concepts on diagnosis and treatment of mastocytosis. Translat Med @ UniSa 8:65–74
Kushnir-Sukhov NM, Brittain E, Scott L, Metcalfe DD (2008) Clinical correlates of blood serotonin levels in patients with mastocytosis. Eur J Clin Investig 38(12):953–958. doi:10.1111/j.1365-2362.2008.02047.x
Lange M, Nedoszytko B, Gorska A, Zawrocki A, Sobjanek M, Kozlowski D (2012) Mastocytosis in children and adults: clinical disease heterogeneity. Arch Med Sci 8(3):533–541. doi:10.5114/aoms.2012.29409
Gilfillan AM, Austin SJ, Metcalfe DD (2011) Mast cell biology: introduction and overview. Adv Exp Med Biol 716:2–12. doi:10.1007/978-1-4419-9533-9_1
Cardet JC, Akin C, Lee MJ (2013) Mastocytosis: update on pharmacotherapy and future directions. Expert Opin Pharmacother 14(15):2033–2045. doi:10.1517/14656566.2013.824424
Galli SJ, Starkl P, Marichal T, Tsai M (2016) Mast cells and IgE in defense against venoms: possible “good side” of allergy? Allergology Int Off J Jpn Soc Allergol 65(1):3–15. doi:10.1016/j.alit.2015.09.002
Elieh Ali Komi D, Shafaghat F, Zwiener RD (2017) Immunology of bee venom. Clin Rev Allergy Immunol. doi:10.1007/s12016-017-8597-4
Overed-Sayer C, Rapley L, Mustelin T, Clarke DL (2013) Are mast cells instrumental for fibrotic diseases? Front Pharmacol 4:174. doi:10.3389/fphar.2013.00174
Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, Asadi S, Vasiadi M, Weng Z, Miniati A, Kalogeromitros D (2012) Mast cells and inflammation. Biochim Biophys Acta 1822(1):21–33. doi:10.1016/j.bbadis.2010.12.014
Galli SJ, Tsai M (2008) Mast cells: versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J Dermatol Sci 49(1):7–19. doi:10.1016/j.jdermsci.2007.09.009
Okayama Y, Kawakami T (2006) Development, migration, and survival of mast cells. Immunol Res 34(2):97–115. doi:10.1385/ir:34:2:97
Dahlin JS, Ding Z, Hallgren J (2015) Distinguishing mast cell progenitors from mature mast cells in mice. Stem Cells Dev 24(14):1703–1711. doi:10.1089/scd.2014.0553
Elieh-Ali-Komi D, Cao Y (2016) Role of mast cells in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Rev Allergy Immunol. doi:10.1007/s12016-016-8595-y
Halova I, Draberova L, Draber P (2012) Mast cell chemotaxis—chemoattractants and signaling pathways. Front Immunol 3:119. doi:10.3389/fimmu.2012.00119
Merz H, Kaehler C, Hoefig KP, Branke B, Uckert W, Nadrowitz R, Cerny-Reiterer S, Herrmann H, Feller AC, Valent P (2010) Interleukin-9 (IL-9) and NPM-ALK each generate mast cell hyperplasia as single ‘hit’ and cooperate in producing a mastocytosis-like disease in mice. Oncotarget 1(2):104–119. doi:10.18632/oncotarget.100602
Migalovich-Sheikhet H, Friedman S, Mankuta D, Levi-Schaffer F (2012) Novel identified receptors on mast cells. Front Immunol 3:238. doi:10.3389/fimmu.2012.00238
da Silva EZ, Jamur MC, Oliver C (2014) Mast cell function: a new vision of an old cell. J Histochem Cytochem Off J Histochem Soc 62(10):698–738. doi:10.1369/0022155414545334
Puri N, Roche PA (2008) Mast cells possess distinct secretory granule subsets whose exocytosis is regulated by different SNARE isoforms. Proc Natl Acad Sci U S A 105(7):2580–2585. doi:10.1073/pnas.0707854105
Wasiuk A, de Vries VC, Hartmann K, Roers A, Noelle RJ (2009) Mast cells as regulators of adaptive immunity to tumours. Clin Exp Immunol 155(2):140–146. doi:10.1111/j.1365-2249.2008.03840.x
Voehringer D (2013) Protective and pathological roles of mast cells and basophils. Nat Rev Immunol 13(5):362–375. doi:10.1038/nri3427
Lundequist A, Pejler G (2011) Biological implications of preformed mast cell mediators. Cell Mol Life Sci 68(6):965–975. doi:10.1007/s00018-010-0587-0
