
Abstract
RNA-binding proteins (RBPs) are pivotal for regulating gene expression as they are involved in each step of RNA metabolism. Several RBPs are essential for viable growth and development in mammals. RNA-binding motif 47 (RBM47) is an RRM-containing RBP whose role in mammalian embryonic development is poorly understood yet deemed to be essential since its loss in mouse embryos leads to perinatal lethality. In this study, we attempted to elucidate the significance of RBM47 in cell-fate decisions of mouse embryonic stem cells (mESCs). Downregulation of Rbm47 did not affect mESC maintenance and the cell cycle but perturbed the expression of primitive endoderm (PrE) markers and increased GATA4 + PrE-like cells. However, the PrE misregulation could be reversed by either overexpressing Rbm47 or treating the knockdown mESCs with the inhibitors of FGFR or MEK, suggesting an implication of RBM47 in regulating FGF-ERK signaling. Rbm47 knockdown affected the multi-lineage differentiation potential of mESCs as it regressed teratoma in NSG mice and led to a skewed expression of differentiation markers in serum-induced monolayer differentiation. Further, lineage-specific differentiation revealed that Rbm47 is essential for proper differentiation of mESCs towards neuroectodermal and endodermal fate. Taken together, we assign a hitherto unknown role(s) to RBM47 in a subtle regulation of mESC differentiation.
Graphical Abstract

Similar content being viewed by others


Introduction
Mammalian embryogenesis is initiated with the formation of a totipotent single-cell zygote, which embarks on a complex sequence of events that involve the establishment of diverse cellular types, intercellular interactions, mechanical and chemical cues that guide the patterning of tissues and organs to generate an entire individual [1, 2]. Mechanistic errors and perturbations during these events lead to embryo defects that sometimes prove to be lethal. Pre-implantation stage mammalian embryos have been successfully cultured in vitro and used to study cell dynamics of the early days of mammalian development. However, the precise course of events in post-implantation stages where the basic body plan is laid down remains the black box of embryonic development due to inaccessibility and associated experimental intricacies within the uterus [2].
Recent advances in the in vitro stem cell-derived embryo models have posed an exceptional avenue for recapitulating in vivo events of mammalian embryogenesis, lineage specification, tissue, and organ formation [1, 3]. The most widely used models are embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), which divide clonally and differentiate into all cell types. Recent studies have demonstrated the potential of ESCs to self-assemble and self-organize into embryo-like structures that mimic in vivo embryonic morphogenesis [1, 4,5,6]. In addition to modeling early embryonic development, the genetically modified ESCs are used for developing efficient differentiation protocols to derive clinically relevant cell types, elucidating the significance of novel factors, and understanding the fundamental processes governing pluripotency. Several studies have thus successfully explained transcriptional factors and their networks governing pluripotency [7]. However, there is increasing evidence for the RNA-based regulation of pluripotency and differentiation through the coordinated interplay between RBPs, cellular mRNAs, and non-coding RNAs [8, 9]. As RNA binding proteins (RBPs) are involved in every process of RNA regulation, several recent studies have emphasized their essential functions in the regulation of pluripotency and differentiation [10,11,12,13,14,15,16].
RNA-binding motif 47 (RBM47) is a novel, vertebrate-conserved RBP that contains three RNA-recognition motifs (RRM) that perform multifaceted roles in RNA editing, early embryonic development, and cancer [17]. In zebrafish, the depletion of rbm47 in the embryos led to the upregulation of wnt8a that affected the head formation [18] and homozygous inactivation of Rbm47 in mouse embryos caused perinatal lethality where embryos were lost particularly after the E10 stage due to fetal resorption [19]. In the present study, to understand the function of Rbm47 in ESCs, we interrogated publicly available datasets and found that RBM47 was preferentially enriched in mRNA interactome of mESCs (Supplementary Fig. 1A) [20]. During the fibroblast reprogramming, there was a surge in Rbm47 expression, particularly in the late phase of reprogramming (day 12 - day 15), which remained high in the stable iPSCs as compared with fibroblasts (Supplementary Fig. 1B and 1C) [21, 22]. A recent study identified two late-phase specific alternative splicing events regulated by RBM47 but did not reveal their importance in maintaining the pluripotent state [23]. It was reported previously that RBM47 binds to Nanog mRNA in mouse ESCs, however the outcome of this interaction was not explained [24]. In the present study, we address some of the current gaps in understanding the function of Rbm47 in mammalian embryonic development using mESC-based models (Fig. 1A). Depletion of Rbm47 in mESCs using specific short hairpin RNAs (shRNAs) did not affect the self-renewal and cell cycle regulation as revealed by a lack of change in pluripotency markers; however various primitive endoderm (PrE) makers were upregulated in these shRbm47 mESCs. The PrE bias was rescued in shRbm47 mESCs by treating with MEK inhibitor or FGFR inhibitor, suggesting a role for RBM47 in modulating the FGF-ERK pathway in mESCs. Moreover, the skew towards PrE persisted when these cells were differentiated into multiple lineages by serum treatment, with a compromised neuroectoderm. Consequently, shRbm47 mESCs displayed a significantly regressed teratoma compared to control mESCs (shlacZ). Lineage-specific differentiation experiments revealed that Rbm47 is necessary for proper differentiation into neuroectodermal and endodermal lineages. Together, our findings reveal a novel role for Rbm47 in cell fate decisions during early embryonic development.

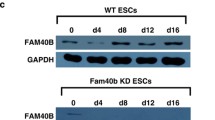
Rbm47 expression is differentially regulated in mESC-based lineage specifications. A Schematic representation of in vitro propagation of stem cells from TE, PrE, and ICM/Epi of a developing blastocyst and their lineage specification potential. TE- trophectoderm, ICM- inner cell mass, PrE- primitive endoderm, Epi- epiblast, TSCs- trophoblast stem cells, ESCs- embryonic stem cell, nEnd- naïve endoderm, cXEN- extra-embryonic endoderm, ExE- extra-embryonic ectoderm, EpiSCs- epliblast stem cells. B RT- qPCR measurement of Rbm47 mRNA levels in indicated cell types (MEF- mouse embryonic fibroblast; mESC- AB2.2 mouse embryonic stem cells). Relative mRNA quantification was performed by normalizing data to Actb, Gapdh, and 18 S rRNA expression from three biological replicates; error bar represents the S.E.M. C Western blot analysis of RBM47 in mentioned cell types. β-Actin (ACTB) was used as the loading control. D Nuclear and cytoplasmic fractionation of mESCs followed by western blotting. GAPDH was used as a cytoplasmic marker and Histone H2A was used as a nuclear marker. E RT-qPCR measurement of Rbm47 mRNA during embryoid body (EB) differentiation at indicated timepoints. Values were normalized to Actb, Gapdh, and 18 S rRNA expression and plotted by assaying three biological replicates. Error bars indicate S.E.M. F Western blot analysis of RBM47 during EB differentiation. Relative RBM47/Total β-Tubulin (TUBB) quantification is represented as mean ± S.E.M from blots with three biological replicates. G RT-qPCR measurement of Rbm47 mRNA in indicated lineages derived from mESCs (EpiLC- epiblast-like cells; cXEN- extra-embryonic endoderm; DE- definitive endoderm, MES- mesoderm, and NE- neuroectoderm). Values were normalized to Actb, Gapdh, and 18 S rRNA expression and relative expression was plotted by assaying three biological replicates. Error bars indicate S.E.M. H Western blot analysis of RBM47 in indicated lineages. Lineage-specific markers were probed to confirm proper differentiation. TUBB was used as the loading control. Relative RBM47/TUBB quantification is represented as mean ± S.E.M from blots with three biological replicates. Statistical test used for B- unpaired student’s t-test ; E, F, G and H- one-way ANOVA followed by Dunnett’s multiple comparison tests, mESC as control sample; ns- non-significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Materials and Methods
Mouse Embryonic Stem Cell Culture
AB 2.2 mESC line was a kind gift from the Wellcome Sanger Institute, Hinxton, UK. mESCs were cultured under feeder-free conditions on 0.2% gelatin as attachment factor in serum-free ESC medium: Knockout DMEM (Cat. no. 10829018) supplemented with 15% Knockout Serum Replacement (KOSR; Invitrogen/10828028), 1x Glutamax (Gibco/35050061), 1x MEM-non-essential amino acids (Gibco/11140050), 0.1 mM 2- Mercaptoethanol (Gibco/21985023), 50U/mL Pen Strep (Gibco/15140122) and 10 ng/mL murine Leukaemia Inhibitory Factor (LIF; Peprotech/AF-250-02). Cells were plated at a density of 2.0-2.5 × 104 live cells per cm2 for routine cultures. The cultures were fed daily and usually passaged on the third day using StemPro Accutase (Gibco/A1110501) as a cell-dissociation agent. All the experiments described in the study were performed using mESCs grown between passages 18 and 30. TaKaRa PCR mycoplasma detection set (cat. no. 6601) was used to screen cultures for mycoplasma contamination regularly. Before lineage-specific differentiation, wild-type ES cells were grown for at least 2 passages in ESC + 2i medium: ESC medium supplemented with 3 μM CHIR99021 (Sigma-Aldrich/SML1046) and 1 μM PD0325901 (Sigma-Aldrich/PZ0162). For FGF4/ERK pathway modulation, ESC medium was supplemented with either DMSO, 1 μM PD0325901, or 100 nM PD173074 (Sigma-Aldrich/341,607). Unless otherwise specified, for all the experiments involving mESCs, the trypan blue-corrected live cell count was considered using countess II automated cell counter (Invitrogen) and a humidified incubator set at 370 C, 5% CO2 was used.
