
Abstract
The neural crest gives rise to progeny as diverse as peripheral neurons, myelinating cells, cranial muscle, bone and cartilage tissues, and melanocytes. Neural crest derivation encompasses complex morphological change, including epithelial-to-mesenchymal transition (EMT) and migration to the eventual target locations throughout the body. Neural crest cultures derived from stem cells provide an attractive source for developmental studies in human model systems, of immediate biomedical relevance for neurocristopathies, neural cancer biology and regenerative medicine, if only appropriate markers for lineage and cell type definition and quality control criteria were available. Implementing a defined, scalable protocol to generate neural crest cells from embryonic stem cells, we identify stage-defining cluster-of-differentiation (CD) surface markers during human neural crest development in vitro. Acquisition of increasingly mesenchymal phenotype was characterized by absence of neuroepithelial stemness markers (CD15, CD133, CD49f) and by decrease of CD57 and CD24. Increased per-cell-expression of CD29, CD44 and CD73 correlated with established EMT markers as determined by immunofluorescence and immunoblot analysis. The further development towards migratory neural crest was associated with decreased CD24, CD49f (ITGA6) and CD57 (HNK1) versus an enhanced CD49d (ITGA4), CD49e (ITGA5) and CD51/CD61 (ITGAV/ITGB3) expression. Notably, a shift from CD57 to CD51/CD61 was identified as a sensitive surrogate surface indicator of EMT in neural crest in vitro development. The reported changes in glycan epitope and integrin surface expression may prove useful for elucidating neural crest stemness, EMT progression and malignancies.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The neural crest (NCR) represents a unique transient population of cells specific to vertebrate ontogenesis [1,2,3,4]. Thought to arise from the neural plate border during neural tube closure, it comprises a transient multipotent population of developmental progenitor cells [1, 2]. In animal model systems, pre-migratory NCR cells transiently reside within the dorsal neural tube, then undergo epithelial-to-mesenchymal transition (EMT) and migrate toward various regions throughout the embryo. NCR derivatives include the peripheral nervous system, craniofacial skeleton, melanocytes, Schwann cells and the enteric nervous system, making NCR cells a particularly attractive and relevant candidate source for applications in regenerative medicine [5,6,7,8]. While an isolation of early primary NCR cells from the endogenous environment in humans is hampered by technical as well as ethical issues [9,10,11], pluripotent stem cell (PSC) systems may provide a prolific and standardizable source for human NCR cells [2, 12,13,14]. Moreover, PSCs offer in vitro tools to investigate these otherwise difficult-to-study early developmental processes in a human model. In vitro culture systems from human PSCs aim to mimic developmental inputs to generate/induce neural cell progeny [15]. Several protocols have become available to generate NCR cells from human PSCs (for review, see [2]). However, most of these either utilize incompletely defined culture conditions [16,17,18,19] or growth as non-adherent cultures and complex cell sorting steps [20,21,22,23]. For therapeutic applicability, NCR cell preparations and other NCR-derived advanced therapy medicinal products (ATMPs) will have to be well defined, standardized and safe for clinical use [13, 14]. Such protocols would greatly profit from a clear definition of NCR differentiation stages according to characteristic, phenotype-specific markers quantifiable in live cells [24,25,26]. Accordingly, a chemically defined method has recently been developed to generate NCR [27]. A better characterization of NCR cell stages will be equally critical for developing clinical cell transplantation paradigms [28, 29], as well as for utilizing in vitro models of NCR-related human disease [30]. Beyond transcriptomic marker analysis [4, 31], surface markers have not yet systematically been characterized or reported. Empirically, since early attempts at human PSC NCR differentiation, surface epitopes have been used to enrich NCR cells, e.g. via fluorescence-activated cell sorting (FACS), from the otherwise heterogeneous differentiation protocols [32] or to separate these from murine stromal co-culture layers and other cellular contaminants. To characterize hematopoietic [33] or mesenchymal stem cell therapeutics [34], rather established and extensive panels of CD surface molecules are routinely applied in clinical cell therapy applications as selection and/or quality control (QC) markers. For NCR(-like) stem cells, the markers applied have been rather simplistic and comparably not well-resolved: most commonly, as also used in classic NCR animal models, the low-affinity nerve growth receptor (LNGFR; neurotrophin receptor p75) CD271 antigen has been utilized as a NCR surface marker in human pluripotent stem cell-derived NCR cell preparations [17, 32] – disregarding its overall limited specificity due to co-expression on neural progenitor cells [35], mature neurons [36,37,38] as well as mesenchymal stem cells [39, 40]. In conjunction with CD271, the CD90 epitope (THY-1) has been used in NC characterization or isolation, while also being expressed on neurons [38, 41] as well as mesenchymal stem cells. As another commonly used label, the human natural killer-1 (HNK1) epitope (CD57; LEU7) is known to be expressed in human, rat and chick (but supposedly not mouse) NCR [42, 43]. However, CD57 is also expressed on striated muscle [44], natural killer (NK) and T-cell subsets [45], underlining how additional markers and marker combinations are required to uniquely identify and better characterize NCR differentiation stages. For example, in PSC cultures β1-integrin (ITGB1, CD29) has been found to be highly expressed in NCR-like “contaminants” in central nervous system-targeting neuronal cell therapeutic preparations [24]. In line with these observations, CD29 as well as the α4-integrin subunit epitope CD49d (ITGA4) have been found to be highly expressed upon initiation of human PSC-derived NCR differentiation [46]. More recently, we identified the transferrin receptor-1 (TFR1, CD71) as an exploitable candidate also for the elimination of NCR cells from human PSC-derived neuronal cell preparations in the differentiation paradigm applied, however, with limited specificity due to its almost ubiquitous expression on other non-neuronal cell types [38]. These data illustrate that a more systematic analysis of NCR marker expression dynamics and combinatorial expression patterns may be warranted. Here, we set out to characterize NCR cells derived from human PSCs according to cluster-of-differentiation (CD) antigens. Based on previous work [17, 46] we share an easy-to-implement, defined feeder-free protocol for the derivation of NCR cells from human embryonic and induced pluripotent stem cells, largely via modulating cellular plating densities. Moreover, we report a detailed signature of cluster-of-differentiation (CD) markers associated with NCR induction, neural EMT and acquisition of mesenchymal features, which will facilitate NCR stage-specific monitoring, quality control and quantitative analysis and may thereby aid in devising clinical-grade NCR cell preparations.
Materials and Methods
Human PSC Propagation
H9 (WA-09) human embryonic stem cells were obtained from WiCell (Madison, WI) and, after initial feeder-based expansion on a layer of mitotically inactive human fibroblasts (D551), routinely cultured under feeder-free conditions on Synthemax II-SC substrate (Corning)-coated cell culture plates (as described previously [46]).
