
Abstract
Introduction
Glycosylation is an important component for a number of biological processes and is perhaps the most abundant and complicated of the known post-translational modifications found on proteins.
Methods
This work combines two-dimensional (2-D) polyacrylamide gel electrophoresis and lectin blotting to map the salivary glycome and mass spectrometry to identity the proteins that are associated with the glycome map. A panel of 15 lectins that recognize six sugar-specific categories was used to visualize the type and extent of glycosylation in saliva from two healthy male individuals. Lectin blots were compared to 2-D gels stained either with Sypro Ruby (protein stain) or Pro-Q Emerald 488 (glycoprotein stain).
Results
Each lectin shows a distinct pattern, even those belonging to the same sugar-specific category. In addition, the glycosylation profiles generated from the lectin blots show that most salivary proteins are glycosylated and that the profiles are more widespread than is demonstrated by the glycoprotein-stained gel. Finally, the coreactivity between lectins was measured to determine what types of glycan structures are associated with one another and also the population variation of the lectin reactivity for 66 individuals were reported.
Conclusions
This starting 2-D gel glycosylation reference map shows that the scientifically accepted, individual oligosaccharide variability is not limited to a few large glycoproteins such as MUC5B, but are found on most members of the salivary proteome. Finally, in order to see the full range of oligosaccharide distribution, multiple reagents or lectins are needed.
Similar content being viewed by others


Introduction
Saliva is the oral fluid that lubricates, buffers, and protects oral tissues against decay, damage, and microbial inflammation and facilitates the remineralization of teeth. Saliva consists primarily of water, minerals, electrolytes, buffer, and proteins that are secreted by three major glands (parotid, submandibular, sublingual) and by numerous minor glands in the lip, cheek, tongue, and palate. In addition, saliva contains microbes, epithelial cells, nasal and bronchial secretions, and serum products. These components can provide clues to local and/or systemic diseases and disorders of the human body [1]. Therefore, numerous studies have sought to develop a complete catalog of the proteins and peptides found in human saliva [2–12]. However, proteins are only one chapter in gaining a comprehensive catalog of all the components in saliva; another is to characterize the post-translational modifications of the proteins and peptides that compose the human salivary catalog.
Glycosylation is the most abundant and among the most complicated of the known post-translational modifications found on proteins [13–15]. It plays an important role in cell adhesion, cell trafficking, protease protection, signaling, protein structure, and many other biological functions [13, 16]. With more than 50% of all proteins estimated to be glycosylated [17] and the mammalian glycome repertoire estimated to be hundreds and thousands of glycan structures [13], it is not surprising that changes in the oligosaccharide structure have been linked to many diseases, including autoimmune diseases [18], cancer [19], pre-term labor [20], rheumatoid arthritis [21], and virus infection [13].
Saliva is a rich source of both N- and O-linked glycoproteins, which play an important role in the maintenance of oral health and protection of teeth [22, 23]. Several glycoproteins bind to a variety of bacteria and either aid or prevent the adherence of bacteria to mucosal and tooth surfaces [24–26]. Other glycoprotein functions include lubricating tooth surfaces [23] and possibly protecting proteins from proteolytic attack [27]. Our laboratory has previously identified a number of Asn-linked glycoproteins in human whole saliva and salivary glandular fluid [28, 29] using the N-linked glycopeptides capture method [30] that employs hydrazide chemistries and resins to capture glycoproteins. Following the enzymatic cleavage of the N-linked glycopeptides with N-glycosidase F and trypsin digestion, glycoproteins of the formerly N-linked glycopeptides were identified by mass spectrometry (MS) analysis. Using this strategy, we identified N-glycopeptides representing 77 unique N-glycoproteins in whole saliva and salivary glandular fluid. However, further work needs to be done in characterizing the glycosylation on those glycoproteins.
Lectins are nonenzymatic, carbohydrate-binding proteins or glycoproteins that are found ubiquitously in nature. These proteins bind reversibly to specific glycan structures. Recognition of a particular monosaccharide is one of the important factors determining the lectin specificity. Other factors may include preference for either the α- or β-anomer along with the recognition of specific linkage(s) for a series of sugars. In addition, most lectins will interact exclusively with the terminal nonreducing position in an oligosaccharide; however, some lectins such as concanavalin A (ConA) and wheat germ agglutinin (WGA) bind to internal sugars [31]. Because lectins are easily purified in large quantities, they have become a useful tool for the detection, isolation, and characterization of glycoconjugates [32]. Besides their primary affinity, lectins do have secondary affinities to different glycan structures. Therefore, the structural information that is provided from the binding of a particular lectin needs to be further validated by other analytical techniques. By using a panel of lectins, other members of the panel can help shed light onto whether or not the binding of one lectin may actually be due to its secondary affinity versus its primary affinity. Even though lectins have both primary and secondary affinities, the use of lectins to study glycoconjugates has a long and productive history [33, 34].