Mouse Embryonic Stem Cell Differentiation
Conversion of mESCs to epiblast-like stem cells (EpiLCs): Briefly, 1 × 105 mESCs were cultured on a fibronectin-coated (15 μg/mL) well of a 6-well plate in ESC + 2i medium. After a day, the ESC + 2i medium was withdrawn and cells were washed with PBS. Cells were allowed to grow in an N2B27 medium supplemented with Activin-A (20 ng/mL) and FGF2 (12 ng/mL) for two more days as previously described [25], before being harvested for RNA and protein extraction.
Conversion of mESCs to extra-embryonic endoderm (cXEN) cells: cXEN cells were generated from mESCs as previously described [26, 27] Briefly, 1 × 105 mESCs were plated onto a gelatin-coated well of a 6-well plate and cultured for 5 days in cXEN derivation medium: RPMI 1640 supplemented with 15% FBS, 0.1 mM 2-Mercaptoethanol, 10ng/mL Activin-A, 0.1 µM all-trans retinoic acid, and 1% Pen Strep. cXEN cells were sub-cultured onto fresh gelatin-coated plates for 2–3 days before being collected for RNA and protein extraction.
Conversion of mESCs to definitive endoderm (DE): mESCs were plated at an initial density of 2 × 104 cells/mL of serum-free endodermal differentiation (SFED) medium to form embryoid bodies (EBs) in a non-treated 6-well dish (Himedia/TPG6-1 × 100NO) as previously described [28]. The composition of SFED medium is as follows: 75% IMDM (Gibco/12440053), 25% Ham’s F12 nutrient solution (Gibco/21700075), 0.5X of both N2 (17502048) and B27 (17504044) supplements, 1% Pen Strep, 0.05% BSA (MP Biomedicals/160069), 1x Glutamax (Gibco/35050061), 0.5 mM Ascorbic acid (MP Biomedicals/100769), and 0.45 mM alpha-Monothioglycerol (MP Biomedicals/155723). The EBs were collected on day 2 by centrifugation at 500 rpm for 3 min followed by gentle dissociation using StemPro Accutase for 2 min at room temperature (100 µL Accutase used for EBs from one well). Dissociated cells were suspended in 2 mL SFED and then harvested by centrifugation at 1000 rpm for 3 min. Next, the cells were reaggregated with SFED supplemented with 50 ng/mL of Activin-A, at twice the volume initially used at day 0, for another two days. The endodermal EBs were harvested on day 4 for RNA and protein and RNA isolation. In an alternative method, mESCs were initially plated at 9 × 103 cells/mL of SFED for EB generation. On day 2, rather dissociating and reaggregating, EBs were collected and resuspended in the same volume of SFED + Activin-A medium (50 ng/mL) for the next two days, before being harvested for analysis.
Conversion of mESCs to mesoderm (MES): mESCs were differentiated into mesoderm as per the protocol stated previously [29, 30]. Briefly, 1 × 105 mESCs were plated per mL of serum-free mesodermal differentiation (SFMD) medium to form EBs in a non-treated 6-well dish (Himedia/TPG6-1 × 100NO). The SFMD was prepared by adding following components: 75% IMDM (Gibco/12440053), 25% Ham’s F12 nutrient solution (Gibco/21700075), 0.05% BSA (MP Biomedicals/160069), 1x Glutamax (Gibco/35050061), 1X B27 supplement without vitamin A (Gibco/12587,010), 1X N2 supplement, 50 µg/mL Ascorbic acid (MP Biomedicals /100769) and 0.45 mM alpha-Monothioglycerol (MP Biomedicals/155723). The EBs were collected on day 2 by centrifugation at 500 rpm for 2 min followed by gentle dissociation using StemPro Accutase for 2 min at room temperature (300 µL Accutase used for EBs from one well). Dissociated cells were suspended in 2 mL SFMD and then harvested by centrifugation at 1000 rpm for 3 min. Next, the cells were reaggregated with SFMD supplemented with human VEGF (5 ng/mL), human Activin-A (5 ng/mL), and human BMP4 (0.25 ng/mL) for another two days to form the mesodermal EBs that were then harvested for RNA and protein and RNA isolation.
Conversion of mESCs to neuroectoderm (NE): Neuroectoderm was derived from mESCs as detailed before [31]. In short, 1 × 105 mESCs were seeded onto 0.2% gelatin-coated 6-well dish and cultured in ESC + 2i medium (wild-type ESCs) or ESC medium (shRNA expressing mESCs) for 16–24 h. Next, the cells were washed with PBS and fed with an N2B27-neuro specific medium (Supplementary Table S3 for recipe) for 6–8 days. The medium was renewed every 2 days.
Differentiation of mESCs into embryoid bodies (EBs): mESCs were plated at a density of 1 × 105 cells/mL on non-treated dishes and aggregated into EBs for six days in the EB medium composed of Knockout DMEM (Cat. no. 10829018), 10% ESC-qualified FBS (Gibco/10439024), 1X Glutamax (Gibco/35050061), 1X MEM-non-essential amino acids (Gibco/11140050), 0.1 mM 2-Mercaptoethanol (Gibco/21985023), and 0.5% Pen Strep. The medium was replenished every alternate day for up to 6 days. Six-day-old EBs were plated onto gelatin-coated dishes and grown for an additional six days with media replenished every alternate day. The cultures were analyzed on day 3, day 6, day 9, and day 12.
Monolayer differentiation of mESCs: mESCs were seeded at a density of 1 × 104 cells/cm2 onto gelatin-coated plates overnight in ESC medium. On the next day, cells were washed with PBS and allowed to differentiate in the EB medium for six days. The medium was replenished every alternate day.