Moon TC, Befus AD, Kulka M (2014) Mast cell mediators: their differential release and the secretory pathways involved. Front Immunol 5:569. doi:10.3389/fimmu.2014.00569
Gri G, Frossi B, D'Inca F, Danelli L, Betto E, Mion F, Sibilano R, Pucillo C (2012) Mast cell: an emerging partner in immune interaction. Front Immunol 3:120. doi:10.3389/fimmu.2012.00120
Rigoni A, Colombo MP, Pucillo C (2015) The role of mast cells in molding the tumor microenvironment. Cancer Microenviron Off J Int Cancer Microenviron Soc 8(3):167–176. doi:10.1007/s12307-014-0152-8
Schafer B, Piliponsky AM, Oka T, Song CH, Gerard NP, Gerard C, Tsai M, Kalesnikoff J, Galli SJ (2013) Mast cell anaphylatoxin receptor expression can enhance IgE-dependent skin inflammation in mice. The Journal of allergy and clinical immunology 131(2):541–548. doi:10.1016/j.jaci.2012.05.009
Balseiro-Gomez S, Flores JA, Acosta J, Ramirez-Ponce MP, Ales E (2015) Identification of a new exo-endocytic mechanism triggered by corticotropin-releasing hormone in mast cells. J Immunol (Baltimore, Md: 1950) 195(5):2046–2056. doi:10.4049/jimmunol.1500253
Ciprandi G, Marseglia GL, Castagnoli R, Valsecchi C, Tagliacarne C, Caimmi S, Licari A (2015) From IgE to clinical trials of allergic rhinitis. Expert Rev Clin Immunol 11(12):1321–1333. doi:10.1586/1744666x.2015.1086645
Rivera J, Fierro NA, Olivera A, Suzuki R (2008) New insights on mast cell activation via the high affinity receptor for IgE. Adv Immunol 98:85–120. doi:10.1016/s0065-2776(08)00403-3
Shin JS, Greer AM (2015) The role of FcepsilonRI expressed in dendritic cells and monocytes. Cell Mol Life Sci 72(12):2349–2360. doi:10.1007/s00018-015-1870-x
Ashman LK (1999) The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol 31(10):1037–1051
Geissler EN, Liao M, Brook JD, Martin FH, Zsebo KM, Housman DE, Galli SJ (1991) Stem cell factor (SCF), a novel hematopoietic growth factor and ligand for c-kit tyrosine kinase receptor, maps on human chromosome 12 between 12q14.3 and 12qter. Somat Cell Mol Genet 17(2):207–214
Bai CG, Hou XW, Wang F, Qiu C, Zhu Y, Huang L, Zhao J, Xu JJ, Ma DL (2012) Stem cell factor-mediated wild-type KIT receptor activation is critical for gastrointestinal stromal tumor cell growth. World J Gastroenterol 18(23):2929–2937. doi:10.3748/wjg.v18.i23.2929
Cruse G, Metcalfe DD, Olivera A (2014) Functional deregulation of KIT: link to mast cell proliferative diseases and other neoplasms. Immunol Allergy Clin N Am 34(2):219–237. doi:10.1016/j.iac.2014.01.002
Besmer P, Manova K, Duttlinger R, Huang EJ, Packer A, Gyssler C, Bachvarova RF (1993) The kit-ligand (steel factor) and its receptor c-kit/W: pleiotropic roles in gametogenesis and melanogenesis. Development (Cambridge, England) Supplement:125–137
Moore S, McDiarmid LA, Hughes TP (2000) Stem cell factor and chronic myeloid leukemia CD34+ cells. Leuk Lymphoma 38(3–4):211–220. doi:10.3109/10428190009087013
Lennartsson J, Ronnstrand L (2012) Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev 92(4):1619–1649. doi:10.1152/physrev.00046.2011
Wang X, Ren H, Zhao T, Chen J, Sun W, Sun Y, Ma W, Wang J, Gao C, Gao S, Lang M, Jia L, Hao J (2014) Stem cell factor is a novel independent prognostic biomarker for hepatocellular carcinoma after curative resection. Carcinogenesis 35(10):2283–2290. doi:10.1093/carcin/bgu162
Metcalfe DD (2008) Mast cells and mastocytosis. Blood 112(4):946–956. doi:10.1182/blood-2007-11-078097
Morales JK, Falanga YT, Depcrynski A, Fernando J, Ryan JJ (2010) Mast cell homeostasis and the JAK-STAT pathway. Genes Immun 11(8):599–608. doi:10.1038/gene.2010.35