Human PSC Differentiation to Neuronal and Neural Crest Lineage
For neural induction, H9 embryonic stem cells were harvested and plated (at 0 days in vitro; DIV0) onto Synthemax II-SC (Corning) coated wells at a density of 200,000 cells/cm2 in E8 medium supplemented with 10 µM Y-27,632 (Sigma). Neural induction was initiated on DIV1 by Neural Induction Medium 1 (NIM1) consisting of E8 medium (Thermo Fisher Scientific) supplemented with 10 µM SB431542 (Tocris) and 1 µM Dorsomorphin (Tocris). Cells were maintained in NIM1 (100%) with daily media changes for a total of 5 days. On DIV6, medium was changed to a 3:1 (part-to-part ratio) combination of NIM1 (75%) and NIM2 (25%; comprising DMEM/F-12 supplemented with 1x N2 supplement, 10 µM SB431542 and 1 µM Dorsomorphin). On DIV7 and DIV8, media were changed to a 1:1 (50/50%) and 1:3 (25/75%) ratio of NIM1:NIM2 respectively. From DIV9 until the end of neural induction (DIV12), the cells were maintained in NIM2 medium (100%) with daily media changes. The resulting high-density neural stem cells (NSCs) were then harvested and plated onto PO/laminin-coated wells in NSC medium comprising DMEM/F-12 supplemented with N2 supplement, 20 ng/ml FGF-2, 20 ng/ml EGF (Peprotech) for maintenance and (for subsequent differentiation towards central nervous system-type neurons or) to direct them towards NCR lineage. The NSCs generated at DIV12 were plated at a high-density (500,000 cells/cm2) for expansion. These NSCs were passaged for up to four times. For neuronal differentiation, P4 NSCs were plated at 200,000 cells/cm2 in differentiation medium comprising Neurobasal medium with 1% B27 supplement (both Thermo Fisher Scientific), 20 ng/ml Brain derived neurotrophic factor (BDNF, Peprotech), 20 ng/ml GDNF, 200 µM Ascorbic acid (Sigma), 0.5 mM cAMP and 2mM L-glutamine (adapted and modified from [47]) for 14 days. For differentiation to NCR lineage, the NSCs at DIV12 were plated at low-density (20,000 cells/cm2) condition and passaged as such for up to nine passages in NSC medium. 2 µM Thiazovivin (Selleck Chemicals) was added to the culture medium at each passaging step.
Differentiation to Peripheral Neurons, Adipocytes, Chondrocytes and Osteocytes
For peripheral neural differentiation, early to mid-stage NCR cells (P2 – P5) were plated on PO/Laminin-coated cover slips at 200,000 cells/cm2 and treated for 14 days with differentiation media DMEM/F-12 supplemented with N2 supplement, 10 ng/ml BDNF, 200 µM Ascorbic acid, 10 ng/ml GDNF, 10 ng/ml Nerve growth factor (Peprotech), 10 ng/ml Neurotrophin 3 (Peprotech) and 0.5 mM cAMP (adapted from [21]). Fresh medium was added to the cells every other day. Late-stage NCR cultures (P6 – P9) were plated on 0.1% gelatin-coated plates for adipocyte (10,000 cells/cm2), osteocyte (5000 cells/cm2) and chondrocyte (droplet plated with 80,000 cells/ 10 µl droplet) culture. Differentiation was carried out using the StemPro Adipogenesis, Osteogenesis and Chondrogenesis (Thermo Fisher Scientific) kits according to the manufacturer’s instructions. Cultures were stained after 3 to 4 weeks using oil red O, alizarin red S and alcian blue for the presence of adipocytes, osteocytes and chondrocytes, respectively.
Immunoblot Analysis
Harvesting for experimental analysis was done by enzymatic digestion using TrypLE Express (Thermo Fisher Scientific) and centrifugation at 2000 g, followed by a wash with 1x PBS (without Ca++ and Mg++, Thermo Fisher Scientific). The resulting pellet was stored at -20 °C or -80 °C for immunoblotting. Cell pellets were lyzed using 3x Sample buffer made up of 1 M Tris HCl (pH 6.8), 15% SDS and 15% Glycerol, boiled at 95°C for 10 min and centrifuged for 10 min. Proteins were size-separated using SDS gel electrophoresis (Mini-PROTEAN TGX precast gels, 4–20% gradient, Biorad) followed by transfer to a PVDF membrane (Merck Millipore). Membranes were incubated for 1 h at room temperature in blocking buffer made up of 4% skimmed milk powder (Fluka) in PBS with 0.2% Tween-20 (AppliChem) followed by incubation with primary antibody diluted in blocking buffer overnight at 4°C. Unbound antibody was washed off using 3 washes with PBST (PBS with 0.2% Tween-20), followed by incubation with HRP-conjugated secondary antibodies for 1 h at room temperature and three washes. Membranes were developed using Amersham ECL Prime western blotting detection reagent (GE Healthcare) or WesternBright Sirius HRP substrate (Advansta) and chemiluminiscence was imaged using ImageQuant LAS 4000 (GE Healthcare) and bands were quantified by ImageJ software. Details of antibodies used are given in Table 1.
Immunocytochemistry
Cells were fixed using 4% paraformaldehyde solution for 30 min, followed by permeabilization in 0.5% Triton X-100 (Sigma) solution for 10 min, and blocking in Ca++/Mg++-free DPBS-buffered blocking solution containing 10% normal donkey serum (Millipore) and 1% bovine serum albumin for 30 min. Cells were then incubated with primary antibody diluted in blocking solution overnight at 4°C. Cells were washed 3 times using DPBS, followed by incubation with secondary antibody (1:500 in blocking solution, Alexa Fluor-488 and − 568 conjugated secondary antibodies; Thermo Fisher Scientific) for 30 min at room temperature. Nuclei were counterstained using Hoechst (1:10,000, Thermo Fisher Scientific) added to the secondary antibody solution. Stained cells were washed and mounted onto glass slides using ProLong Gold or ProLong Diamond (both Thermo Fisher Scientific) antifade mountant. Images were acquired using AxioImager-M2 and AxioPlan-2 fluorescence microscopes and analyzed using Zen Blue software (Zeiss). Details and concentrations of all primary antibodies used for immunocytochemistry are given in Table 1.
Flow Cytometric Analysis
Cells harvested using TrypLE were re-suspended to a concentration of 0.5 to 2 × 106 cells/ml in flow cytometry (FC) buffer made up of DPBS (Ca++ and Mg++-free, Gibco) and 2% FBS [48,49,50]. 100 µl of cell suspension was used for each staining sample and 2 µl of fluorophore-conjugated primary CD antibodies were added prior to 30 min incubation on a shaker protected from light. After 3 washes using FC buffer for 3 min at 376 g on a centrifuge maintained at 4°C, the resulting pellet was re-suspended in 100 µl of FC buffer and analyzed using a BD Accuri C6 (BD Biosciences) benchtop flow cytometer and corresponding software. Details on antibodies used for flow cytometry are given in Table 2. Live population gates, doublet exclusion and determination of percent positivity was done as described before [48, 49]. Co-expression and Spearman correlation was performed via a combined and integrated analysis across NCR differentiation, including P0, P3, P6, P9 as well as P3, P4, P5, P6 experimental series.