Plasmids, Lentivirus Preparation, and Transduction
Plasmids used for lentivirus preparation were as follows: pLKO.1-TRC cloning vector was a gift from David Root (RRID:Addgene_10878); psPAX2- packaging plasmid & pMD2.G- envelop encoding plasmid were gifts from Didier Trono (RRID: Addgene_12260 and Addgene_12259 respectively). pLKO.1-TRC plasmid was digested with AgeI and EcoRI and the 7 kb fragment was gel purified and quantified. Sequences of shRNA oligos were obtained from The RNAi Consortium (TRC) public portal from Broad Institute (https://portals.broadinstitute.org/gpp/public/). Forward and reverse oligos were reconstituted at a concentration of 0.1nmol/µL and the annealing mixture of 25 µL was prepared by adding 11.25 µL of both the oligos and 2.5 µL 10x annealing buffer (1 M NaCl, 100mM Tris-HCl pH 7.4 or NEB Buffer 2). The annealing mixture was placed in boiling water and gradually cooled to room temperature. The annealing mixture was diluted 100 times using 0.5x annealing buffer. 1 µL of 1:100 diluted mixture along with 10–20 ng of linear pLKO.1-TRC was used for 20 µL ligation reaction using rapid ligation kit (Roche/NEB). Competent E. coli stbl3 strain was transformed using 10 µL ligation mixture. Sequences encoding shRNAs were confirmed by Sanger DNA sequencing using U6 promoter-specific primer. Sequence targeting lacZ (shlacZ: 5’-TCGTATTACAACGTCGTGACT-3’) was used as a non-targeting control. Three shRNAs were used to target Rbm47 in mouse ESCs, however, shRbm47#1 (5’-CCCGCGTTCATACATTTCTAA-3’) and shRbm47#3 (5’-CCGTCCAATAACTCCTGTGTA-3’) showed better knockdown efficiency both at RNA and protein levels and hence considered further. The complete sequence of the oligos used are mentioned in the supplementary Table S1.
Lentiviruses were produced in HEK 293T cells grown in DMEM/F12 supplemented with 10% FBS. These cells were reverse transfected with lentiviral plasmids using lipofectamine 3000 (Invitrogen). DNA mix was prepared by diluting 2.5 µg pLKO.1-shRNA plasmid, 2 µg psPAX2 and 1 µg pMD2.G in Opti-MEM (Gibco) or any serum-free basal medium to a volume of 243 µL and followed by 7 µL P3000 reagent. The lipid mix was prepared in a separate tube by diluting 7 µL Lipofectamine 3000 reagent in 243 µL Opti-MEM. DNA-lipid mix was obtained by dropwise addition of the DNA mix to the lipid mix. The contents were gently mixed with a pipette and incubated inside the hood for 10 min. Meanwhile, HEK 293T cells were dissociated with trypsin, harvested, and re-suspended in the complete medium. DNA-lipid mix along with 5–6 × 106 293T cells were plated per transfection in a 60 mm dish pre-coated with 0.2% gelatin and incubated overnight. The next day, the spent medium was replaced with 3–4 mL fresh complete medium. Supernatants were collected at 24 and 48 h time points and pooled. The pooled supernatant was next passed through a 0.45 μm filter to remove cell debris and concentrated using Lenti-X concentrator (TaKaRa cat. no. 631,231) following the manufacturer’s instructions. The viral pellet was suspended in a 300 µL ESC medium and used for transduction, or aliquots were stored in -80oC.
mESCs were transduced on a 6-well dish at the time of plating with different doses of virus concentrate suspended in ESC medium containing 6 µg/ mL polybrene and incubated for 16–24 h. Cultures were fed with fresh medium and grown for another day. Next, cells were selected with a medium containing 1 µg/ mL puromycin until the mock-transduced cells (no virus) completely died. MOI was determined by counting the virus-transduced and control cells (no virus/no puromycin) as previously described [32]. The polyclonal population that yielded MOI of ~ 1 was used for further studies.
RNA Isolation and Reverse Transcription-Quantitative Real-Time PCR (RT-qPCR)
Total RNA was isolated from all the cell types by adding the appropriate amount of TRIzol Reagent (Cat. No. 15596-018; Ambion, life technologies) following the manufacturer’s protocol. Quantification of isolated total RNA was done using a DeNovix spectrophotometer. Total RNA (0.5 -1.0 µg) was reverse transcribed into cDNA using Verso cDNA synthesis kit (Thermo Scientific). RT-qPCR was set up with two technical repeats per biological sample in a reaction volume of 10 µL containing 2.5 µL of 1:25 diluted cDNA as template, 0.4 µM each forward and reverse primer and PowerUp SYBR green master mix (Applied biosystems A25743) and run in Quantstudio 6 Flex (Thermo Scientific). A four-stage thermocycling protocol (fast cycling mode) was employed. Briefly, stage I (hold) – 2 min at 500 C for activation of UDG, Stage II (hold)- 2 min at 950 C for activation of dual-lock DNA polymerase, stage III (40 cycles)- 950 C melt for 3s and 600 C anneal/extend for 30 s and Stage IV (post-amplification melt curve analysis)- the ramp (auto mode) was initiated from 600 to 950 C. Melting curve was monitored in each run to determine the specificity of amplification. The details of the primers used in the study are mentioned in the supplementary Table S2. Unless otherwise stated, all the data were normalized to multiple endogenous controls- 18 S rRNA, Actb, and Gapdh and relative quantification (RQ) of target gene expression were performed as described previously [33]. RQ plots were represented as mean \(\pm\) S.E.M as described in the figure legends.
Immunocytochemistry
mESCs were plated in 96-well optical-bottom plates (Nunc 165,305) coated with 0.2% gelatin. The spent medium was removed and cells were washed with PBS. Cells were fixed in 4% phosphate-buffered formaldehyde for 15 min at room temperature and washed thrice with PBS. Cells were permeabilized and blocked for 30 min in PBS containing 5% BSA + 0.2% Triton-X. Cells were incubated with primary antibodies (Supplementary Table S3) diluted with antibody dilution buffer (PBS containing 1% BSA + 0.05% Triton-X) for one hour at room temperature. Excess antibodies were removed by rinsing with PBS thrice, cells were incubated with fluorophore-conjugated appropriate secondary antibodies diluted in antibody dilution buffer, washed thrice with PBS and nuclear staining was done with mounting medium containing DAPI and anti-fade agent DABCO (Sigma). Samples were imaged in Thermo Scientific CellInsight CX7 LZR High Content Analysis (HCA) platform using wide-field or confocal applications. Images were processed further using ImageJ software.
Teratoma Formation
In vivo differentiation potential of mESCs stably expressing either shlacZ or shRbm47 was tested by subcutaneously injecting 1 × 106 cells suspended in 100 µL knockout-DMEM into the dorsal flank of 8-week old NOD/SCID mice.
Mice were regularly monitored for teratoma formation and euthanized between 4 and 5 weeks for harvesting teratomas. Teratoma dimensions were recorded using a Vernier caliper and the ellipsoid volume, V= ½ (Length*Width2) was calculated [34]. Teratomas were fixed in 4% formaldehyde in PBS at 40 C overnight, embedded in paraffin and the sections were stained with hematoxylin and eosin for histological analysis.
Electroporation Using Neon Transfection System
For Neon transfections, pCAG-EGFP-N1 (A pEGFP-N1 backbone modified by replacing CMV promoter with CAG promoter) and pCAG-Rbm47-EGFP-N1 plasmid DNAs of high purity were prepared at a concentration of 2–4 µg/µL using Purelink HiPure plasmid midiprep kit (K210005). Briefly, mESCs were dissociated with accutase and harvested. The cells were re-suspended PBS and cell count was determined in Countess automated cell counter. For electroporation in 12w format using Neon 10 µL tip, 1 × 105 cells were suspended gently in buffer R containing 1 µg plasmid DNA to obtain the cell-DNA mix. The Neon tube was filled with 3 mL electrolytic buffer E and placed in the Neon pipette station. The Neon pipette with a 10 µL tip was then immersed in the cell-DNA mix and slowly aspirated. Next, the Neon pipette was inserted into the neon tube with buffer E and electroporated with the following conditions: 1200 V / 10 ms / 3 pulses. Once the electric pulses are completed, transfer the cells into the gelatin pre-coated 12w plate containing pre-warmed ESC medium. The plate was gently rocked to ensure uniform distribution and incubated for 48 h in a humidified 370 C, 5% CO2 incubator, before being used for analysis.