Chatterjee A, Ghosh J, Kapur R (2015) Mastocytosis: a mutated KIT receptor induced myeloproliferative disorder. Oncotarget 6(21):18250–18264. doi:10.18632/oncotarget.4213
Zappulla JP, Dubreuil P, Desbois S, Letard S, Hamouda NB, Daeron M, Delsol G, Arock M, Liblau RS (2005) Mastocytosis in mice expressing human Kit receptor with the activating Asp816Val mutation. J Exp Med 202(12):1635–1641. doi:10.1084/jem.20050807
Tefferi A, Levine RL, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Finke CM, Mullally A, Li CY, Pardanani A, Gilliland DG (2009) Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia 23(5):900–904. doi:10.1038/leu.2009.37
Pullarkat ST, Pullarkat V, Kroft SH, Wilson CS, Ahsanuddin AN, Mann KP, Thein M, Grody WW, Brynes RK (2009) Systemic mastocytosis associated with t(8;21)(q22;q22) acute myeloid leukemia. J Hematop 2(1):27–33. doi:10.1007/s12308-009-0023-2
Kataoka TR, Fujimoto M, Moriyoshi K, Koyanagi I, Ueshima C, Kono F, Tsuruyama T, Okayama Y, Ra C, Haga H (2013) PD-1 regulates the growth of human mastocytosis cells. Allergol Int Off J Jpn Soc Allergol 62(1):99–104. doi:10.2332/allergolint.12-OA-0450
Kuklinski LF, Kim J (2016) Expression of PD-L1 in mastocytosis. J Am Acad Dermatol 74(5):1010–1012. doi:10.1016/j.jaad.2015.09.029
Bibi S, Arslanhan MD, Langenfeld F, Jeanningros S, Cerny-Reiterer S, Hadzijusufovic E, Tchertanov L, Moriggl R, Valent P, Arock M (2014) Co-operating STAT5 and AKT signaling pathways in chronic myeloid leukemia and mastocytosis: possible new targets of therapy. Haematologica 99(3):417–429. doi:10.3324/haematol.2013.098442
Chan EC, Bai Y, Bandara G, Simakova O, Brittain E, Scott L, Dyer KD, Klion AD, Maric I, Gilfillan AM, Metcalfe DD, Wilson TM (2013) KIT GNNK splice variants: expression in systemic mastocytosis and influence on the activating potential of the D816V mutation in mast cells. Experimental hematology 41(10):870–881.e872. doi:10.1016/j.exphem.2013.05.005
Castells M, Austen KF (2002) Mastocytosis: mediator-related signs and symptoms. International archives of allergy and immunology 127(2):147–152
Shim WS, Oh U (2008) Histamine-induced itch and its relationship with pain. Mol Pain 4:29. doi:10.1186/1744-8069-4-29
Yang L, Murota H, Serada S, Fujimoto M, Kudo A, Naka T, Katayama I (2014) Histamine contributes to tissue remodeling via periostin expression. J Invest Dermatol 134(8):2105–2113. doi:10.1038/jid.2014.120
Castells M (2006) Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin N Am 26(3):465–485. doi:10.1016/j.iac.2006.05.005
Jian T, Yang N, Yang Y, Zhu C, Yuan X, Yu G, Wang C, Wang Z, Shi H, Tang M, He Q, Lan L, Wu G, Tang Z (2016) TRPV1 and PLC participate in histamine H4 receptor-induced itch. Neural Plast 2016:1682972. doi:10.1155/2016/1682972
Bonadonna P, Zanotti R, Muller U (2010) Mastocytosis and insect venom allergy. Curr Opin Allergy Clin Immunol 10(4):347–353. doi:10.1097/ACI.0b013e32833b280c
Hannah-Shmouni F, Stratakis CA, Koch CA (2016) Flushing in (neuro)endocrinology. Rev Endocr Metab Disord 17(3):373–380. doi:10.1007/s11154-016-9394-8
Sukrithan VK, Salamon JN, Berulava G, Sibinga NE, Verma A (2016) Systemic mastocytosis presenting as cardiac tamponade with CD25(+) pericardial mast cells. Clin Case Rep 4(3):279–281. doi:10.1002/ccr3.505
Arock M, Akin C, Hermine O, Valent P (2015) Current treatment options in patients with mastocytosis: status in 2015 and future perspectives. Eur J Haematol 94(6):474–490. doi:10.1111/ejh.12544