Results
Generation of Neural Crest Cells from Human Pluripotent Stem Cells
We developed and employed an easy-to-use, fully defined protocol (Fig. 1a) for the generation of NCR cells versus central nervous system neurons from a common rosette forming neural stem/ progenitor cell pool primarily via manipulating the cellular plating density. High-density (200,000 cells/cm2) neuronal differentiation conditions resulted in the sequential acquisition of typical morphological features from pluripotent cell colonies to polarized neuroepithelial stem cells capable of forming rosette-like structures and towards process-bearing neurons (Fig. 1b). Comparing the surface marker profile of NSCs (passage 1) versus 14 days of neuronal differentiation, we observed a downregulation in markers associated with proliferation and neural stemness (CD15, CD49f, CD184) and an upregulation of markers associated with neuronal phenotype (CD24 and CD200high), in keeping with prior studies. The efficacy of neuronal differentiation was most notably underlined by the significant enrichment (11.8-fold) of the neuronal subset defined by the known surface marker code CD49f−/200+ in neuronal differentiation (ND) compared to NSCs (Fig. 1c and Suppl. Fig. 1). In contrast, using the same original neuroepithelial progenitor pool as starting material, upon adjusting cell plating densities to ten-fold lower levels (20,000 cells/cm2), phenotypic changes towards increasingly mesenchymal morphology with high cytoplasmic-nuclear ratio were observed over the course of propagation and passaging time in vitro (Fig. 1d). Through consecutive rounds of passaging, three distinct stages could be morphologically categorized. Cells at early low-density passages (P0 – P2) were smaller with little cytoplasmic rim and partially still polarized morphology. Mid passages (P3 – P5) showed larger spreading, flat cell NCR morphology, while late stage cells (P6 – P9) displayed increasingly mesenchymal phenotypes.

Generation of neural crest cells from human embryonic stem cell-derived neural progenitors. (a) Schematic outlining the steps for generation of neuronal (ND) and different stages of neural crest (NCR) progeny (NCR precursors, NCPs; migratory NCR; increasingly mesenchymal stem cell-like cells, MSCs) from hPSCs. (b) Phase contrast images of hESCs (H9), NSCs (P0) and neurons (ND) after 14 days of differentiation, depicting morphology of the respective stages (Scale bars: 100 μm). (c) Flow cytometric analysis revealing the presence of a population exhibiting the surface marker code CD49f− /200+ underlining that the neuronal fraction is significantly enriched in ND compared to NSCs (also see [48] and Suppl. Fig. 1). (d) Phase contrast images of various passages of NCR differentiation, illustrating the morphology of P0-P2 NCR, P3-P5 NCR and late stage (P6-P9) mesenchymal passaging stages (Scale bars: 50 μm)
Neural Crest Stage Phenotypic Characterization and Multipotency
NCR (P3 – P5) and mesenchymal stem cell-like (P6 – P9) phenotypes generated in this low density-based protocol were differentiated into brain-specific homeobox/POU domain protein 3 A (BRN3A) and microtubule-associated protein-2 (MAP2)-positive peripheral neurons (Fig. 2a), oil red O-positive adipocytes (Fig. 2b), alizarin red-positive osteocytes (Fig. 2c) and alcian blue-positive chondrocytes (Fig. 2d), thus confirming NCR multipotency and lineage differentiation potential. Protein expression analysis by immunoblot (Fig. 2e) showed the expression of neuroepithelium-associated SOX2 and the bona fide EMT activator ZEB1 at P0, thus consistent with pre-migratory neural phenotypes. EMT-associated TWIST1, SNAI1, SLUG (SNAI2) and migratory NCR-associated HNK1 (CD57) were most closely associated with P3, while the post-migratory marker TFAP2A (AP2-α) and the NCR and mesenchymal marker CD29 (ITGB1) were strongly upregulated over the course of passages P6 and P9, thus confirming the sequential presence of pre-migratory (P0 – P2), EMT/ early migratory NCR (P3 – P5) and increasingly mesenchymal stem cell-like (P6-P9) populations during the course of differentiation in low-density culture conditions.

Protein expression changes and epithelial-to-mesenchymal (EMT) markers in neural crest differentiation. (a) Multipotency of generated NCR cells (P5 to P9) was confirmed by its ability to differentiate towards peripheral neuronal lineage (co-expressing BRN3A and MAP2), as well as into (b) adipocyte (stained positive for oil red O), (c) osteocyte (stained positive for alizarin red) and (d) chondrocyte phenotypes (stained positive for alcian blue). (e) Immunoblot analysis comparing the expression of neural stemness (SOX2, ZEB1), EMT (TWIST1, SNAI1, SLUG/SNAI2, HNK1) and NCR and MSC-related proteins (ITGB1/CD29; TFAP2A/AP2α) at neural stem cell (NSC) stage and across different passages of NCR cell culture (NCR P0 to P9)
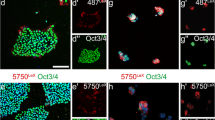
In agreement with the immunoblot data, the microscopical protein expression analyses of these cells using immunocytochemistry (Fig. 3a) for doublecortin, SOX2, HNK1, CD44 and vimentin confirmed the sequential changes of phenotypes from still polarized, stem cell marker-bearing NCR precursors towards mesenchymal phenotypes over time in vitro (P0 – P9). Clearly, P3 stood out as a transitional stage, where NSC-like HNK1 expression was jointly present with early CD44 and vimentin, while SOX2 and doublecortin were clearly already downregulated. Based on morphological and protein expression data (see Figs. 1, 2 and 3), passages P3 to P5 were designated as the stages most closely representing neuroepithelial-to-mesenchymal transitions in this protocol. To better understand these stages at the level of surface marker signatures enabling quantitative live-cell monitoring of intermediate developmental stages in culture, we next performed a flow cytometric analyses of the various passages of NCR cell cultures (P0, P3, P6 and P9) using stage-specific cluster of differentiation (CD) markers.

Lineage marker analysis confirms the presence of distinct developmental stages in long-term neural crest culture. (a) Immunocytochemical analysis of different passages throughout NCR differentiation (P0, P3, P6 and P9) for neural progenitors (DCX and SOX2), NCR (HNK1/CD57) and mesenchymal cells (CD44, Vimentin) confirms the presence of clearly distinct phenotypic stages in long-term NCR culture in congruence with morphological changes (Scale bar: 50 μm)
Surface Marker Profiles of Neural Crest Differentiation
In accordance with categories identified by morphology and analysis based on established marker proteins by immunoblot and immunofluorescence, flow cytometric analyses revealed three clearly distinct stages in terms of CD molecule expression patterns (Fig. 4a, b). Over the course of early passages (P0 – P3) markers associated with stemness and neuronal phenotype were progressively downregulated. Specifically, CD15 (Lewis-X antigen, SSEA-1), a known neuroepithelial marker decreased from 22.7 ± 3.1% at P0 to close to 5.1 ± 2.8% in P3 NCR (while expressed at ca. 60% in NSCs also see Suppl. Fig. 1). Similarly, CD49f (ITGA6), previously identifed by our group on PSC-derived NSCs was reduced from almost ubiquitous expression at P0 (> 95.3 ± 1.9%) to ca. 45.9 ± 26.0% at P3. In contrast, CD133 (prominin-1) a widely used stem cell marker in various tissues throughout the body including NCR-derived tumors remained highly expressed without significant reduction from P0 to P3 (77.1 ± 5.6% to 70.1 ± 12.2%, respectively). Notably, CD271 (p75/LNGFR) as a rather commonly used marker of NCR phenotype was consistently expressed at stable levels throughout NCR in vitro expansion, without significant change. CD24, present on NSCs and neurons similarly continued to be expressed on the cell surface, as was the established putative NCR marker HNK1 (CD57) at this stage, prior to a considerable downregulation from P3 to P6. In contrast to these previous glycosylated epitopes, CD49d and CD51/CD61 surface antigens were significantly upregulated over the course of NCR differentiation. In addition to overall percentual changes, a number of surface molecules enhanced per-cell levels of expression over the course of the later passages in vitro, reflected by the increased presence of the CD29high, CD44high, CD73high and CD90high populations (enhanced mean fluorescence intensity; Fig. 4b).