Statistics
Biological replicates in cell culture experiments refer to samples collected from independent experiments. RT-qPCR reactions were set-up with two technical repeats per biological sample. For western blotting experiments, a representative image from independent blotting experiments with three biological replicates is shown. Graph plots and statistical analyses were performed using GraphPad Prism 8.2.1 (441) software. Student’s t-test was employed for comparisons between two groups whereas one-way ANOVA followed by a Dunnett’s multiple comparison test was utilized for comparisons between three or more groups. Each figure legend includes relevant information about precision measures, the number of replicates, and the statistical tests conducted. The statistical significance was defined as follows: p > 0.05 as non-significant (ns), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Rbm47 is Abundantly Expressed in Pluripotent Stem Cells (PSCs) and Localizes to Both Nucleus and Cytoplasm
We found that Rbm47 transcript and protein were abundantly expressed in mESCs as compared with differentiated cells such as mouse embryonic fibroblasts (MEF) (Fig. 1B, C). To determine the cellular localization of RBM47 protein, we fractionated mouse ESCs into cytoplasmic and nuclear extracts and analyzed them by immunoblotting (Fig. 1D). RBM47 was found to be expressed as both, a nuclear and a cytoplasmic RBP with significant enrichment in the nucleus of mESCs, indicating its discrete roles in nuclear and cytoplasmic RNA metabolism. Similar data were obtained with human iPSC lines and human dermal fibroblasts suggesting closely related functions in human counterparts (Supplementary Fig. S1D, E, and F).
Rbm47 Expression Profile in Specific Lineages Derived In Vitro From mESCs
Early mouse development involves the specification of three lineages at the blastocyst stage. As shown in Fig. 1A, the early blastocyst stage (E3.25) has a fully specified outer layer of cells called trophectoderm (TE), while the inner cell mass (ICM) retains pluripotency. In the late blastocyst stage (E4.5), ICM undergoes lineage specification into the pluripotent epiblast (Epi) and primitive endoderm (PrE). As the blastocyst enters the subsequent stages of development, TE gives rise to the extraembryonic ectoderm (ExE), and the PrE gives rise to visceral endoderm surrounding both the Epi and ExE. Epi gives rise to tri-lineages of the embryo proper. Fortunately, the self-renewing ability of these cells enabled us to derive and maintain stem cell lines from each of these lineages and utilize them to study blastocyst development in vitro [3]. ESCs can be derived from both the ICM and Epi and cultured indefinitely in vitro under defined growth conditions. Currently, protocols are available to convert mESCs into naive endoderm (nEnd) or extraembryonic endoderm (EXEn; similar to PrE) and epiblast-like cells (EpiLCs, similar to E5.5 epiblast cells) in addition to the traditional germ layer differentiation of embryo proper.
To explore the function of Rbm47 during early embryonic development using mESCs, we subjected mESCs to various differentiation strategies and analyzed Rbm47 expression. First, we differentiated mESCs as embryoid bodies (EBs), which undergo spontaneous differentiation into the three germ layers and mimic post-implantation embryos in vitro. RT-qPCR profiling revealed a 2–3 fold upregulation of Rbm47 expression in differentiating EBs compared with mESCs (Fig. 1E). In contrast, there was a gradual reduction in RBM47 protein levels as EB differentiation progressed. However, the correlation between protein and mRNA levels was seen only in day-3 EBs (Fig. 1F). Expression of Nanog, a core pluripotency marker, significantly reduced both at mRNA and protein levels, served as an indicator for EB differentiation (Fig. 1E, F).
Next, to test whether Rbm47 expression is subjected to any lineage-specific regulation, we directed mESCs into epiblast-like cells (EpiLCs) and extra-embryonic endoderm (cXEN) cells, that mimic the successive cell fates of inner cell mass in the post-implantation embryos in vitro. Furthermore, mESCs were specified into derivatives of definitive endoderm (DE), mesoderm (MES), and neuroectoderm (NE), which mimic the primitive lineages of the embryo proper. To ensure proper differentiation, all the lineage derivatives were profiled for expression of specific-lineage markers besides monitoring Rbm47 expression (Fig. 1G, H and Supplementary Fig. S2). We observed a differential expression of Rbm47 in EpiLCs and cXEN cells, with mRNA being significantly downregulated in EpiLCs, but upregulated nearly 1.6-fold in cXEN cells (Fig. 1G). RBM47 protein level correlated substantially with mRNA level in EpiLC but not in cXEN as protein level was reduced compared to mESCs (Fig. 1H). We speculated that Rbm47 expression might undergo post-transcriptional regulation in cXEN cells. We assessed the half-life of Rbm47 mRNA by treating mESCs and cXEN cells with alpha-amanitin, followed by the determination of mRNA abundance at different time points. We observed only a slight increase in the half-life of Rbm47 mRNA (mESC t1/2= 10.01 h and cXEN t1/2= 10.83 h) in cXEN cells (Supplementary Fig. S3). To gain insights into Rbm47 expression in the lineages of the embryo proper, we measured its mRNA and protein expression in DE, MES, and NE derived from mESC differentiation (Fig. 1G, H). We found that Rbm47 expression is indeed subjected to lineage-specific regulation, with its expression remained unchanged in DE but compromised largely in MES and NE compared to mESCs.
Taken together these results suggest that RBM47 is abundantly expressed in mESCs and its expression undergoes lineage-specific regulation. As described in previous studies, Rbm47 expression was strongly detected in the endoderm (primitive gut, liver, and pancreas) and ExEn appendages (yolk sac) in E8.5 mouse embryos, but poorly detected in developing tissues of other lineages [19, 35], demonstrating that our expression analysis utilizing mESCs differentiation has fairly recapitulated the in vivo expression patterns.
Rbm47 Depletion Doesn’t Affect In Vitro mESC Maintenance
We used the RNAi approach for loss-of-function studies to investigate the significance of Rbm47 expression in mESCs. We transduced ESCs with lentivirus that expresses shRNAs targeting either Rbm47 mRNA or lacZ mRNA (non-targeting control) and established stable cell lines (Rbm47 depleted mESCs designated as shRbm47#1 and shRbm47#3; control ESCs as shlacZ in the figures). There was an efficient knockdown of Rbm47 at mRNA (~ 80%) as well as protein levels (~ 50%) (Fig. 2A and B). Rbm47-depleted mESCs displayed no apparent change in the undifferentiated state as they expressed similar levels of pluripotency markers as control mESCs as demonstrated by RT-qPCR, western blotting, and immunocytochemistry (Fig. 2E, F, and G). Furthermore, shRbm47 mESCs could be propagated continuously (for at least 15 passages) in the ESC medium supplemented with leukaemia inhibitory factor (LIF) and retained the ability to form colonies. Rbm47 knockdown mESCs had a cell profile that was indistinguishable to control mESCs, as demonstrated by cell cycle analysis (Fig. 2H). In terms of morphology and alkaline phosphatase (ALP) activity, shRbm47#3 mESCs were comparable to control mESCs, whereas shRbm47#1 mESCs showed a slightly diffused morphology with a reduced intensity of ALP staining (Fig. 2C and D). However, these cells display a similar pluripotency marker profile as control mESCs. Overall, our data suggest that Rbm47 is not necessary to maintain the pluripotent state of mESCs.

Rbm47 depleted mESCs are pluripotent and can self-renew. A-B Rbm47 knockdown efficiency by indicated shRNAs at RNA (Error bar represents the S.E.M.) and protein levels from three biological replicates. shlacZ was used as a non-targeting control shRNA. Relative RBM47/TUBB protein quantification is represented as mean ± S.E.M from three independent blots. C Phase-contrast images of shlacZ mESCs and shRbm47 mESCs, scale-100 μm, (Top). Cells were fixed and stained for alkaline phosphatase (ALP) activity, scale-100 μm, (Bottom). D ALP activity was quantified by calculating integrated densities (I.D) of images processed in ImageJ software. Mean integrated densities ± S.E.M from three staining experiments were plotted. E Relative mRNA expression of indicated markers in control and shRbm47 mESCs. Log2 normalized values from three biological replicates were used for heatmap generation. F Western blot analysis of indicated pluripotency markers. Relative quantification is represented as mean ± S.E.M from three independent blots. TUBB was used as the loading control. G Widefield fluorescence images of mESCs stained for pluripotency markers (Scale-100 μm). H Cell cycle profiling of control and shRbm47 mESCs from three biological replicates. The graph represents mean percent population ± S.E.M. Statistical test used for A, B, D, F and H: unpaired student’s t-test; ns- non-significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Downregulation of Rbm47 Increases Primitive Endoderm (PrE)-Like Cells in mESC Culture
Since Rbm47 depletion did not affect the pluripotency and self-renewal of mESCs, we next considered analyzing the expression of differentiation markers specific to PrE, mesendoderm, TE, and neuroectoderm. Previously, it was established that mESC cultures display heterogeneity and comprise a subpopulation of lineage-committed cells [36, 37]. RT-qPCR revealed a significant upregulation of PrE markers Gata6, Gata4, Sox17, Dab2, Pdgfra, and Foxa2 in shRbm47 mESCs as compared with control mESCs (Fig. 3A and B). In agreement to RT-qPCR data, immunostaining with GATA4 antibody revealed that Rbm47 knockdown mESCs had a greater fraction of PrE-like cells (shRbm47#1 mESCs: 6.2% ± 1%; shRbm47#3 mESCs: 6% ± 0.4%) than control mESCs (shlacZ mESCs: 2.2% ± 0.5%) (Fig. 3C and D). The expression of mesendodermal markers remained mostly unchanged, but the expression of a few NE markers (Nestin, Pax6 and Emx1) was downregulated marginally.