Molderings GJ, Haenisch B, Brettner S, Homann J, Menzen M, Dumoulin FL, Panse J, Butterfield J, Afrin LB (2016) Pharmacological treatment options for mast cell activation disease. Naunyn Schmiedeberg's Arch Pharmacol 389(7):671–694. doi:10.1007/s00210-016-1247-1
Picard M, Giavina-Bianchi P, Mezzano V, Castells M (2013) Expanding spectrum of mast cell activation disorders: monoclonal and idiopathic mast cell activation syndromes. Clin Ther 35(5):548–562. doi:10.1016/j.clinthera.2013.04.001
Nurmatov UB, Rhatigan E, Simons FE, Sheikh A (2015) H1-antihistamines for primary mast cell activation syndromes: a systematic review. Allergy 70(9):1052–1061. doi:10.1111/all.12672
Akin C (2015) Mast cell activation syndromes presenting as anaphylaxis. Immunol Allergy Clin N Am 35(2):277–285. doi:10.1016/j.iac.2015.01.010
Ratner V (2015) Mast cell activation syndrome. Transl Androl Urol 4(5):587–588. doi:10.3978/j.issn.2223-4683.2015.09.03
Verstovsek S (2013) Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. Eur J Haematol 90(2):89–98. doi:10.1111/ejh.12043
Fernandes IC, Teixeira Mdos A, Freitas I, Selores M, Alves R, Lima M (2014) Adult mastocytosis: a review of the Santo Antonio Hospital’s experience and an evaluation of World Health Organization criteria for the diagnosis of systemic disease. An Bras Dermatol 89(1):59–66. doi:10.1590/abd1806-4841.20141847
Quintas-Cardama A, Jain N, Verstovsek S (2011) Advances and controversies in the diagnosis, pathogenesis, and treatment of systemic mastocytosis. Cancer 117(24):5439–5449. doi:10.1002/cncr.26256
Sanchez-Munoz L, Alvarez-Twose I, Garcia-Montero AC, Teodosio C, Jara-Acevedo M, Pedreira CE, Matito A, Morgado JM, Sanchez ML, Mollejo M, Gonzalez-de-Olano D, Orfao A, Escribano L (2011) Evaluation of the WHO criteria for the classification of patients with mastocytosis. Mod Pathol Off J US Can Acad Pathol Inc 24(9):1157–1168. doi:10.1038/modpathol.2011.84
Hartmann K, Escribano L, Grattan C, Brockow K, Carter MC, Alvarez-Twose I, Matito A, Broesby-Olsen S, Siebenhaar F, Lange M, Niedoszytko M, Castells M, Oude Elberink JN, Bonadonna P, Zanotti R, Hornick JL, Torrelo A, Grabbe J, Rabenhorst A, Nedoszytko B, Butterfield JH, Gotlib J, Reiter A, Radia D, Hermine O, Sotlar K, George TI, Kristensen TK, Kluin-Nelemans HC, Yavuz S, Hagglund H, Sperr WR, Schwartz LB, Triggiani M, Maurer M, Nilsson G, Horny HP, Arock M, Orfao A, Metcalfe DD, Akin C, Valent P (2016) Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol 137(1):35–45. doi:10.1016/j.jaci.2015.08.034
Theoharides TC, Valent P, Akin C (2015) Mast cells, mastocytosis, and related disorders. N Engl J Med 373(19):1885–1886. doi:10.1056/NEJMc1510021
Alvarez-Twose I, Vano-Galvan S, Sanchez-Munoz L, Morgado JM, Matito A, Torrelo A, Jaen P, Schwartz LB, Orfao A, Escribano L (2012) Increased serum baseline tryptase levels and extensive skin involvement are predictors for the severity of mast cell activation episodes in children with mastocytosis. Allergy 67(6):813–821. doi:10.1111/j.1398-9995.2012.02812.x
Matito A, Morgado JM, Alvarez-Twose I, Sanchez-Munoz L, Pedreira CE, Jara-Acevedo M, Teodosio C, Sanchez-Lopez P, Fernandez-Nunez E, Moreno-Borque R, Garcia-Montero A, Orfao A, Escribano L (2013) Serum tryptase monitoring in indolent systemic mastocytosis: association with disease features and patient outcome. PLoS One 8(10):e76116. doi:10.1371/journal.pone.0076116
Tuysuz G, Ozdemir N, Apak H, Kutlubay Z, Demirkesen C, Celkan T (2015) Childhood mastocytosis: results of a single center. Turk pediatri arsivi 50(2):108–113. doi:10.5152/tpa.2015.2332
Wöhrl S, Moritz KB, Bracher A, Fischer G, Stingl G, Loewe R (2013) A c-kit mutation in exon 18 in familial mastocytosis. J Invest Dermatol 133(3):839–841. doi:10.1038/jid.2012.394