Since post-translational modifications alter the apparent molecular weight and isoelectric point of a protein, two-dimensional (2-D) gel electrophoresis is a powerful tool for examining variation of these types of modifications. For example, gain or loss of glycosylation sites and differences in the extent of branching and/or modifications in the number of sialic acid residues bound to a particular protein will change the spot pattern during 2-D gel electrophoresis. Many times these modifications are observed as a “train” of spots in the 2-D gels. By combining 2-D gel electrophoresis, lectin blotting, and identification by mass spectrometry, we can correlate changes in the migration pattern of a particular protein to possible changes in the glycoforms. Any interesting spots would need further detailed analysis (i.e., mass spectrometry) to identify all of the post-translational modifications associated with a change in the migration pattern.
A panel of 15 lectins (Table 1) from six monosaccharide-specific categories (fucose (lectins AAL, PSA, and UEA I), galactose (lectins JAC and Mal I), mannose (lectins ConA, GNL, and HHL), N-acetyl-galactosamine (lectins BPL and VVA), N-acetyl-glucosamine (lectins DSL, LEL, and WGA), and sialic acid (lectins MAL II and SNA)) was used to visualize the type and the extent of glycosylation. The lectin blots were then compared to 2-D gels stained either with Sypro Ruby total protein stain or Pro-Q Emerald 488 glycoprotein stain. Although lectin blotting has been previously used in combination with 2-D gel electrophoresis [64–69], this is the first report using a comprehensive lectin panel to profile the glycosylation pattern of a particular proteome. Comparison of lectin 2-D blotting maps provides an important first step in identifying interesting candidate spots that could be used in the discovery of disease-related biomarkers from human saliva. Further studies would need to be carried out to fully characterize the entire glycan moiety associated with these candidates and to determine whether other post-translational modifications are also associated with the proteins identified from that spot.
Materials and Methods
Whole Saliva Collection and Preparation
Saliva collection was performed under UCLA IRB approval protocol number 04-09-051 titled “Human Salivary Proteome”. The subjects in this study were two healthy, Asian males ranging in ages of 33–35 and representing both A and B blood types. These individuals rinsed their mouths three times with bottled water to remove any food particles. Five minutes after the oral rinse, the subjects were then asked to spit 10 ml of resting (unstimulated) whole saliva into a 50-ml conical tube containing 20 μl protease inhibitor cocktail for mammalian tissues (Sigma-Aldrich, St. Louis, MO, USA). The tube was kept on ice during the collection. Equal volumes of saliva from these two individuals were pooled and then centrifuged for 10 min at 17,000×g, 4°C to remove epithelial cells and other debris. The salivary proteins were precipitated by adding 5× volume of ice-cold 100% ethanol. The tubes were mixed by inversion and incubated overnight at −20°C. The precipitant was collected the next day by centrifugation for 15 min at 20,000×g, 4°C. The pellet was washed twice with room temperature (RT) 70% ethanol and centrifuged between each wash as described in the precipitation step. The residual ethanol was removed before the pellet was air-dried for 5 min at RT. The pellet was resuspended by pipetting up and down in 7 M urea, 2 M thiourea, 4% CHAPS, 0.2% Bio-Lyte 3/10 ampholyte, 0.0002% bromophenol blue, and 0.4% DTT. Any insoluble material was removed by centrifugation for 5 min at 16,000×g, 4°C. The sample was aliquoted and stored immediately at −80°C. One of the aliquots was used to measure the protein concentration using a scaled-down version of the Modified Bradford described by Ramagli [70].
Two-Dimensional Gel Electrophoresis
A volume of 185 μl of the saliva sample was passively loaded overnight at RT onto an 11 cm Bio-Rad (Hercules, CA, USA) pH 3–10 NL ReadyStrip IPG strip. The loaded samples contained either 50 μg (only for lectins AAL, ConA, JAC, PSA, SNA, and WGA) or 250 μg of total protein. The isoelectric focusing was performed for a total of 36 kV h (30 μA limit) using a Bio-Rad PROTEAN IEF cell. The program for isoelectric focusing was the following: 100 V (linear) for 4 h, 250 V (linear) for 2 h, 4,000 V (linear) for 5 h, 4,000 V (rapid) for 25,000 V h, and 250 V (rapid) for approximately 5 h. After focusing, the IPG strips were first equilibrated with rocking for 20 min at RT in 50 mM Tris–HCl, pH 8.8, 7 M urea, 20% glycerol, 2% SDS, and 2% DTT followed by 20 min with the same buffer with 2.5% iodoacetamide replacing DTT. The strips were then rinsed with SDS-PAGE running buffer (25 mM Tris, pH 8.3, 192 mM glycine, and 0.1% SDS). The second dimension was performed on a Bio-Rad 12.5% precast Criterion gel using a Criterion Dodeca Cell at 10°C. The gels were electrophoresed at a constant 40 V for 10 min followed by a constant 200 V until the bromophenol blue front reached the bottom of the gel (∼65 min).