Rbm47 depletion primes mESCs towards a PrE fate which can be reversed by FGF-ERK pathway inhibition or Rbm47 overexpression. A Relative mRNA expression of indicated markers in control and shRbm47 mESCs. Log2 normalized values from three biological replicates were used for heatmap generation. B Relative mRNA levels of PrE markers in linear scale, the plotted values represent mean ± S.E.M. C Quantification of GATA4+ cell population in indicated cultures using cell counter plugin of ImageJ software. Mean cell percentage ± S.E.M were plotted by analyzing images from three immunostaining experiments with > 1000 nuclei (DAPI) analyzed per experiment. D Widefield fluorescence images of mESCs immunostained for GATA4 (Scale-100 μm). E Schematic representation of the development of early blastocyst to late blastocyst. Autocrine FGF4-ERK signaling is majorly responsible for the salt-pepper distribution of NANOG+ Epi and GATA6+ PrE specification. F Relative mRNA levels of Gata6 and Gata4 in DMSO treated, PD17 (Pan-FGFR inhibitor) treated and PD032 (MEK1/2 inhibitor) treated control and Rbm47 knockdown mESCs (48 h treatment) from three independent experiments. Error bars indicate S.E.M. Values were normalized to DMSO treated shlacZ mESCs. G Immunostaining of GATA4 in DMSO treated and inhibitor-treated mESCs (Scale-100 μm). H Relative mRNA levels of PrE markers in shRbm47#1 mESCs that were Neon transfected with EGFP control vector and RBM47-EGFP overexpression vector; the plotted values represent mean ± S.E.M from three Neon transfection experiments. I Immunostaining of GATA4 in EGFP and RBM47-EGFP overexpressing shRbm47#1 mESCs (Scale-50 μm). J Quantification of GATA4+ cell population in indicated mESCs using cell counter plugin of ImageJ software. Mean cell percentage ± S.E.M were plotted by analyzing images from three immunostaining experiments with > 1000 nuclei (DAPI) analyzed per experiment. Statistical test used for: B, C, H and J- unpaired student’s t-test; F- ordinary one-way ANOVA followed by Dunnett’s multiple comparison test, DMSO treated cells used as calibrator; ns- non-significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
FGF-ERK Pathway Inhibitors or Overexpression of Rbm47 Can Reverse the PrE Priming in Rbm47 Knockdown mESCs
Since ESCs are in vitro models of the ICM, these cells are routinely cultured in a medium supplemented with leukemia inhibitory factor (LIF). However, they can be converted to a hypomethylated ground state of pluripotency (similar to preimplantation ICM) by culturing in a medium containing inhibitors of GSK3ß and MEK1/2 (termed as ‘2i’) [38]. To determine whether a return to the ground state would reverse Rbm47-depletion induced priming towards the PrE lineage, we cultured control and Rbm47-depleted mESCs for two passages in ‘2i’ supplemented ESC medium and analyzed these for PrE marker expression. We indeed observed that culture condition capturing the ground state was sufficient to reverse the PrE priming in Rbm47-depleted mESCs (Supplementary Fig. S4).
It is well-documented that FGF4-ERK signaling is the central pathway in cell-fate determination of the ICM into NANOG-positive Epi and GATA6-positive PrE at the E4.5 stage of the mouse blastocyst (Fig. 3E). Blocking FGF signaling with inhibitors of FGF receptor (FGFR) and ERK is reported to convert ICM into Epi [39]; in contrast, overactivation of FGF signaling can transform ICM to PrE [40]. To assess the possible effect of Rbm47 depletion in modulating the FGF-ERK pathway, we treated shRbm47 mESCs with PD0325901 (MEK1/2 inhibitor, MEKi) or PD173074 (Pan-FGFR inhibitor, FGFRi) for 48 h and profiled for expression of PrE markers by RT-qPCR and GATA4 + PrE-like cell population by immunostaining. The use of either inhibitor reduced the PrE marker levels significantly, suggesting the involvement of FGF-ERK signaling in increasing PrE-like subpopulation in Rbm47 knockdown mESCs (Fig. 3F and G). Further, to confirm the activation of this pathway, we quantified the expression of Fgfr1, Fgfr2, and Fgf4 mRNAs in Rbm47-depleted mESCs; however, there was no significant change in the mRNA levels of the FGF receptors and the ligand as compared with control cells (Supplementary Fig. S5). Additionally, we did not observe a spike in phospho-ERK1/2 levels in Rbm47-depleted mESCs, a direct measure of FGF signaling (Supplementary Fig. S5).
To overrule any off-target effect of Rbm47 knockdown, we overexpressed Rbm47 in mESCs. We electroporated shRbm47#1 mESCs (we did not use shRbm47#3 as it targets CDS) with pCAG-EGFP-N1 (empty vector) and pCAG-RBM47-EGFP-N1 using Neon transfection system and cultured for 48 h. We then measured the expression of PrE markers in these cells. We found that overexpression of RBM47 in knockdown cells significantly reduced the expression of PrE markers and GATA4 + PrE-like cells, indicating the rescue of the phenotype (Fig. 3H, I and J). Collectively, our findings demonstrate that Rbm47-depleted mESCs displayed upregulated PrE related genes and contained an increased population of GATA4 + cells as compared with control mESCs. Overexpression of Rbm47 in knockdown cells or supplementing them with either 2i, MEKi, or FGFRi could reverse the priming towards PrE, suggesting the involvement of RBM47 in regulating the FGF-ERK pathway in mESCs. However, further determining the direct targets of RBM47 that are associated with the FGF-ERK pathway would provide more insights into its mechanism of action.
Rbm47 Depleted mESCs Do Not Retain a Complete Multi-lineage Differentiation Potential
To evaluate the role Rbm47 on differentiation potential of mESCs in vivo, we xenografted shlacZ and shRbm47 mESCs subcutaneously into NSG mice for teratoma formation. Rbm47-depleted mESCs formed significantly smaller teratomas (mean teratoma volume: shRbm47#1 = 251.63 mm3; shRbm47#3 = 324.6 mm3) as compared with control mESC-derived teratoma (mean teratoma volume: 2081.36 mm3), indicating that Rbm47 might be necessary for self-renewal during the differentiation of mESCs (Fig. 4A and B). We then subjected the harvested teratomas for sectioning and histological observation following HE staining to look for any skews in the formation of primitive tissues from tri-lineage. Despite the notable gross reduction in the size of Rbm47 knockdown teratoma, we did not find any conclusive changes as primitive structures belonging to all three germ layers were identified (Fig. 4C). Because teratoma assays cannot solely explain the multi-lineage differentiation potential of PSCs due to inconsistencies in injection sites and data reporting formats, as well as the difficulty of quantifying in vivo differentiation kinetics, alternative assays in the form of spontaneous and directed in vitro differentiation have been suggested [41]. We employed an in vitro spontaneous differentiation assay where mESCs were differentiated in the presence of serum for six days as monolayer cultures and the expression of many lineage-specific markers was analysed. We observed a consistent upregulation of extraembryonic endoderm (ExEn) markers such as Gata6, Gata4, Pdgfra, Sox17, and Foxa2 in shRbm47 differentiation cultures as shown in Fig. 4D. Likewise, the neuroectoderm precursor markers Nestin, Pax6 and Cdh2 were downregulated; however, the expression of mesodermal markers was inconsistent when compared to control mESC differentiation (Fig. 4D). Together, results from in vitro spontaneous differentiation experiment indicate that Rbm47 knockdown enhances the tendency of mESCs to differentiate into ExEn lineage with compromised neuroectodermal fate, thereby affecting their multi-lineage differentiation capacity.