Horny HP, Sotlar K, Valent P, Hartmann K (2008) Mastocytosis: a disease of the hematopoietic stem cell. Dtsch Arztebl Int 105(40):686–692. doi:10.3238/arztebl.2008.0686
Sotlar K, Cerny-Reiterer S, Petat-Dutter K, Hessel H, Berezowska S, Mullauer L, Valent P, Horny HP (2011) Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol Off J US Can Acad Pathol Inc 24(4):585–595. doi:10.1038/modpathol.2010.224
Moura DS, Sultan S, Georgin-Lavialle S, Barete S, Lortholary O, Gaillard R, Hermine O (2012) Evidence for cognitive impairment in mastocytosis: prevalence, features and correlations to depression. PLoS One 7(6):e39468. doi:10.1371/journal.pone.0039468
Zanotti R, Bonadonna P, Bonifacio M, Artuso A, Schena D, Rossini M, Perbellini O, Colarossi S, Chilosi M, Pizzolo G (2011) Isolated bone marrow mastocytosis: an underestimated subvariant of indolent systemic mastocytosis. Haematologica 96(3):482–484. doi:10.3324/haematol.2010.034553
Lee JK, Whittaker SJ, Enns RA, Zetler P (2008) Gastrointestinal manifestations of systemic mastocytosis. World J Gastroenterol 14(45):7005–7008
Kovalszki A, Weller PF (2014) Eosinophilia in mast cell disease. Immunol Allergy Clin N Am 34(2):357–364. doi:10.1016/j.iac.2014.01.013
Morgado JM, Sanchez-Munoz L, Teodosio CG, Jara-Acevedo M, Alvarez-Twose I, Matito A, Fernandez-Nunez E, Garcia-Montero A, Orfao A, Escribano L (2012) Immunophenotyping in systemic mastocytosis diagnosis: ‘CD25 positive’ alone is more informative than the ‘CD25 and/or CD2’ WHO criterion. Mod Pathol Off J US Can Acad Pathol Inc 25(4):516–521. doi:10.1038/modpathol.2011.192
Ustun C, Reiter A, Scott BL, Nakamura R, Damaj G, Kreil S, Shanley R, Hogan WJ, Perales MA, Shore T, Baurmann H, Stuart R, Gruhn B, Doubek M, Hsu JW, Tholouli E, Gromke T, Godley LA, Pagano L, Gilman A, Wagner EM, Shwayder T, Bornhauser M, Papadopoulos EB, Bohm A, Vercellotti G, Van Lint MT, Schmid C, Rabitsch W, Pullarkat V, Legrand F, Yakoub-Agha I, Saber W, Barrett J, Hermine O, Hagglund H, Sperr WR, Popat U, Alyea EP, Devine S, Deeg HJ, Weisdorf D, Akin C, Valent P (2014) Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol Off J Am Soc Clin Oncol 32(29):3264–3274. doi:10.1200/jco.2014.55.2018
Zhrebker L, Cooper B, Krause JR (2014) Systemic mastocytosis with associated acute myelogenous leukemia. Proc (Baylor Univ Med Cent) 27(1):22–24
Tschandl P, Mullauer L, Kittler H (2012) Systemic mastocytosis associated with chronic myelomonocytic leukemia and xanthogranuloma. Dermatology practical & conceptual 2(3):203a203. doi:10.5826/dpc.0203a03
Hermans MA, Broijl A, van Daele PL (2016) A unique presentation of pulmonary disease in advanced systemic mastocytosis, proven by the presence of mast cells in bronchoalveolar lavage: a case report. J Med Case Rep 10(1):283. doi:10.1186/s13256-016-1066-5
Addepally NS, Klair JS, Girotra M, Jones J, Aduli F (2016) Systemic mastocytosis causing refractory pruritus in a liver disease patient. ACG case reports journal 3(4):e152. doi:10.14309/crj.2016.125
Lerner M, Pal RS, Borici-Mazi R (2017) Kounis syndrome and systemic mastocytosis in a 52-year-old man having surgery. CMAJ: Canadian Medical Association journal = journal de l’Association medicale canadienne 189(5):E208–e211. doi:10.1503/cmaj.151314
Ehara Y, Yoshida Y, Tahira M, Yamamoto O (2014) The expression of melanoma inhibitory activity on mast cells in child patients with cutaneous mastocytosis. Yonago Acta Med 57(3):99–101