Gel Staining and Image Acquisition
Following electrophoresis, one of the gels was fixed for 2 h in 100 ml 10% methanol and 7% acetic acid. This gel was stained overnight in Sypro Ruby stain (Invitrogen-Molecular Probes, Carlsbad, CA USA), diluted 1:2 in previously used Sypro Ruby stain. The gel was then destained in fixer solution for 48 h with two solution changes. Another gel was fixed in 100 ml 50% methanol and 5% acetic acid. After 2 h, the fixing solution was replaced, and the gel continued fixing overnight. This gel was then stained with Pro-Q Emerald 488 (Molecular Probes) according to the instructions provided by the manufacturer. The maximum time lengths were used in all the steps, and the last wash step was carried out overnight.
The gels were imaged on a Typhoon 9410 Variable Mode Imager (GE Healthcare, Piscataway, NJ, USA) at 200 nm resolution using the following excitation/emission filters: 457 nm/610 nm BP 30 (Sypro Ruby) and 532 nm/520 nm BP 40 (Pro-Q Emerald 488).
Spot Retrieval and Trypsin Digestion
Protein spots from the Sypro Ruby-stained gel were excised by a Proteome Works Spot Cutter (Bio-Rad). The proteins were in-gel trypsin digested overnight at 30°C using Montage In-Gel DigestZP kit according to the instructions from Millipore (Billerica, MA, USA). Peptides were eluted twice in 20 μl 0.1% trifluoroacetic acid and 50% acetonitrile (ACN) by centrifugation for 15 s at 1,750×g, 4°C. The eluted peptides were then dried down and stored at −20°C until they were analyzed by liquid chromatography-tandem MS (LC-MS/MS) with electrospray ionization.
Protein Identification by Liquid Chromatography-Mass Spectrometry
The dried peptides from spot numbers 19, 23, 24, 27, 28, 51, 62, 64, 65, 67–69, 76, 78, 81, 82, 88, 96, and 106–174 from Fig. 1a and Supplemental Table were resuspended in 0.1% formic acid and identified on an Applied Biosystems (Foster City, CA, USA) QSTAR® XL (QqTOF) mass spectrometer equipped with a nanoelectrospray (Protana, Odense, Denmark) interface to a Dionex-LC Packings (Sunnyvale, CA, USA) nanoflow HPLC system. The nano-LC was equipped with a set of custom pre-columns (75 μm × 10 mm) and column (75 μm × 150 mm) packed with Jupiter Proteo C12 resins (particle size 4 μm, Phenomenex, Torrance, CA, USA). For each LC-MS/MS run, typically 6 μl sample solution was first loaded onto the pre-column and washed with the loading solvent, water containing 0.1% formic acid, for 4 min before injection to the LC column. The eluants used for the LC were (A) water containing 0.1% formic acid and (B) 95% ACN/water containing 0.1% formic acid. The flow was 200 nl/min, and the following gradient was used: 3% B to 35% B in 25 min, 35% B to 80% B in 4 min and held at 80% B for 8 min. The column was finally reequilibrated with 3% B for 15 min before the next run. For online MS and MS/MS analyses, a Proxeon (Odense, Denmark) nano-bore stainless steel online emitter (i.d. 30 μm) was used for spraying with the voltage set at 1,900 V. Peptide product ion spectra were automatically recorded during the LC-MS/MS runs by the information-dependent analysis on the QSTAR® XL mass spectrometer. Argon was employed as collision gas. Collision energies for maximum fragmentation were automatically calculated using empirical parameters based on the charge and mass-to-charge ratio of the peptide.

Total protein and glycoprotein-stained 2-D gels. Whole saliva was subjected to isoelectric focusing on a pH 3–10 nonlinear IPG strip, which was then resolved in the second dimension on a 12.5% SDS-PAGE gel. The resulting gels were either stained with SYPRO Ruby total protein stain (a) or Pro-Q Emerald 488 glycoprotein stain (b). Labeled are the protein spots that were identified from the Sypro Ruby-stained gel either by MALDI-TOF MS or LC-MS/MS in our previous work [8] or by LC-MS/MS in this work
Peptides separated by HPLC underwent fragmentation in the tandem mass spectrometer, and the resulting MS/MS spectra were searched against the International Protein Index for human database (version 3.03) using the Mascot search engine (Matrix Science, London, UK). MudPIT scoring with a threshold <0.05 ppm and ion score cut-off of 20 was used in the search parameters for positive protein identification. The peptide assignments for all significant hits were then manually validated. All other spots were previously cut and identified by Hu et al. [8].
Lectin Blotting
The gels were transferred for 2 h/4°C coldbox onto nitrocellulose using a Hoefer (San Francisco, CA, USA) TE62 transfer unit containing pre-chilled 25 mM Tris, 192 mM glycine, and 20% methanol. After transfer, the nitrocellulose was allowed to air dry and was stored at RT until use. The dried blots were incubated with continuous agitation for 2 h at RT in TBST (50 mM Tris buffer at pH 7.6, 150 mM NaCl, 0.1% Tween 20, and 0.01% sodium azide) containing biotinylated lectin at the manufacturer’s recommended concentration (Vector Laboratories, Burlingame, CA, USA and E-Y Laboratories, San Mateo, CA, USA). Following two 10-min washes with TBST, the blot was reacted for 20 min with avidin D–alkaline phosphatase (Vector Laboratories). Two additional 10-min washes with TBST were followed by two 10-min washes with TNM (100 mM Tris buffer at pH 9.5, 100 mM NaCl, and 5 mM MgCl2). Color development was accomplished using one-step NBT/BCIP (Pierce, Rockford, IL, USA) with agitation at RT and was allowed to proceed until sample-free areas of the blot began to exhibit a weak background. The blot was then placed in a stop solution (20 mM Tris buffer at pH 2.9) and subsequently allowed to air dry. All of the reagents contributing to the reporter system were reconstituted according to manufacturer’s specifications. The developed lectin blots were scanned and saved as high-resolution tiff-files.