Rbm47 knockdown compromises the multi-lineage differentiation potential of mESCs. A An image of the teratomas harvested from indicated NOD/SCID mice groups (Number of mice in each group: shlacZ = 6; shRbm47#1 = 6 and shRbm47#3 = 6). B Mean teratoma volumes were calculated for each group and plotted as mean ± S.D. C Representative images of HE-stained sections of teratomas from control and Rbm47 knockdown mESCs displaying structures from three lineages: Mesoderm- osteoid/primitive bone-like (i); Ectoderm- primitive neuroepithelium (ii), a squamous epithelium with keratin pearls (iv); Endoderm- columnar epithelium/secretory glands (iii & v). Scale bar, 20 μm. D RT-qPCR profiling of control and Rbm47 knockdown mESCs differentiated for 6-days as a monolayer culture in a serum-supplemented medium. Mean log2 relative mRNA expression ± S.E.M values were plotted from three biological replicates. Statistical test used for B- unpaired student’s t-test; D- ordinary one-way ANOVA followed by Dunnett’s multiple comparison test; ns- non-significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Lineage-Specific Differentiation Reveals Rbm47 is Essential of Neuroectoderm and Endoderm Fate of mESCs
To further probe the effects of Rbm47 depletion on differentiation, we profiled the expression of lineage-specific markers in shRbm47 and control mESCs differentiated into definitive endoderm (DE), mesoderm (MES), and neuroectoderm (NE). As shown in Fig. 1G, H, Rbm47 expression undergoes lineage-specific modulation, and the mRNA and protein levels were notably remained stable in wild-type mESC-derived DE. To determine whether Rbm47 is essential for DE formation, we plated control and shRbm47 mESCs to form EBs for 2-days in the serum-free formulation and then directed to DE lineage by supplementing the medium with Activin A. We observed that Rbm47-depleted EBs were significantly smaller and irregularly shaped than the control EBs (Fig. 5A and B). RT-qPCR profiling revealed a consistent downregulation of definitive endoderm markers (Sox17, Foxa2, and Hhex) (Fig. 5E), suggesting Rbm47 is necessary for proper differentiation of mESCs to DE.

Rbm47 is essential for neuroectoderm and endoderm differentiation of mESCs. A, C and G Phase-contrast images of indicated mESCs directed to DE, MES, and NE lineages. Scale bar − 200 μm. B and D Mean diameter ± S.E.M of the DE-EBs and MES-EBs were calculated using ImageJ software from images acquired from three independent experiments and compared. Unpaired t-test was used to test significance; ns- non-significant; *p < 0.05; **p < 0.01; ***p < 0.001. E, F,and I RT-qPCR profiling of control and shRbm47 mESCs differentiated into indicated lineage. Mean log2 relative expression ± S.E.M values were plotted from three biological replicates. One-way ANOVA followed by Dunnett’s test was used for comparisons; ns- non-significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. H Immunostaining of control NE and shRbm47 NE with PAX6, TUBB3, and GATA4 antibodies. Widefield fluorescent images were acquired in the ThermoFisher CellInsight-high content screening platform. Scale bar, 500 μm
We next specified Rbm47-depleted mESCs to MES by treating 2-day old EBs with Activin A, VEGF, and BMP4 for two days. A role for Rbm47 in the mesodermal specification was not expected since it was significantly downregulated in wild-type mESC-derived MES (Fig. 1G, H) and undetected in mesodermal tissues embryo and adult mouse [35]. However, Rbm47-depleted mESCs not only formed significantly smaller EBs (Fig. 5C and D) but also displayed enhanced expression of mesodermal markers such as Mixl1, Kdr, Gsc, Mesp1, and Hand1 (Fig. 5F).
On the other hand, the involvement of Rbm47 in neuroectodermal differentiation was indicated since critical NE progenitor markers (Pax6, Nestin, and Cdh2) were suppressed upon Rbm47 knockdown (Fig. 3A and D), we expected to be necessary for proper NE differentiation. As expected, we observed a significant decrease in self-renewal of Rbm47-depleted mESCs on day 2 of NE induction with N2B27-high insulin medium, and the effect persisted throughout the duration (day 8) (Fig. 5G). RT-qPCR analysis of NE-differentiated shRbm47 mESCs revealed a significant downregulation of most NE markers, with a clear bias towards ExEn markers as compared with control mESCs (Fig. 5I). Immunostaining of these cultures with NE markers PAX6 and TUBB3 (B-3-Tubulin) and ExEn marker GATA4 correlated with the RT-qPCR data (Fig. 5H). Collectively, these findings from the lineage-specific differentiation models successfully demonstrated that Rbm47 is essential for fine-tuning the cell-fate decisions and lineage specification of mESCs. Rbm47 knockdown strongly affects the DE and NE differentiation programs.
Discussion
RBM47 is a multifunctional RNA-binding protein conserved in vertebrates, which has been suggested to be essential for development, C to U RNA editing, and tumor suppression [17]. Amongst these, previous investigations have clearly defined the role of RBM47 as a tumor suppressor and as a cofactor for APOBEC1-mediated C to U RNA editing in mammals [35, 42]. However, its role in mammalian development is poorly elucidated. In mouse embryos, Fossat et al. showed that one functional allele of Rbm47 is necessary for viability and postnatal growth of the embryo proper [19]. Inactivation of both Rbm47 alleles resulted in loss of embryos due to fetal resorption after mid-gestation.
The present study used mouse ESCs and lineage-specific differentiation methods as in vitro tools to understand the principal causes for perinatal lethality in Rbm47-inactivated mouse embryos. Profiling of Rbm47 expression in mESC-derived lineages indicated downregulation of both Rbm47 mRNA and protein in EpiLCs, while in cXEN cells, mRNA level increased significantly, but protein level did not correlate with mRNA, suggesting a possibility post-transcriptional/translational regulation in ExEn derivatives that needs further exploration. Differentiation of mESCs to lineages of embryo proper revealed that Rbm47 expression remained high in definitive endoderm but downregulated in mesoderm and neuroectoderm derivatives. In earlier studies with E8.5 embryos, Rbm47 mRNA was strongly expressed in endoderm and ExEn appendages like foregut and yolk sac but weakly detected in other developing tissues [35], indicating that our study using mESCs indeed recapitulated in vivo expression patterns. Furthermore, this spatial expression also correlated with specific adult mouse tissues as this protein was highly detected in the small intestine, pancreas, and liver but undetected in the brain, heart, and skeletal muscles [35]. The Genotype-Tissue Expression (GTEx) data ([43], accessed on 2021-08-13) suggests that this tissue-specific expression pattern is conserved for human RBM47.
By generating Rbm47 knockdown mESCs, our study demonstrated that insufficient levels of RBM47 did not affect mESC maintenance as the cells displayed a similar profile for pluripotency markers and cell cycle. However, profiling these cells for lineage-specific differentiation markers indicated a dysregulation of PrE lineage marker expression. It is well-known that mESC cultures are heterogeneous and express a small fraction of lineage-committed metastable cells [36, 37]. We observed that Rbm47 depletion induced a significant increase in GATA4 + PrE-like cells. There is clear evidence that FGF4-ERK signaling is the central pathway that converts pluripotent Epi (NANOG+) to PrE (GATA6/GATA4+) in developing embryos [39, 40]. We thus tested whether this pathway is implicated in Rbm47 depleted mESCs. These cells indeed were responsive to FGFRi, or MEKi supplemented medium and displayed reduced PrE markers and GATA4 + cells in culture upon treatment. Further, overexpression of Rbm47 in these cells rescued them from PrE bias by significantly reducing the levels of PrE-related mRNAs and GATA4 + PrE-like cells, asserting the implication of RBM47 in regulating FGF-ERK pathway in mESCs. To understand this implication we searched RBP-RNA interactome databases like ATtRACT [44] and RBPDB (http://rbpdb.ccbr.utoronto.ca/) to find potential targets of RBM47 that are associated with the FGF-ERK pathway. However, there is no study conducted yet to determine the targets in silico. Since there is a lack of clear information on the specific binding motifs that RBM47 recognizes on RNA, a de novo motif discovery approach is required to identify targets associated with developmentally crucial pathways such as the FGF-ERK signaling, which will be a step ahead in unraveling the mechanisms of action of RBM47 [17].