Hochhaus A, Baccarani M, Giles FJ, le Coutre PD, Muller MC, Reiter A, Santanastasio H, Leung M, Novick S, Kantarjian HM (2015) Nilotinib in patients with systemic mastocytosis: analysis of the phase 2, open-label, single-arm nilotinib registration study. J Cancer Res Clin Oncol 141(11):2047–2060. doi:10.1007/s00432-015-1988-0
Sokol KC, Ghazi A, Kelly BC, Grant JA (2014) Omalizumab as a desensitizing agent and treatment in mastocytosis: a review of the literature and case report. J Allergy Clin Immunol Pract 2(3):266–270. doi:10.1016/j.jaip.2014.03.009
Vega-Ruiz A, Cortes JE, Sever M, Manshouri T, Quintas-Cardama A, Luthra R, Kantarjian HM, Verstovsek S (2009) Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk Res 33(11):1481–1484. doi:10.1016/j.leukres.2008.12.020
Napier RJ, Norris BA, Swimm A, Giver CR, Harris WA, Laval J, Napier BA, Patel G, Crump R, Peng Z, Bornmann W, Pulendran B, Buller RM, Weiss DS, Tirouvanziam R, Waller EK, Kalman D (2015) Low doses of imatinib induce myelopoiesis and enhance host anti-microbial immunity. PLoS Pathog 11(3):e1004770. doi:10.1371/journal.ppat.1004770
Pereira PR, Odashiro AN, Marshall JC, Correa ZM, Belfort R Jr, Burnier MN Jr (2005) The role of c-kit and imatinib mesylate in uveal melanoma. J Carcinog 4:19. doi:10.1186/1477-3163-4-19
Yacoub A, Prochaska L (2016) Ruxolitinib improves symptoms and quality of life in a patient with systemic mastocytosis. Biomarker Res 4:2. doi:10.1186/s40364-016-0056-5
Prignano F, Troiano M, Lotti T (2010) Cutaneous mastocytosis: successful treatment with narrowband ultraviolet B phototherapy. Clin Exp Dermatol 35(8):914–915. doi:10.1111/j.1365-2230.2010.03825.x
Baumgartner C, Cerny-Reiterer S, Sonneck K, Mayerhofer M, Gleixner KV, Fritz R, Kerenyi M, Boudot C, Gouilleux F, Kornfeld JW, Sillaber C, Moriggl R, Valent P (2009) Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: subcellular distribution and role of the transforming oncoprotein KIT D816V. Am J Pathol 175(6):2416–2429. doi:10.2353/ajpath.2009.080953
Sharma N, Everingham S, Zeng LF, Zhang ZY, Kapur R, Craig AW (2014) Oncogenic KIT-induced aggressive systemic mastocytosis requires SHP2/PTPN11 phosphatase for disease progression in mice. Oncotarget 5(15):6130–6141. doi:10.18632/oncotarget.2177
Ferrante G, Scavone V, Muscia MC, Adrignola E, Corsello G, Passalacqua G, La Grutta S (2015) The care pathway for children with urticaria, angioedema, mastocytosis. World Allergy Organ J 8(1):5. doi:10.1186/s40413-014-0052-x
Edwards AM, Capkova S (2011) Oral and topical sodium cromoglicate in the treatment of diffuse cutaneous mastocytosis in an infant. BMJ Case Rep. doi:10.1136/bcr.02.2011.3910
Ryan RJ, Akin C, Castells M, Wills M, Selig MK, Nielsen GP, Ferry JA, Hornick JL (2013) Mast cell sarcoma: a rare and potentially under-recognized diagnostic entity with specific therapeutic implications. Mod Pathol Off J US Can Acad Pathol Inc 26(4):533–543. doi:10.1038/modpathol.2012.199
Acknowledgements
Open access funding provided by Medical University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
I hereby state that none of the coauthors and the corresponding author of this paper have a conflict of interest and it has been prepared for publication without using any funding. Moreover, the paper does not contain any studies with human participants or animals performed by any of the authors.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Komi, D.E.A., Rambasek, T. & Wöhrl, S. Mastocytosis: from a Molecular Point of View. Clinic Rev Allerg Immunol 54, 397–411 (2018). https://doi.org/10.1007/s12016-017-8619-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-017-8619-2