None of the 2-D analysis software tested by us could accurately match and/or quantitate the spots from the lectin blots. The spots from the lectin blots were thus visually ranked as having low, medium, high, or no reactivity to the various lectins or stain. This ranking was done as a comparison for all the spots across a single gel. Some of the spots migrated to areas of the lectin blot that showed nonspecific reactivity, meaning no definitive spot could be observed. These spots were ranked as having a positive reactivity (+). This same ranking system of high, medium, and low reactivity was also carried out for both the Sypro Ruby and the Pro-Q Emerald-stained gels.
Population Variation
The range of population variation for each lectin affinity was determined from resting whole saliva from a group of 66 healthy adults ranging 24–36 years of age and covering a broad racial spectrum. Assay conditions and quantitation methods were performed as previously described [71]. A dilution equivalent to 0.2 μl of whole saliva was spotted onto nitrocellulose along with 0.12 and 0.3 μg of the commercially available blood glycoprotein, glycophorin (Sigma-Aldrich, St. Louis, MO, USA). These dot blots were developed and visualized as described above in “Lectin Blotting”. The bound lectin was then quantitated using SigmaScan (Systat Software, Chicago, IL, USA). Finally, the measured reactivity was normalized against the glycophorin standard in order to determine the population variation.
Results
Protein Identification from 2-D Gels
Resting whole saliva was collected from two healthy male individuals. The saliva was pooled, clarified of epithelia cells and other debris, and precipitated with ethanol. The salivary proteins were then separated on a two-dimensional gel that was stained with Sypro Ruby total protein stain (Fig. 1a). The spots from this gel image were matched to the 2-D gel image that our group had previously published [8, 28]. Of the 105 spots that were previously identified from whole saliva of one healthy female individual, we were able to match all the spots from our pooled males except for nine spots (numbers 17, 18, 54, and 100–105). To verify that we had accurately matched the gel spots and to examine for individual differences, 18 spots (numbers 19, 23, 24, 27, 28, 51, 62, 64, 65, 67–69, 76, 78, 81, 82, 88, and 96), which were previously identified, were excised along with 78 additional spots that had not previously been identified. After in-gel digestion with trypsin, the resulting peptides were analyzed by LC-MS/MS. The complete list of protein identifications is provided in the Supplemental Table. The higher abundant proteins in our repeated spot identification matched our previous work. As expected, we were able to identify a number of new lower abundant proteins using LC-MS/MS, compared to our previous work that primarily employed MALDI-TOF (matrix-assisted laser desorption ionization-time of flight) MS. As commonly observed by others, rarely did a single gel spot yield a single protein identification. However, for many of the spots, based on the number of unique peptides that were identified through MS/MS, there was one higher abundant protein with a number of much lower abundant proteins. Of the 78 additional spots picked, 69 spots (numbers 106–174) yielded a positive identification. When comparing these protein identifications to our previous work with whole saliva that used both 2-D gel electrophoresis and bottom-up LC-MS/MS and MALDI-TOF MS approaches [8], 50 new proteins were identified in this study. The data were submitted to the database (www.hspp.ucla.edu) that houses the compiled data from the Human Salivary Proteome Project, representing work from the Scripps Research Institute/University of Rochester, the University of California—Los Angeles/University of Southern California, and the University of California—San Francisco.
Generation of a Glyoprofile Catalog
In order to determine the glycosylation profile of the spots observed on the total protein stained 2-D gel, the same sample was run on additional gels that were stained either with Pro-Q Emerald 488 glycoprotein stain (Fig. 1a) or transferred and probed with a panel of 15 lectins representing six monosaccharide-specific categories (Fig. 2 and Supplemental Figures). The primary monosaccharide preference and the binding specificity for each of the lectins are described in Table 1. For most of the 2-D gels used in this study, 250 μg total protein was loaded onto the isoelectric focusing strip. However, some of the lectin blots appeared overloaded and had a high background at this concentration, thus making it very difficult to distinguish individual spots. For these blots (lectin AAL, ConA, JAC, PSA, SNA, and WGA), the total protein loading was 50 μg to reduce the background and nonspecific binding.