Since Rbm47 knockdown did not affect mESC maintenance in vitro, we sought whether its depletion would affect multi-lineage differentiation potential. Although Rbm47 knockdown severely reduced the gross size of teratomas, there were no overt skews among lineages and the knockdown teratomas contained primitive tissues from all three lineages. It is possible that the abnormalities or skews were not conclusively defined as we did not obtain knockdown teratoma of similar size and maturation as control. Nevertheless, in vitro multi-lineage differentiation experiments hinted that Rbm47 depletion affects multi-lineage potential of mESCs. Knockdown cells showed a predisposition towards ExEn lineage with an increased number of GATA4 + ExEn-like cells but affected neuroectodermal fate by decreasing the generation of PAX6 + neural progenitor cells. Further, by applying a definitive endoderm conversion protocol, we proved the relevance of Rbm47 in the endoderm. As it is expressed mainly in endodermal lineages in vivo, we found that a sufficient level of RBM47 is necessary for endoderm cell-fate choice. Conversely, mesoderm conversion revealed that Rbm47 knockdown could enhance the expression of mesodermal markers, suggesting its crucial role in fine-tuning cell fate decisions during mESC differentiation.
Our study effectively recapitulated the significance of RBM47 in early mammalian development using mESCs. We demonstrated its critical role in mESC differentiation into neuroectoderm and endoderm lineages. Further, in vivo studies are required to thoroughly understand the underlying aberrations that led to the fetal resorption and perinatal lethality due to Rbm47 inactivation. Although our study is limited to deciphering its role in cell-fate decisions of mESCs, these additional insights would open new avenues for future investigations. RBM47 is a classical RNA-binding protein known to exert its function by regulating cellular mRNAs post-transcriptionally in various biological contexts. It is a novel regulator of alternative splicing besides stabilizing mRNA by binding to 3’-UTRs. Earlier studies have reported that undifferentiated ESCs possess a surplus of free ribosomes and subunits but are relatively poor in polysome abundance [45]. However, ESCs display a rapid surge in translation rate correlated with increased polysome activity in response to differentiation cues. Consequently, ESCs contain a reserve of lineage-related mRNAs that are not translated but are poised for a rapid translational leap. We believe RBM47 might play an essential role in regulating the quality of this mRNA pool and fine-tunes ES cell-fate decisions to the differentiation milieu. We envisage future studies that determine the target mRNA network of RBM47 in ESCs for a better understanding of critical molecular events it regulates during early embryonic development.
Data Availability
The manuscript and its supplementary information file contain all the relevant data generated during this study.
References
Rossant, J., & Tam, P. P. L. (2021). Opportunities and challenges with stem cell-based embryo models. Stem Cell Reports, 16, 1031–1038. https://doi.org/10.1016/j.stemcr.2021.02.002
Shahbazi, M. N., & Zernicka-Goetz, M. (2018). Deconstructing and reconstructing the mouse and human early embryo. Nature Cell Biology, 20, 878–887. https://doi.org/10.1038/s41556-018-0144-x
Zylicz, J. J. (2020). Defined stem cell culture conditions to model mouse blastocyst development. Current Protocols in Stem Cell Biology, 52, 1–11. https://doi.org/10.1002/cpsc.105
Veenvliet, J. V., Bolondi, A., Kretzmer, H., et al. (2020). Mouse embryonic stem cells self-organize into trunk-like structures with neural tube and somites. Science (80-) 370. https://doi.org/10.1126/science.aba4937
Rivron, N. C., Frias-Aldeguer, J., Vrij, E. J., et al. (2018). Blastocyst-like structures generated solely from stem cells. Nature, 557(7703), 106–111. https://doi.org/10.1038/s41586-018-0051-0
Sahu, S., & Sharan, S. K. (2020). Translating embryogenesis to generate organoids: novel approaches to personalized medicine. iScience, 23, 101485. https://doi.org/10.1016/j.isci.2020.101485
Orkin, S. H., & Hochedlinger, K. (2011). Chromatin connections to pluripotency and cellular reprogramming. Cell, 145, 835–850. https://doi.org/10.1016/J.CELL.2011.05.019
Wright, J. E., & Ciosk, R. (2013). RNA-based regulation of pluripotency. Trends in Genetics, 29, 99–107. https://doi.org/10.1016/J.TIG.2012.10.007
Corsini, N. S., Peer, A. M., Moeseneder, P., et al. (2018). Coordinated control of mRNA and rRNA processing controls embryonic stem cell pluripotency and differentiation. Cell Stem Cell, 22, 543–558. https://doi.org/10.1016/J.STEM.2018.03.002/ATTACHMENT/30C9A0AE-E728-4A09-9CE0-C689C7C233E1/MMC4.XLSX
Fagoonee, S., Bearzi, C., Di Cunto, F., et al. (2013). The RNA binding protein ESRP1 fine-tunes the expression of pluripotency-related factors in mouse embryonic stem cells. PLoS One, 8, e72300. https://doi.org/10.1371/JOURNAL.PONE.0072300
Han, H., Irimia, M., Ross, P. J., et al. (2013). MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature, 498, 241–245. https://doi.org/10.1038/NATURE12270
Elatmani, H., Dormoy-Raclet, V., Dubus, P., et al. (2011). The RNA-binding protein Unr prevents mouse embryonic stem cells differentiation toward the primitive endoderm lineage. Stem Cells, 29, 1504–1516. https://doi.org/10.1002/STEM.712
Dvir, S., Argoetti, A., Lesnik, C., et al. (2021). Uncovering the RNA-binding protein landscape in the pluripotency network of human embryonic stem cells. Cell Reports, 35, 109198. https://doi.org/10.1016/J.CELREP.2021.109198
Li, D., Kishta, M. S., & Wang, J. (2020). Regulation of pluripotency and reprogramming by RNA binding proteins. Current Topics in Developmental Biology, 138, 113. https://doi.org/10.1016/BS.CTDB.2020.01.003
Ye, J., Jin, H., Pankov, A., et al. (2017). NF45 and NF90/NF110 coordinately regulate ESC pluripotency and differentiation. RNA, 23, 1270–1284. https://doi.org/10.1261/RNA.061499.117/-/DC1
Chen, G., Zhang, D., Zhang, L., et al. (2018). RBM14 is indispensable for pluripotency maintenance and mesoderm development of mouse embryonic stem cells. Biochemical and Biophysical Research Communications, 501, 259–265. https://doi.org/10.1016/J.BBRC.2018.04.231
Shivalingappa, P. K. M., Sharma, V., Shiras, A., & Bapat, S. A. (2021). RNA binding motif 47 (RBM47): emerging roles in vertebrate development, RNA editing and cancer. Molecular and Cellular Biochemistry, 2021, 1–13. https://doi.org/10.1007/S11010-021-04256-5
Guan, R., El-Rass, S., Spillane, D., et al. (2013). rbm47, a novel RNA binding protein, regulates zebrafish head development. Developmental Dynamics, 242, 1395–1404. https://doi.org/10.1002/dvdy.24039
Fossat, N., Radziewic, T., Jones, V., et al. (2016). Conditional restoration and inactivation of Rbm47 reveal its tissue-context requirement for viability and growth. Genesis, 54, 115–122. https://doi.org/10.1002/dvg.22920
Kwon, S. C., Yi, H., Eichelbaum, K., et al. (2013). The RNA-binding protein repertoire of embryonic stem cells. Nature Structural & Molecular Biology, 20, 1122–1130. https://doi.org/10.1038/nsmb.2638
Hansson, J., Rafiee, M. R., Reiland, S., et al. (2012). Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Reports, 2, 1579–1592. https://doi.org/10.1016/J.CELREP.2012.10.014/ATTACHMENT/BD2B3958-D120-410A-A770-B7621D69ED8A/MMC6.XLSX