Surface marker expression changes in PSC-derived neural crest development. (a) Flow cytometric characterization of surface markers across four different stages of NCR differentiation (Passages 0, 3, 6 and 9) shows the temporal changes in surface marker expression as the cells progress along NCR differentiation. Error bars represent arithmetic mean ± SD (n = 3; CD90high at P9 n = 2). (b) Figure depicts the arbitrary threshold (indicated by red gate line) set and consistently applied across the various passages of NCR differentiation to identify CD29dhigh, CD44high, CD73high and CD90high cells. (c) Heatmap representation of CD antigen levels (percentage of expression) in relation to established intracellular NCR and EMT protein expression (lines of squares indicating passage no. at P0, P3, P6, P9; also see Fig. 2e and Suppl. Fig. 2), illustrating a critical transition stage around P3 and association of CD markers with either NSC (left, top) or post-EMT (right, bottom) phenotypes. (d) Time-dependent correlation of percentage of CD57 versus CD51/CD61 expression underlining a gradual decrease of HNK1 accompanied by a gain of CD51/61 expression when focusing on P3 to P6 as a critical transition period (left panel), with the clearest drop occuring between P3 to P4 (right panel). (e) Spearman correlation matrix of flow cytometric expression data across NCR differentiation (percentage positivity at P0, P3, P4, P5, P6, P9) highlights the largest negative correlation of surface antigen expression of CD57 vs. CD51/61 (–0.89; see Suppl. Table 1) over the course of NCR EMT
By surface marker analysis, NCR EMT could thus be defined by the respective expression patterns of initially losing neural stemness (CD15, CD49f, CD133). Subsequently, more profoundly, pre-migratory neuroepithelial features defined by other glycan epitopes (CD24, CD57) were lost, followed by enhanced expression of migration-promoting integrin subunits (CD29, CD49d, CD51/CD61) and, eventually, increasingly mesenchymal surface molecular features (CD44, CD90; CD73high; see Fig. 4c). Additional CD expression studies at a greater temporal resolution of the critical P3 to P6 period confirmed a shift from glycan epitopes such as CD24 and to CD57 towards CD49e and CD51/CD61 integrins (Fig. 4c-e and Suppl. Fig. 2). Finally, an integrated correlation analysis across NCR differentiation (including P0, P3, P4, P5, P6, P9 data points) showed that the loss of CD57 and occurrence of CD51/CD61 was the marker shift most closely associated with and most sensitive for detecting the phenotypic transition occurring at this stage (NCR EMT): identifying clearly distinct clusters in bivariate expression analysis of these markers over time (Fig. 4d), and also showing the clearest expression disparity in NCR development across all passages (Spearman correlation coefficient of − 0.89; Fig. 4e, see Suppl. Table 1).
Discussion
The ability of human PSCs to differentiate to derivatives of all three germ layers has proven to be a valuable tool for elucidating human neural development and for modeling diseases in vitro [51, 52]. Here, we report a simplified, scalable protocol for the generation of NCR lineage from human PSC-derived neural progenitors, based on our understanding of the cell surface-mediated biological processes that modulate human neural development [2, 4, 46, 53]. Previous studies described the generation of neuroepithelial precursors from human PSCs via dual SMAD inhibition: the synergistic action of the two small molecules noggin and SB431542 have long been known to produce mostly PAX6+ neuroectodermal progenitors with high efficiency in adherent culture conditions. At low cell density, however, the same neural induction conditions were reported to result in a mixture of PAX6+ and p75+/HNK1+ NCR cells [54]. In line with this, another report showed that the plating density of PSA-NCAM+ neural precursor cells determined their differentiation propensity, with lower density favoring cells of NCR character [22]. Further, our own research implicated the cell density-dependent Hippo/YAP-TAZ signaling pathway as a critical regulator of NCR development [46]. In the protocol applied here, we show that rosette-forming neuroepithelial precursors can be successfully maintained as NSCs and subsequently differentiated to neurons when plated at a high density (200,000 cells/cm2). On the other hand, at low density (20,000 cells/cm2), they may be efficiently differentiated to various stages of NCR lineage (see [46]). In addition to cost-efficiency and scalability, a key feature of this current protocol is that the cells generated can emulate various stages of NCR development. Upon repeated passaging at low density conditions, three distinct sequential phases could be discerned – an initial pre-migratory neuroepithelial NCR precursor stage (exhibiting SOX1, ZEB1; P0 – P2); an EMT-phase NCR stage with migratory properties and positivity for HNK1, SNAI1, SLUG/SNAI2 and TWIST (P3 – P5); and a later migratory mesenchymal stem cell-like stage positive for CD73, CD90, CD44, CD49d, CD29 and CD51/61 (P6 – P9).
In congruence with our previous work [24, 48] and other prior studies [55, 56] P0 NCR cells still bear neuroepithelial markers (CD15, CD49f, CD133 [57, 58]) on their surface as well as by immunocytochemical analysis (e.g., SOX2, ZEB1). Other groups have shown that SOX2 is a hallmark of pre-migratory NCR cells [59] and that SOX2 downregulation is sufficient to induce and control EMT. Moreover, ZEB1 expression may be associated with maintaining neural stemness [60, 61]. On the other hand, low levels of p75/CD271 [21, 62] and HNK1/CD57 are also expressed here, in conjunction with ZEB1/TWIST1/AP2A and thereby already clearly distinct from NSC phenotype. In comparison to the preceding NSC stage, according to surface markers, this stage (P0 NCR) is characterized by maintained expression of stemness markers CD49f and CD133, the bona fide pan-neural marker CD24 as well as low-level positivity for CD90 paired with increasing levels of CD57 and CD49d. Notably, CD133 (a stem cell marker in various tissue systems including neural stem cells [58]) has previously been used to isolate stem cells of NCR origin [55]. Thus, in the stem cell system studied here, P0 NCR, represents an early pre-migratory NCR precursor stage that is distinguishable from NSC and later NCR stages (beyond P3) by co-expression of CD24, CD49f and CD133 with a combinatorial expression of CD57. The clear morphological and overall phenotypic change occurring with P3 NCR is accompanied by acquisition of more prominent vimentin and CD44 expression as well as a profound upregulation of the known EMT activators ZEB1, TWIST and SLUG [59, 61, 63, 64]. While overall CD44 expression is clearly increased, an excessive CD44 expression per cell (CD44high) has not yet been reached at this stage. In addition to own prior work [24], CD44 has recently been implicated as a marker for NCR stem-like cells [65]. Positivity for CD24, CD49d and negativity for CD49f and CD15 characterize this NCR transitional stage (pre-migratory to EMT; also see [46]). As is also the case in vivo, SNAI1 expression (P0, P3) precedes CD51/CD61 induction (at P6) [66].