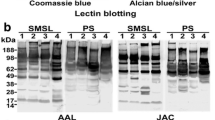
2-D lectin blots. Two-dimensional gels containing whole saliva were transferred and probed with either lectin DSL (a), LEL (b), UEA I (c), AAL (d), or WGA (e). The spots were matched to the Sypro Ruby-stained gel shown in Fig. 1a and scored for having high, medium, low, or no reactivity (Supplemental Table)
The spots from the Pro-Q Emerald-stained gels and the lectin blots (Figs. 1b, 2, and Supplemental Figures) were matched to the Sypro-stained gel (Fig. 1a) from which the spots had been identified. In order to generate a catalog (see Supplemental Table), the spots on the lectin blots and the Pro-Q Emerald-stained gel were visually rated as having a high, medium, low, or no reactivity to a particular lectin or to the stain. The lectin blots were repeated multiple times and showed that the 2-D pattern of the individual lectin reactivity was reproducible with regard to the relative ranking of individual spots. The reproducibility of ranking is due to the assay conditions, which typically can be quantitated over a tenfold range of concentration [71]. However, any spot demonstrating color saturation would narrow the quantitation range. For certain lectins described above, this problem was addressed by reducing the total amount of protein loaded in the 2-D gel.
For some of the lectin blots, there is a large area of nonspecific reactivity near the acidic and/or basic end of the gel (i.e., Fig. 2e, basic region). Spots which migrated to these areas of nonspecific reactivity were rated as having a positive reactivity (+) in the Supplemental Table, even though they were not clearly defined. Since these areas did not stain with Sypro Ruby and in the absence of salivary proteins none of the reagents used individually or in concert yielded a visible background, we speculate that these areas of reactivity result from free, charged oligosaccharides of varying chain lengths and branching.
This type of scoring system, which is a comparison for all the spots across a single gel or lectin blot, does not take into account the degree of protein staining. Therefore, the Sypro Ruby-stained gel was also scored the same way (Supplemental Table) so that the lectin reactivity can also be compared to the Sypro Ruby staining. The degree of protein staining is dependent on at least two factors: molecular weight and the availability and accessibility of the stain to its binding sites. For instance, proteins at high molecular weight that stain darkly have a lower molar concentration than lower molecular proteins that stain at the same darkness. In addition, Sypro Ruby stain binds to basic amino acids and the polypeptide backbone [72, 73], and therefore, the effects of amino acid content and glycosylation on stain binding are, in theory, thought to be minimal. However, the manufacturers of newer fluorescent stains claim that their stain binds better to glycoproteins than Sypro Ruby. In regards to staining proteins from whole saliva, we found that two of these newer stains resulted in lower staining intensity (GE Healthcare Deep Purple and Bio-Rad Flamingo), and only one stain worked slightly better than Sypro Ruby (Pierce Krypton). Though this type of scoring system provides only a semi-quantitative analysis, it is important because it helps provide insight to the secondary affinity of a particular lectin and/or the abundance of a particular oligosaccharide. This type of analysis using a total blot comparison has been very useful for developing a dental caries risk assessment test [71, 74]. However, since each lectin has a different binding constant to its substrate, this type of quantitation system is only useful for comparing spots from the same blot and for comparing the reactivity of a particular lectin between various individuals.
Lectin Blot Results
Generally, the best way to quantitate all of the lectin blots in a large scale study would be to normalize against one universal, housekeeping protein such as the use of actin in quantitative western blot analysis. This type of normalization helps to account for differences in protein loads and development times for the lectin blots. The predominant, housekeeping protein in whole saliva is α-amylase. However, the copy number of the salivary amylase gene was shown to vary for different populations of individuals [75], and we, so far, have not identified a good candidate. In addition, neither this protein nor any other protein possesses equal amounts of all possible glycan structures. For example, lectins DSL, LEL, and WGA all recognize (β-1,4)-linked N-acetylglucosamine, but each lectin recognizes a different glycan structure containing this oligosaccharide and the (β-1,4)-linkage. Therefore, when comparing the spot pattern from the various lectin blots, each lectin shows a distinct pattern even if they belong to the same sugar-specific category (Fig. 2 and Supplemental Figures). To illustrate, spot 9 (serotransferrin) demonstrates a low reactivity to lectins DSL and WGA but a high reactivity to lectin LEL (Fig. 2a, b, and e). The power of this type of scoring is that it indicates that serotransferrin is predicted to have predominately (β-1,4)-linked N-acetylglucosamine trimer or tetramer as part of the attached oligosaccharide.
Another example of how this type of scoring provides an added dimension of knowledge is the reactivity of the lectin panel to a train of zinc-alpha-2-glycoprotein (ZA2G) spots (numbers 60–63). Each spot showed a different pattern of reactivity to the lectin panel (Supplemental Table). Work by Löster and Kannicht [76] demonstrated that enzymatic desialylation shifts the pI toward the basic pH along with decreasing the molecular weight, while chemical deglycosylation only decreases the molecular weight. These spots do show differential reactivity to the lectins (Mal II and SNA) that primarily bind to sialic acid (Supplemental Table). However, other post-translational modifications may also be responsible for the shift in pI. Although other types of glycosylations are highly unlikely responsible for the shift in pI, we do observe differential reactivity to these spots for other members of the lectin panel. For example, both lectins AAL and UEA I recognize α-monofucosyl oligomers, but only lectin AAL reacted strongly to all of the spots, suggesting that ZA2G contains fucose (α-1,6)-N-acetylglucosamine and/or fucose (α-1,3)-N-acetyllactosamine residue(s) (Fig. 2c–d). On the other hand, lectin UEA I displayed differential recognition to these same spots, in which spot 60 had no reactivity, spots 61–62 had strong reactivity, and spot 63 had weak reactivity (Supplemental Table). Therefore, the proteins in spots 61 and 62 are likely to have a second α-monofucosyl oligomer consisting of fucosyl (α-1,2) galactosyl (β-1,4) N-acetylglucosamine (β-1,6)-R. Interestingly, lectin UEA I preferentially binds blood group O cells and has been used to determine secretor status.