Polo, J. M., Anderssen, E., Walsh, R. M., et al. (2012). A molecular roadmap of reprogramming somatic cells into iPS cells. Cell, 151, 1617–1632. https://doi.org/10.1016/j.cell.2012.11.039
Cieply, B., Park, J. W., Nakauka-Ddamba, A., et al. (2016). Multiphasic and dynamic changes in alternative splicing during induction of pluripotency are coordinated by numerous RNA-binding proteins. Cell Reports, 15, 247–255. https://doi.org/10.1016/j.celrep.2016.03.025
Yeganeh, M., Seyedjafari, E., Kamrani, F. A., & Ghaemi, N. (2013). RNA-binding protein Rbm47 binds to Nanog in mouse embryonic stem cells. Molecular Biology Reports, 40, 4391–4396. https://doi.org/10.1007/s11033-013-2528-0
Samanta, M., & Kalantry, S. (2020). Generating primed pluripotent epiblast stem cells: A methodology chapter. Current Topics in Developmental Biology, 138, 139–174. https://doi.org/10.1016/bs.ctdb.2020.01.005
Ngondo, R. P., Cirera-Salinas, D., Yu, J., et al. (2018). Argonaute 2 is required for extra-embryonic endoderm differentiation of mouse embryonic stem cells. Stem Cell Reports, 10, 461–476. https://doi.org/10.1016/j.stemcr.2017.12.023
Niakan, K. K., Schrode, N., Cho, L. T. Y., & Hadjantonakis, A. K. (2013). Derivation of extraembryonic endoderm stem (XEN) cells from mouse embryos and embryonic stem cells. Nature Protocols, 8, 1028–1041. https://doi.org/10.1038/nprot.2013.049
Gouon-Evans, V., Boussemart, L., Gadue, P., et al. (2006). BMP-4 is required for hepatic specification of mouse embryonic stem cell-derived definitive endoderm. Nature Biotechnology, 24, 1402–1411. https://doi.org/10.1038/nbt1258
Kattman, S. J., Witty, A. D., Gagliardi, M., et al. (2011). Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell, 8, 228–240. https://doi.org/10.1016/J.STEM.2010.12.008
Klattenhoff, C. A., Scheuermann, J. C., Surface, L. E., et al. (2013). Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell, 152, 570–583. https://doi.org/10.1016/J.CELL.2013.01.003
Ying, Q. L., Stavridis, M., Griffiths, D., et al. (2003). Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nature Biotechnology, 21, 183–186. https://doi.org/10.1038/nbt780
Wiederschain, D. The “all-in-one”system for the inducible expression of shRNA. https://media.addgene.org/data/41/67/165920fc-af64-11e0-90fe-003048dd6500.pdf
Taylor, S. C., Nadeau, K., Abbasi, M., et al. (2019). The ultimate qPCR experiment: Producing publication quality, reproducible data the first time. Trends Biotechnol, 37, 761–774. https://doi.org/10.1016/J.TIBTECH.2018.12.002
Tomayko, M. M., & Reynolds, C. P. (1989). Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 1989, 243 24, 148–154. https://doi.org/10.1007/BF00300234
Fossat, N., Tourle, K., Radziewic, T., et al. (2014). C to U RNA editing mediated by APOBEC 1 requires RNA -binding protein RBM 47. Embo Reports, 15, 903–910. https://doi.org/10.15252/embr.201438450
Hayashi, K., Lopes, S. M. C., de Tang, S., & Surani, F. (2008). Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell, 3, 391–401. https://doi.org/10.1016/J.STEM.2008.07.027/ATTACHMENT/062F78BA-63D5-4437-9C98-39F447B153FB/MMC1.PDF
Toyooka, Y., Shimosato, D., Murakami, K., et al. (2008). Identification and characterization of subpopulations in undifferentiated ES cell culture. Development, 135, 909–918. https://doi.org/10.1242/DEV.017400
Ying, Q. L., Wray, J., Nichols, J., et al. (2008). The ground state of embryonic stem cell self-renewal. Nature, 453, 519–523. https://doi.org/10.1038/nature06968
Nichols, J., Silva, J., Roode, M., & Smith, A. (2009). Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development, 136, 3215–3222. https://doi.org/10.1242/DEV.038893
Yamanaka, Y., Lanner, F., & Rossant, J. (2010). FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development, 137, 715–724. https://doi.org/10.1242/DEV.043471
Buta, C., David, R., Dressel, R., et al. (2013). Reconsidering pluripotency tests: Do we still need teratoma assays? Stem Cell Res, 11, 552–562. https://doi.org/10.1016/J.SCR.2013.03.001
Vanharanta, S., Marney, C. B., Shu, W., et al. (2014). Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. Elife 2014. https://doi.org/10.7554/eLife.02734.001
GTEx Portal. https://gtexportal.org/home/. Accessed 13 Aug 2021
Giudice, G., Sánchez-Cabo, F., Torroja, C., & Lara-Pezzi, E. (2016). ATtRACT—a database of RNA-binding proteins and associated motifs. Database, 2016, 35. https://doi.org/10.1093/DATABASE/BAW035
Sampath, P., Pritchard, D. K., Pabon, L., et al. (2008). A hierarchical network controls protein translation during murine embryonic stem cell self-renewal and differentiation. Cell Stem Cell, 2, 448–460. https://doi.org/10.1016/J.STEM.2008.03.013
Acknowledgements
We dedicate this research paper in the loving memory of our colleague the late Dr. Anjali Shiras, who unfortunately succumbed to COVID-19 on 2nd October 2020. We sincerely thank Prof. Alan Bradley & Wellcome Sanger Institute for providing AB2.2 mouse ESCs. We acknowledge Dr. Akaram Bagal, for his comments on teratoma sections. We thank all the central facilities of NCCS and their staff for the support during the investigation.
Funding
This work was supported by the Department of Biotechnology, Government of India (BT/PR15178/MED/31/313/2015) and intramural funds from National Centre for Cell Science (NCCS). Junior and senior research fellowships were granted to: PKMS by University Grants Commission; DKS by Department of Biotechnology; VS by Indian Council of Medical Research. VA was supported by project grant BT/PR 10708.
Author information
Authors and Affiliations
Contributions
Conceptualization: SAB, AS & PKMS; Funding acquisition: AS; Supervision: SAB & AS; Methodology: SAB, AS & PKMS; Investigation and formal analysis: PKMS (ES cell culture & experiments, RNA isolation, RT-qPCR, western blotting, immunostaining, molecular cloning) DKS (RNA isolation, RT-qPCR, cell cycle & western botting), VS (RNA isolation & RT-qPCR) & VA (RT-qPCR & plasmid screening); Writing - original draft preparation: PKMS and SAB; Writing - review and editing: SAB, PKMS. All authors commented on previous versions of the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical Approval
Experiments on pluripotent stem cells (ESCs/iPSCs) were in compliance with the National Guidelines for Stem Cell Research (2013 & 2017) and ethical approval was granted by NCCS Institutional Committee for Stem Cell Research (ID: NCCS/IC-SCR/2016-I/3). Animal experiments were carried out as per CPCSEA guidelines and ethical approval was granted by the Institute’s Animal Ethics Committee (ID: EAF/2015/B-253 & EAF/2019/B-351). Recombinant DNA technology used in the study was in compliance with Guidelines for Biosafety from Department of Biotechnology and ethical approval was granted by the Institutional Biosafety Committee (ID: 2015033 & 2018111).
Conflict of Interest
The authors have no financial or proprietary interests in any material discussed in this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1.69 MB)
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shivalingappa, P.K.M., Singh, D.K., Sharma, V. et al. RBM47 is a Critical Regulator of Mouse Embryonic Stem Cell Differentiation. Stem Cell Rev and Rep 19, 475–490 (2023). https://doi.org/10.1007/s12015-022-10441-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-022-10441-w