The mesenchymal migratory NCR phenotype is consolidated over P6 to P9 NCR, reflected by loss of CD24, CD133 and CD57 and acquisition of CD29high and CD44high eventually across the entire cell population. Notably, also increased per-cell levels of CD73high and CD90high accompany this development as known mesenchymal stem cell markers [41, 67,68,69,70]. Late-passage NCR derivatives generated here were comparable to mesenchymal phenotypes obtained from NCR cells by repeated passaging in low density conditions in terms of morphology, surface marker profile and cell proliferation potential [41, 71]. We have previously observed a correlation of CD49d expression and nuclear localization of the Hippo pathway transcriptional regulator YAP in low-density human neural differentiation culture systems [46]. Similarly, loss of CD15 expression had previously been observed [24, 46]. Historically, others [17, 32] had characterized human NCR as p75+/HNK1+ (CD271+/CD57+) and we have previously seen CD15−/CD44+/CD49d+ as NCR signatures comprising both pre-migratory and migratory NCR sub-populations [46]. The results of this current study underline how NCR EMT can be identified, specifically and sensitively, by the surface expression shift from CD57 and other glycan moieties (CD15, CD24) towards a pro-migratory integrin panel of surface molecules including, most notably, CD51/61 (αVβ3). These results are corroborated by the alterations of glycan moities during epithelial-to-mesenchymal transition and neural development [72,73,74,75]. Our data show that TWIST and SLUG as established intracellular NCR markers are most closely associated with a population uniquely characterized by shifting from CD57 to CD51/CD61 at the cell surface level. CD51/CD61 seems to be regulated through complex signaling networks including the Hippo pathway downstream effector YAP [76]. Further, in the context of angiogenesis, the CCN1 protein, an upstream regulator of YAP was found to also enhance the expression of CD51/CD61 [77]. These results are in keeping with our findings of low-density culture conditions promoting YAP and its nuclear translocation in NCR [46]. While the direct signaling routes remain to be experimentally dissected, it is plausible that the reported low-density NCR differentiation protocol involves YAP as well as EMT-associated regulatory networks (ZEB1, TWIST, SNAI1, SLUG), thereby controlling a functionally relevant shift of glycan and integrin cell surface epitopes. The specific heterodimers most prominently upregulated at NCR EMT in the human stem cell system described here are congruent with the classically described involvement of primarily α4β1 and αvβ3 in cellular migration [78]. Beyond the inherent utility of stage-specific live-cell markers of human NCR EMT, the systematic, stage-specific analysis of CD surface antigen expression patterns of NCR in vitro development provides an enhanced resolution of NCR phenotype dynamics and transitions. Thereby the results obtained might prove useful not only for in vitro monitoring of NCR differentiation and quality control (QC) of NCR cell therapeutics, but also as histopathological markers in the context of NCR-associated tumors [79]. Interestingly, one of the most important proteins associated with melanoma potential is ITGB3/CD61 [80,81,82], and CD90 and CD51/CD61 have already been shown to functionally interact in melanoma cell adhesion [83].
In summary, this current analysis amends our and others NCR-associated marker codes (CD15/CD24/CD29 [24]; CD44/CD49d [46] with critical additions. Complementing established NCR transcriptional markers, the flow cytometrically assessable shift from CD57 epitopes towards CD51/CD61 surface expression further resolves the stages of neuroepithelial EMT. Such enhanced insights into NCR stemness and phenotypic regulation may contribute to more efficaciously realizing the broad therapeutic potential of human NCR in vitro models and stem cell preparations [2, 5].
Data Availability
The authors declare that all data supporting the findings of this study are available within the article (and its supplemental information) or from the corresponding author on reasonable request.
Code Availability
Not applicable.
References
Prasad, M. S., Charney, R. M., & García-Castro, M. I. (2019). Specification and formation of the neural crest: Perspectives on lineage segregation. Genesis (New York N Y: 2000), 57(1), e23276. https://doi.org/10.1002/dvg.23276
Srinivasan, A., & Toh, Y. C. (2019). Human pluripotent stem cell derived neural crest cells for tissue regeneration and disease modeling. Frontiers in Molecular Neuroscience, 12, 39. https://doi.org/10.3389/fnmol.2019.00039
York, J. R., & McCauley, D. W. (2020). The origin and evolution of vertebrate neural crest cells. Open Biology, 10(1), 190285. https://doi.org/10.1098/rsob.190285
Bronner, M. E., & Simões-Costa, M. (2016). The neural crest migrating into the twenty-first century. Current Topics in Developmental Biology, 116, 115–134. https://doi.org/10.1016/bs.ctdb.2015.12.003
Liu, J. A., & Cheung, M. (2016). Neural crest stem cells and their potential therapeutic applications. Developmental Biology, 419(2), 199–216. https://doi.org/10.1016/j.ydbio.2016.09.006
Lim, J. Y. (2021). In Park, S., Park, S. A., Jeon, J. H., Jung, H. Y., Yon, J.-M., Jeun, S.-S., Lim, H. K., & Kim, S. W. Potential application of human neural crest-derived nasal turbinate stem cells for the treatment of neuropathology and impaired cognition in models of Alzheimer’s disease. Stem Cell Research & Therapy, 12(1), 402. https://doi.org/10.1186/s13287-021-02489-1
Soto, J., Ding, X., Wang, A., & Li, S. (2021). Neural crest-like stem cells for tissue regeneration. Stem Cells Translational Medicine, 10(5), 681–693. https://doi.org/10.1002/sctm.20-0361
Achilleos, A., & Trainor, P. A. (2012). Neural crest stem cells: Discovery, properties and potential for therapy. Cell Research, 22(2), 288–304. https://doi.org/10.1038/cr.2012.11
Ramos-Zúñiga, R., González-Pérez, O., Macías-Ornelas, A., Capilla-González, V., & Quiñones-Hinojosa, A. (2012). Ethical implications in the use of embryonic and adult neural stem cells. Stem Cells International, 2012, 470949. https://doi.org/10.1155/2012/470949
Etchevers, H. (2011). Primary culture of chick, mouse or human neural crest cells. Nature Protocols, 6(10), 1568–1577. https://doi.org/10.1038/nprot.2011.398
Gonzalez Malagon, S. G., Dobson, L., Muñoz, A. M. L., Dawson, M., Barrell, W., Marangos, P. … Liu, K. J. (2019). Dissection, culture and analysis of primary cranial neural crest cells from mouse for the study of neural crest cell delamination and migration. Journal of Visualized Experiments: JoVE, 152. https://doi.org/10.3791/60051