Of the 166 2-D gel spots, which have been identified, all but eight spots reacted to our lectin panel (Supplemental Table). This indicates that most of the abundant salivary proteins are glycosylated and is consistent with the fact that the majority of the proteins in saliva are secreted as extracellular proteins. Spot 31 showed the highest reactivity to the lectin panel and was identified as α-amylase. This protein is the most abundant protein found in saliva and was found in 88 of the 2-D gel spots examined. Other highly reactive spots contained carbonic anhydrase VI or polymeric-immunoglobulin receptor. The second most abundant protein in terms of spot number was serum albumin. Since serum albumin is only glycosylated in a few of the genetic variants [77], it will not normally contribute to the glycoprofile.
By observing the reactivity of all of the lectins to the various spots (Supplemental Table), the following general trend was observed. As the molecular weight increases, the more likely that a particular spot reacts to the lectin panel. Larger proteins may have more potential glycosylation sites and may have more heterogeneity or more complex oligosaccharides than smaller proteins. Only a few spots showed no reactivity to any of the lectins; this includes spot numbers 64, 83, 84, 95, 170, and 172–174. Interestingly, all of the “unreactive” spots had a molecular weight of 40 kDa or less. Since most of the spots reacted to the lectin panel, several members of the lectin panel were used on extracts from the proteobacteria Myxococcus xanthus to test the specificity of the lectins (data not shown). The degree and pattern of reactivity of these lectins was distinct for human whole saliva and was vastly different when comparing results shared with us on the archaeon Methanosarcina acetivorans (Leon-Russell D and Denny PC, 2007, personal communication), indicating that the reactivity pattern was specific for human whole saliva.
Lectins bind to only the exposed glycan structures of a complex oligosaccharide. These structures are involved in biological activity. By comparing the total number of spots that reacted with a particular lectin, Mal II demonstrated the most reactivity by reacting with 128 of the 166 2-D gel spots (Table 2). This suggests that most of the salivary proteins contain a (α-2,3)-linked sialic acid residue. On the other end of the scale are lectins PSA and AAL, which reacted with the least number of spots (54 of 166 spots), indicating that fucose (α-1,6)-N-acetylglucosamine, fucose (α-1,3)-N-acetyllactosamine, and α-mannose oligosaccharides containing an N-acetylchito-biose-linked α-fucose residues are the least likely of the glycan structures to be found attached to salivary proteins. Since both of these blots had the lower protein loads (see “Materials and Methods” section), each of the low loaded blots were compared to the original high load blots to find new spots that could be detected in the higher loaded blots. No new spots were detected in the higher loaded blots, and a number of spots blended into the higher background or merged with neighboring spots.
Coreactivity Between Two Lectins
To determine the glycan structures that are most and least often found together, the number of spots showing coreactivity between two lectins was counted (Table 2), and the percent of coreactivity between two lectins was calculated (Table 3). Comparing the data from the various rows in Table 3, the top three lectins showing the most coreactivity with each of the lectins from the panel were BPL, MAL I, and MAL II. Not surprisingly, these were among the top four lectins reacting to the most spots (Table 2). LEL, JAC, and PSA coreacted with the least number of spots. Although JAC and PSA were among the least reactive lectins (Table 2), LEL has relatively moderate total coreactivity. This profile becomes more apparent by comparing the data from the various columns in Table 3, where typically the spots from lectins demonstrating the least reactivity (Table 2) were more likely to be recognized by multiple lectins from the panel. If LEL was consistent with this trend, then it should fall in the middle. Instead, the spots recognized by LEL were near the least recognizable by multiple members of the lectin panel. This suggests that the (β-1,4)-linked N-acetylglucosamine trimers and tetramers primarily recognized by LEL are less likely to be associated with other glycan structures or that other glycan structures maybe inhibiting the binding of LEL. Additional studies using deglycosylating enzymes would help to provide insight as to whether the latter is true, and other studies are necessary to determine if the LEL coreactivity pattern for these two individuals is also true for a broader population.
Population Variation
To address the possibility that the oligosaccharides cataloged in this study exhibit levels of variation which might lead to correlations with other diseases, we assessed the range of population variation for each of the lectins (Table 1). Based on dot-blot quantification of whole resting saliva, the population variation for the individual oligosaccharide motifs ranged from 9,000-fold for the LEL ligand to 3.5-fold for the ConA ligand. Previous work showed that normal individual variation for the concentration of mucins in resting saliva [78] and the concentration of oligosaccharide ligands for DSL, BPL, HHL, and MALI (unpublished) rarely exceed ±30% of an individual’s mean. Thus, individual variation is generally less than population variation and supports the possibility that the amount of variation of an oligosaccharide in the population may have functional significance.
A comparison of population variation (Table 1) with coreactivity (Tables 2 and 3) reveals that two of the three lectins (PSA and JAC) showing the least coreactivity also exhibited the least population variation. One can speculate that while humans appear to have substantial amounts of these oligosaccharides, the distribution of the oligosaccharides is more selective than for most other lectin ligands. Furthermore, since PSA and JAC demonstrated the least amount of coreactivity with each other, it suggests that this selectivity is targeted, in part, to different groups of proteins. In contrast, LEL, which is also in the least coreactive group, showed the greatest amount of population variation and appeared to be rare in some individuals. The fact that LEL exhibited the least coreactivity with PSA and JAC suggests that its selectivity is also targeted to different glycoproteins than either of them.
Glycoprotein Stain Comparison
In comparing the lectin blots to the Pro-Q Emerald glycoprotein-stained gel (Supplemental Figures), the lectins reacted with more spots than did the glycoprotein stain (Supplemental Table) and, therefore, demonstrated that the glycosylation pattern is more widespread than suggested by the glycoprotein-stained gel. This difference may be due in part to the fact that lectin probing is more sensitive than Pro-Q Emerald staining, as the alkaline phosphatase reporter is amplifying the lectin blot signal. Also, the Pro-Q Emerald stain appeared to have preference for particular glycan structures. For example, Pro-Q Emerald-stained spots recognized by lectins AAL and ConA more than 90% of the time but only stained spots recognized by lectin LEL less than 50% of the time (Table 3). Although the overall sensitivity may be weaker, the Pro-Q Emerald stain was very specific for glycoproteins because every spot which was stained also reacted with at least one lectin.
Discussion
The power of 2-D gel electrophoresis is that it can resolve post-translationally modified protein entities into separate, multiple spots. These differences are observed often as a “train” of spots, e.g., salivary α-amylase (spot numbers 29–31, 33–34, and 36–54) and carbonate dehydratase VI (numbers 70–75). Our work demonstrates that more information can be obtained on protein glycosylation when 2-D gels are combined with glycoprotein staining and lectin blotting. A number of studies have used glycoprotein staining to evaluate whether a protein within a spot is glycosylated [69, 79–82]. However, for our human whole saliva study, the glycoprotein staining method missed 40% of the spots that were recognized by the lectins.
The glycosylation for several of the proteins identified from the 2-D gel have been previously characterized by other groups using a number of different techniques. Larsen et al. [83] used titanium dioxide chromatography and mass spectrometry to identify 45 proteins from human whole saliva that had 97 glycosylation sites that were modified with sialic acid. From 2-D gels, we too identified 11 of the 45 proteins: polymeric-immunoglobulin receptor, salivary α-amylase, zinc alpha-2-glycoprotein 1, DMBT1 (glycoprotein 340), lactotransferrin precursor, mucin 5-B, lactotransferrin, lactoperoxidase, immunoglobulin Mu chain C region, kallikrein 1, and myeloperoxidase. All of the spots containing these proteins reacted with one or both of the sialic acid recognizing lectins, SNA and MAL II, though the level of reactivity may have varied for the various isoforms. Others have used lectin affinity chromatography to purify glycoproteins/glycopeptides from complex mixtures such as serum, bile, or saliva. These same lectins used for affinity purification also reacted to some of the spots from our 2-D gel that contained the same proteins. Examples include the following proteins: polymeric-immunoglobulin receptor was purified from bile using lectins ConA and WGA [84]; α-amylase was purified from whole saliva using ConA [85]; immunoglobulin Mu chain C region was purified from serum using multi-lectin affinity chromatography and was found in the ConA and WGA displacement fractions [86]; and immunoglobulin alpha-1 chain C region was purified from serum using ConA, UEA I, and SNA [87]. Since the glycan structure can be tissue- or cell-type specific, the glycosylation for a particular protein found in saliva may not always correlate with those found on that same protein when it comes from another source.
Lectin affinity chromatography is a popular method to capture glycoproteins for proteomic identification and characterization [88–90]. For this strategy, the most commonly used lectins are ConA and WGA, which bind to a broad range of monosaccharides and glycan structures. Our work suggests that ConA and WGA do not capture all glycoproteins from human whole saliva, and other lectins have higher reactivity for salivary glycoproteins. In terms of recognizing the most spots, ConA was moderately reactive and WGA was poor (Tables 2 and 3). Lectin Mal II reacted to the most spots and coreacted at least 83% of the time to the spots that were recognized by the majority of the other lectins. However, for the lectins VVA, DSL, and LEL, the ability to coreact dropped down to 51–66%. Therefore, in order to truly capture the majority of all the glycoproteins or glycopeptides, a multi-affinity column would be needed.