Huang, M., Miller, M. L., McHenry, L. K., Zheng, T., Zhen, Q., Ilkhanizadeh, S., & Weiss, W. A. (2016). Generating trunk neural crest from human pluripotent stem cells. Scientific Reports, 6, 19727. https://doi.org/10.1038/srep19727
Zhu, Q., Lu, Q., Gao, R., & Cao, T. (2016). Prospect of human pluripotent stem cell-derived neural crest stem cells in clinical application. Stem Cells International, 2016, 7695836. https://doi.org/10.1155/2016/7695836
Mehler, V. J., Burns, C. J., Stauss, H., Francis, R. J., & Moore, M. L. (2020). Human iPSC-derived neural crest stem cells exhibit low immunogenicity. Molecular Therapy Methods & Clinical Development, 16, 161–171. https://doi.org/10.1016/j.omtm.2019.12.015
Solozobova, V., Wyvekens, N., & Pruszak, J. (2012). Lessons from the embryonic neural stem cell niche for neural lineage differentiation of pluripotent stem cells. Stem Cell Reviews and Reports, 8(3), 813–829. https://doi.org/10.1007/s12015-012-9381-8
Jiang, X., Gwye, Y., McKeown, S. J., Bronner-Fraser, M., Lutzko, C., & Lawlor, E. R. (2009). Isolation and characterization of neural crest stem cells derived from in vitro-differentiated human embryonic stem cells. Stem Cells and Development, 18(7), 1059–1070. https://doi.org/10.1089/scd.2008.0362
Lee, G., Kim, H., Elkabetz, Y., Al Shamy, G., Panagiotakos, G., Barberi, T., & Studer, L. (2007). Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nature Biotechnology, 25(12), 1468–1475. https://doi.org/10.1038/nbt1365
Li, W., Huang, L., Zeng, J., Lin, W., Li, K., Sun, J., & Xiang, A. P. (2018). Characterization and transplantation of enteric neural crest cells from human induced pluripotent stem cells. Molecular Psychiatry, 23(3), 499–508. https://doi.org/10.1038/mp.2016.191
Liu, Q., Swistowski, A., & Zeng, X. (2014). Human neural crest stem cells derived from human pluripotent stem cells. Methods in Molecular Biology (Clifton N J), 1210, 79–90. https://doi.org/10.1007/978-1-4939-1435-7_7
Kerosuo, L., Nie, S., Bajpai, R., & Bronner, M. E. (2015). Crestospheres: Long-term maintenance of multipotent, premigratory neural crest stem cells. Stem Cell Reports, 5(4), 499–507. https://doi.org/10.1016/j.stemcr.2015.08.017
Lee, G., Chambers, S. M., Tomishima, M. J., & Studer, L. (2010). Derivation of neural crest cells from human pluripotent stem cells. Nature Protocols, 5(4), 688–701. https://doi.org/10.1038/nprot.2010.35
Lee, D. R., Yoo, J. E., Lee, J. S., Park, S., Lee, J., Park, C. Y., & Kim, D. W. (2015). PSA-NCAM-negative neural crest cells emerging during neural induction of pluripotent stem cells cause mesodermal tumors and unwanted grafts. Stem Cell Reports, 4(5), 821–834. https://doi.org/10.1016/j.stemcr.2015.04.002
Menendez, L., Kulik, M. J., Page, A. T., Park, S. S., Lauderdale, J. D., Cunningham, M. L., & Dalton, S. (2013). Directed differentiation of human pluripotent cells to neural crest stem cells. Nature Protocols, 8(1), 203–212. https://doi.org/10.1038/nprot.2012.156
Pruszak, J., Ludwig, W., Blak, A., Alavian, K., & Isacson, O. (2009). CD15, CD24, and CD29 define a surface biomarker code for neural lineage differentiation of stem cells. Stem Cells (Dayton Ohio), 27(12), 2928–2940. https://doi.org/10.1002/stem.211
Pruszak, J. (2015). Chapter 19 - Synopsis and epilogue: Neural surface antigen studies in biology and biomedicine - What we have learned and what the future may hold (J. B. T.-N. S. A. Pruszak (ed.); pp. 223–228). Academic. https://doi.org/10.1016/B978-0-12-800781-5.00019-0
Simões-Costa, M., & Bronner, M. E. (2015). Establishing neural crest identity: a gene regulatory recipe. Development (Cambridge England), 142(2), 242–257. https://doi.org/10.1242/dev.105445
Serrano, F., Bernard, W. G., Granata, A., Iyer, D., Steventon, B., Kim, M., & Sinha, S. (2019). A novel human pluripotent stem cell-derived neural crest model of treacher collins syndrome shows defects in cell death and migration. Stem Cells and Development, 28(2), 81–100. https://doi.org/10.1089/scd.2017.0234
Greiner, J. F. W., Grunwald, L. M., Müller, J., Sudhoff, H., Widera, D., Kaltschmidt, C., & Kaltschmidt, B. (2014). Culture bag systems for clinical applications of adult human neural crest-derived stem cells. Stem Cell Research & Therapy, 5(2), 34. https://doi.org/10.1186/scrt422
Zeuner, M. T., Didenko, N. N., Humphries, D., Stergiadis, S., Morash, T. M., Patel, K., & Widera, D. (2018). Isolation and characterization of neural crest-derived stem cells from adult ovine palatal tissue. Frontiers in Cell and Developmental Biology, 6, 39. https://doi.org/10.3389/fcell.2018.00039
Lee, G., Papapetrou, E. P., Kim, H., Chambers, S. M., Tomishima, M. J., Fasano, C. A., & Studer, L. (2009). Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature, 461(7262), 402–406. https://doi.org/10.1038/nature08320
Chong-Morrison, V., & Sauka-Spengler, T. (2021). The cranial neural crest in a multiomics era. Frontiers in Physiology, 12, 634440. https://doi.org/10.3389/fphys.2021.634440
Yuan, S. H., Martin, J., Elia, J., Flippin, J., Paramban, R. I., Hefferan, M. P., & Carson, C. T. (2011). Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One1, 6(3), e17540. https://doi.org/10.1371/journal.pone.0017540
Orfao, A., Matarraz, S., Pérez-Andrés, M., Almeida, J., Teodosio, C., Berkowska, M. A., & van Dongen, J. J. (2019). Immunophenotypic dissection of normal hematopoiesis. Journal of Immunological Methods, 475, 112684. https://doi.org/10.1016/j.jim.2019.112684
Lv, F. J., Tuan, R. S., Cheung, K. M. C., & Leung, V. Y. L. (2014). Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells (Dayton Ohio), 32(6), 1408–1419. https://doi.org/10.1002/stem.1681
van Strien, M. E., Sluijs, J. A., Reynolds, B. A., Steindler, D. A., Aronica, E., & Hol, E. M. (2014). Isolation of neural progenitor cells from the human adult subventricular zone based on expression of the cell surface marker CD271. Stem Cells Translational Medicine, 3(4), 470–480. https://doi.org/10.5966/sctm.2013-0038
Cohen-Cory, S., Elliott, R. C., Dreyfus, C. F., & Black, I. B. (1993). Depolarizing influences increase low-affinity NGF receptor gene expression in cultured Purkinje neurons. Experimental Neurology, 119(2), 165–175. https://doi.org/10.1006/exnr.1993.1018
Pérez, V., Bermedo-Garcia, F., Zelada, D., Court, F. A., Pérez, M., Fuenzalida, M., & Henríquez, J. P. (2019). The p75(NTR) neurotrophin receptor is required to organize the mature neuromuscular synapse by regulating synaptic vesicle availability. Acta Neuropathologica Communications, 7(1), 147. https://doi.org/10.1186/s40478-019-0802-7
Menon, V., Thomas, R., Elgueta, C., Horl, M., Osborn, T., Hallett, P. J., & Pruszak, J. (2019). Comprehensive cell surface antigen analysis identifies transferrin receptor protein-1 (CD71) as a negative selection marker for human neuronal cells. Stem Cells (Dayton Ohio), 37(10), 1293–1306. https://doi.org/10.1002/stem.3057
Barilani, M., Banfi, F., Sironi, S., Ragni, E., Guillaumin, S., Polveraccio, F., & Lazzari, L. (2018). Low-affinity nerve growth factor receptor (CD271) heterogeneous expression in adult and fetal mesenchymal stromal cells. Scientific Reports, 8(1), 9321. https://doi.org/10.1038/s41598-018-27587-8
Álvarez-Viejo, M., Menéndez-Menéndez, Y., & Otero-Hernández, J. (2015). CD271 as a marker to identify mesenchymal stem cells from diverse sources before culture. World Journal of Stem Cells, 7(2), 470–476. https://doi.org/10.4252/wjsc.v7.i2.470
Ouchi, T., Morikawa, S., Shibata, S., Fukuda, K., Okuno, H., Fujimura, T., & Okano, H. (2016). LNGFR(+)THY-1(+) human pluripotent stem cell-derived neural crest-like cells have the potential to develop into mesenchymal stem cells. Differentiation; Research in Biological Diversity, 92(5), 270–280. https://doi.org/10.1016/j.diff.2016.04.003
Yanagisawa, M., & Yu, R. K. (2007). The expression and functions of glycoconjugates in neural stem cells. Glycobiology, 17(7), 57R – 74. https://doi.org/10.1093/glycob/cwm018
Itokazu, Y., & Yu, R. K. (2015). Chapter 8 - Glycolipid antigens in neural stem cells (J. B. T.-N. S. A. Pruszak (ed.); pp. 91–102). Academic. https://doi.org/10.1016/B978-0-12-800781-5.00008-6
Mechtersheimer, G., Staudter, M., & Möller, P. (1991). Expression of the natural killer cell-associated antigens CD56 and CD57 in human neural and striated muscle cells and in their tumors. Cancer Research, 51(4), 1300–1307
Kared, H., Martelli, S., Ng, T. P., Pender, S. L. F., & Larbi, A. (2016). CD57 in human natural killer cells and T-lymphocytes. Cancer Immunology Immunotherapy: CII, 65(4), 441–452. https://doi.org/10.1007/s00262-016-1803-z
Hindley, C. J., Condurat, A. L., Menon, V., Thomas, R., Azmitia, L. M., Davis, J. A., & Pruszak, J. (2016). The Hippo pathway member YAP enhances human neural crest cell fate and migration. Scientific Reports, 6(1), 23208. https://doi.org/10.1038/srep23208
Schöndorf, D. C., Ivanyuk, D., Baden, P., Sanchez-Martinez, A., De Cicco, S., Yu, C., & Deleidi, M. (2018). The NAD + precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Reports, 23(10), 2976–2988. https://doi.org/10.1016/j.celrep.2018.05.009
Turaç, G., Hindley, C. J., Thomas, R., Davis, J. A., Deleidi, M., Gasser, T., & Pruszak, J. (2013). Combined flow cytometric analysis of surface and intracellular antigens reveals surface molecule markers of human neuropoiesis. PLoS One1, 8(6), e68519. https://doi.org/10.1371/journal.pone.0068519
Menon, V., Thomas, R., Ghale, A. R., Reinhard, C., & Pruszak, J. (2014). Flow cytometry protocols for surface and intracellular antigen analyses of neural cell types. Journal of Visualized Experiments: JoVE, 94. https://doi.org/10.3791/52241
Ferlemann, F. C., Menon, V., Condurat, A. L., Rößler, J., & Pruszak, J. (2017). Surface marker profiling of SH-SY5Y cells enables small molecule screens identifying BMP4 as a modulator of neuroblastoma differentiation. Scientific Reports, 7(1), 13612. https://doi.org/10.1038/s41598-017-13497-8
Liu, C., Oikonomopoulos, A., Sayed, N., & Wu, J. C. (2018). Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development (Cambridge England), 145(5). https://doi.org/10.1242/dev.156166
Lee, K. M., Hawi, Z. H., Parkington, H. C., Parish, C. L., Kumar, P. V., Polo, J. M., & Tong, J. (2020). The application of human pluripotent stem cells to model the neuronal and glial components of neurodevelopmental disorders. Molecular Psychiatry, 25(2), 368–378. https://doi.org/10.1038/s41380-019-0495-0
Simões-Costa, M., & Bronner, M. E. (2013). Insights into neural crest development and evolution from genomic analysis. Genome Research, 23(7), 1069–1080. https://doi.org/10.1101/gr.157586.113
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., & Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology, 27(3), 275–280. https://doi.org/10.1038/nbt.1529
Shmelkov, S. V., St Clair, R., Lyden, D., & Rafii, S. (2005). AC133/CD133/Prominin-1. The International Journal of Biochemistry & Cell Biology, 37(4), 715–719. https://doi.org/10.1016/j.biocel.2004.08.010
Shi, H., Li, X., Yang, J., Zhao, Y., Xue, C., Wang, Y., & Ding, F. (2019). Bone marrow-derived neural crest precursors improve nerve defect repair partially through secreted trophic factors. Stem Cell Research & Therapy, 10(1), 397. https://doi.org/10.1186/s13287-019-1517-1
Tong, Q. S., Zheng, L. D., Tang, S. T., Ruan, Q. L., Liu, Y., Li, S. W., & Cai, J. B. (2008). Expression and clinical significance of stem cell marker CD133 in human neuroblastoma. World Journal of Pediatrics: WJP, 4(1), 58–62. https://doi.org/10.1007/s12519-008-0012-z
Jászai, J., Corbeil, D., & Fargeas, C. A. (2015). Chapter 10 - Comprehensive overview of CD133 biology in neural tissues across species (J. B. T.-N. S. A. Pruszak (ed.); pp.13–129). Academic. https://doi.org/10.1016/B978-0-12-800781-5.00010-4
Brazel, C. Y., Limke, T. L., Osborne, J. K., Miura, T., Cai, J., Pevny, L., & Rao, M. S. (2005). Sox2 expression defines a heterogeneous population of neurosphere-forming cells in the adult murine brain. Aging Cell, 4(4), 197–207. https://doi.org/10.1111/j.1474-9726.2005.00158.x
Cimadamore, F., Fishwick, K., Giusto, E., Gnedeva, K., Cattarossi, G., Miller, A., & Terskikh, A. V. (2011). Human ESC-derived neural crest model reveals a key role for SOX2 in sensory neurogenesis. Cell Stem Cell, 8(5), 538–551. https://doi.org/10.1016/j.stem.2011.03.011
Mandalos, N., Rhinn, M., Granchi, Z., Karampelas, I., Mitsiadis, T., Economides, A. N., & Remboutsika, E. (2014). Sox2 acts as a rheostat of epithelial to mesenchymal transition during neural crest development. Frontiers in Physiology, 5, 345. https://doi.org/10.3389/fphys.2014.00345
Li, H. Y., Say, E. H. M., & Zhou, X. F. (2007). Isolation and characterization of neural crest progenitors from adult dorsal root ganglia. Stem Cells (Dayton Ohio), 25(8), 2053–2065. https://doi.org/10.1634/stemcells.2007-0080
Krebs, A. M., Mitschke, J., Lasierra Losada, M., Schmalhofer, O., Boerries, M., Busch, H., & Brabletz, T. (2017). The EMT-activator ZEB1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nature Cell Biology, 19(5), 518–529. https://doi.org/10.1038/ncb3513
Banerjee, P., Xiao, G. Y., Tan, X., Zheng, V. J., Shi, L., Rabassedas, M. N. B., & Kurie, J. M. (2021). The EMT activator ZEB1 accelerates endosomal trafficking to establish a polarity axis in lung adenocarcinoma cells. Nature Communications, 12(1), 6354. https://doi.org/10.1038/s41467-021-26677-y
Vega, F. M., Colmenero-Repiso, A., Gómez-Muñoz, M. A., Rodríguez-Prieto, I., Aguilar-Morante, D., Ramírez, G., & Pardal, R. (2019). CD44-high neural crest stem-like cells are associated with tumour aggressiveness and poor survival in neuroblastoma tumours. eBioMedicine, 49, 82–95. https://doi.org/10.1016/j.ebiom.2019.10.041
Qiao, L., Gao, H., Zhang, T., Jing, L., Xiao, C., Xiao, Y., & Mo, X. (2014). Snail modulates the assembly of fibronectin via α5 integrin for myocardial migration in zebrafish embryos. Scientific Reports, 4(1), 4470. https://doi.org/10.1038/srep04470
Cappellari, A. R., Rockenbach, L., Dietrich, F., Clarimundo, V., Glaser, T., Braganhol, E., & Battastini, A. M. (2012). Characterization of ectonucleotidases in human medulloblastoma cell lines: ecto-5’NT/CD73 in metastasis as potential prognostic factor. PLoS One1, 7(10), e47468. https://doi.org/10.1371/journal.pone.0047468
Ulrich, H., do Nascimento, I. C., Bocsi, J., & Tárnok, A. (2015). Immunomodulation in stem cell differentiation into neurons and brain repair. Stem Cell Reviews and Reports, 11(3), 474–486. https://doi.org/10.1007/s12015-014-9556-6
Ramos, L., Sánchez-Abarca, T., Muntión, L. I., Preciado, S., Puig, S., López-Ruano, N., & del Cañizo, C. (2016). MSC surface markers (CD44, CD73, and CD90) can identify human MSC-derived extracellular vesicles by conventional flow cytometry. Cell Communication and Signaling: CCS, 14, 2. https://doi.org/10.1186/s12964-015-0124-8
Pham, H., Tonai, R., Wu, M., Birtolo, C., & Chen, M. (2018). CD73, CD90, CD105 and Cadherin-11 RT-PCR screening for mesenchymal stem cells from cryopreserved human cord tissue. International Journal of Stem Cells, 11(1), 26–38. https://doi.org/10.15283/ijsc17015
Chijimatsu, R., Ikeya, M., Yasui, Y., Ikeda, Y., Ebina, K., Moriguchi, Y. … Norimasa, N. (2017). Characterization of mesenchymal stem cell-like cells derived from human iPSCs via neural crest development and their application for osteochondral repair. Stem Cells International, 2017, 1960965. https://doi.org/10.1155/2017/1960965
Pucci, M., Malagolini, N., & Dall’Olio, F. (2021). Glycobiology of the epithelial to mesenchymal transition. Biomedicines, 9(7), 770. https://doi.org/10.3390/biomedicines9070770
Li, X., Wang, X., Tan, Z., Chen, S., & Guan, F. (2016). Role of glycans in cancer cells undergoing epithelial-mesenchymal transition. Frontiers in Oncology, 6, 33. https://doi.org/10.3389/fonc.2016.00033
Xu, Q., Niu, X., Wang, W., Yang, W., Du, Y., Gu, J., & Song, L. (2017). Specific N-glycan alterations are coupled in EMT induced by different density cultivation of MCF 10A epithelial cells. Glycoconjugate Journal, 34(2), 219–227. https://doi.org/10.1007/s10719-016-9754-3
Williams, S. E., Noel, M., Lehoux, S., Cetinbas, M., Xavier, R. J., Sadreyev, R. I., & Mealer, R. G. (2022). Mammalian brain glycoproteins exhibit diminished glycan complexity compared to other tissues. Nature Communications, 13(1), 275. https://doi.org/10.1038/s41467-021-27781-9
Nardone, G., Oliver-De La Cruz, J., Vrbsky, J., Martini, C., Pribyl, J., Skládal, P., & Forte, G. (2017). YAP regulates cell mechanics by controlling focal adhesion assembly. Nature Communications, 8, 15321. https://doi.org/10.1038/ncomms15321
Park, M. H., Kim, A. K., Manandhar, S., Oh, S. Y., Jang, G. H., Kang, L. … Lee, Y. M. (2019). CCN1 interlinks integrin and Hippo pathway to autoregulate tip cell activity. ELife, 8. https://doi.org/10.7554/eLife.46012
Testaz, S., Delannet, M., & Duband, J. (1999). Adhesion and migration of avian neural crest cells on fibronectin require the cooperating activities of multiple integrins of the (beta)1 and (beta)3 families. Journal of Cell Science, 112(Pt 2), 4715–4728
Powell, D. R., Blasky, A. J., Britt, S. G., & Artinger, K. B. (2013). Riding the crest of the wave: parallels between the neural crest and cancer in epithelial-to-mesenchymal transition and migration. Wiley Interdisciplinary Reviews Systems Biology and Medicine, 5(4), 511–522. https://doi.org/10.1002/wsbm.1224
Nemlich, Y., Baruch, E. N., Besser, M. J., Shoshan, E., Bar-Eli, M., Anafi, L., & Markel, G. (2018). ADAR1-mediated regulation of melanoma invasion. Nature Communications, 9(1), 2154. https://doi.org/10.1038/s41467-018-04600-2
Gaggioli, C., Hooper, S., Hidalgo-Carcedo, C., Grosse, R., Marshall, J. F., Harrington, K., & Sahai, E. (2007). Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nature Cell Biology, 9(12), 1392–1400. https://doi.org/10.1038/ncb1658
Orgaz, J. L., & Sanz-Moreno, V. (2013). Emerging molecular targets in melanoma invasion and metastasis. Pigment Cell & Melanoma Research, 26(1), 39–57. https://doi.org/10.1111/pcmr.12041
Saalbach, A., Wetzel, A., Haustein, U. F., Sticherling, M., Simon, J. C., & Anderegg, U. (2005). Interaction of human Thy-1 (CD 90) with the integrin alphavbeta3 (CD51/CD61): An important mechanism mediating melanoma cell adhesion to activated endothelium. Oncogene, 24(29), 4710–4720. https://doi.org/10.1038/sj.onc.1208559
Acknowledgements
The authors thank S. Frerix, C. Reinhard and T.-Y. Su for technical assistance and members of the Pruszak lab for comments on the manuscript.
Funding
Open access funding provided by Paracelsus Medical University. This project was funded by institutional funds (PMU; JP), by the Spemann Graduate School of Biology and Medicine (SGBM; DFG Excellence Initiative; RT, VM), and the Emmy Noether program of the German Research Foundation (DFG, grant PR1132/3 − 1; JP).
Author information
Authors and Affiliations
Contributions
R.T: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing. V.M: collection and assembly of data. R.M.: data analysis and interpretation, manuscript writing. J.P: conception and design, collection and assembly of data, data analysis and interpretation, financial support, manuscript writing. All authors contributed to the article and critically reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest/ Competing Interests
The authors report no conflict of interest.
Ethics Approval
All stem cell work conducted was conducted with long-established publicly available ES cell lines (source: WiCell, Madison, WI, USA). Human ES cell work had been approved by the Zentrale Ethik-Kommission für Stammzellenforschung, Germany (ZES No. 59, 2011).
Consent to Participate
Not applicable.
Consent to Publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
ESM 1
(PDF 658 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thomas, R., Menon, V., Mani, R. et al. Glycan Epitope and Integrin Expression Dynamics Characterize Neural Crest Epithelial-to-Mesenchymal Transition (EMT) in Human Pluripotent Stem Cell Differentiation. Stem Cell Rev and Rep 18, 2952–2965 (2022). https://doi.org/10.1007/s12015-022-10393-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-022-10393-1