The advantage of using a panel of lectins versus an individual lectin is that one can gain insight about which glycan structures are most and least frequently associated together. Since many glycoproteins contain more than one glycosylation site, the association of staining with multiple lectins may originate from different glycosylation sites on the same glycoprotein. For instance, one protein we identified was immunoglobulin A1 (IgA), which is the principle antibody found in external secretions and thought to be responsible for the protection of the mucosal surfaces from the external environment. Human IgA1 contains two N-linked glycosylation sites in the second domain and in the secreted tailpiece along with three to five O-linked oligosaccharides in the hinged region. Structural analysis of human IgA1 revealed that there are three major N-linked disialylated biantennary complex-type structures containing Gal (β-1,4) GlcNAc [91–93]. A subpopulation (7%) of IgA1 has N-linked oligosaccharides that are triantennary with the outer branched substituted with fucose (α-1,3) [94]. The O-linked oligosaccharides consist of N-acetylgalactosamine with or without a terminal d-galactose in the (β-1,3) configuration [95].
Aberrant glycosylated IgA has been associated with nephropathy and liver diseases. In patients with IgA nephropathy, IgA1 shows increased levels of sialylation, and the O-glycans were found to be incompletely galactosylated [95, 96], thereby exposing a terminal N-acetylgalactosamine. Lectins have been used to characterize the glycoforms from serum of these patients. To study O-linked glycans, lectin VVA was used to detect the ungalactosylated N-acetylgalactosamine moiety, while the lectins Arachis hypoaea (peanut agglutinin (PNA)) and Amaranthus caudatus were employed to detect the complete galactosylated Gal (β-1,3) GalNAc moiety [97]. N-linked glycosylations were characterized with lectin Triticum vulgaris to detect the terminal GlcNAc moiety and Erythrina crystagalli to detect terminal d-Gal of N-linked moieties [97]. Sialylation was characterized with MAL II to detect (α-2,3)-sialic acid and SNA to detect (α-2,6)-sialic acid [96]. Although not all of these lectins were utilized in the current study, the value of this approach is supported by use of other lectins, which have a similar primary binding specificity. By comparing spot numbers 35 and 112, which both contain IGA1 and demonstrate a difference in both MW and pI, this alteration appears to be the result of variation in both N- and O-linked glycosylations.
Beyond their use as carbohydrate recognition tools, lectins provide valuable insight to the potential function of a particular oligosaccharide moiety. One such function is the oligosaccharide-mediated interaction that is involved in cell adhesion of normal and pathogenic processes [98–100]. Therefore, it is not surprising that carbohydrate-binding specificities have been demonstrated for a variety of microbes (viruses, bacteria, fungi, and parasites). The most actively studied bacteria is Helicobacter pylori, one of the pathogens responsible for gastric ulcers, and has been associated with at least seven carbohydrate-binding specificities, including the fucosylated Lewis B blood group antigen [99, 101–103]. Another example is the galactosyl (β-1,4) N-acetylglucosamine ligand preferred by oral bacterium Fusobacterium nucleatum, a potential pathogen for periodontal disease [104]. These specific, exposed, host-cell oligosaccharides will promote colonization of teeth and the mucosal surfaces of the oral cavity and digestive tract, a critical factor in initiating infection. Therefore, lectins identifying these host-cell oligosaccharides may be useful to titrate a host’s potential for interacting with a microbe’s surface lectins or to act as surrogates for the microbe’s binding preferences in screening operations.
Salivary mucins and agglutinins bind a variety of oral microbes [104–107] and thereby play a fundamental role in protection against oral pathogens. Mucins have been shown to possess an enormous repertoire of glycan structures [108–111]. Lectins typically have been used to identify or verify the interacting glycoforms on these large glycoproteins present in saliva [104–107]. Thus, it was somewhat surprising that in the present study, many of the same glycoforms are widely distributed among the salivary proteome. This suggests that the host glycoprotein interactions with planktonic microbes might be more broadly based than expected and could provide protection from colonization by competing with the solid-phase receptors and/or promoting agglutination.
Differences in the total amount of selected oligosaccharides, as quantitated in resting whole saliva by dot-blot, has been correlated with levels of caries disease in young adults and is believed to be useful as a predictor of caries susceptibility in children [71, 74]. During development of a test for assessing this caries susceptibility, it was shown that certain oligosaccharides are strongly positively correlated with caries history, while others are negatively correlated. It is the ratio of these two groups of oligosaccharides that determines an individual’s susceptibility to caries. A question that remains is whether there are a few glycoproteins in saliva possessing the appropriate oligosaccharides, which are positively or negatively involved in the caries process, or if the mechanism is founded on a broadly based, mass action as suggested by the 2-D gel lectin blots. Further studies are needed to resolve this issue.
By understanding global changes in the salivary glycosylation profile, we will gain important insight on the glycoforms present in saliva. This knowledge could provide potential targets for diagnostic tools and therapeutic treatments. In addition, specific glycolyl-targets identified could be used to monitor the success of a treatment for a particular disease. For example, cancer has been shown to cause altered patterns of glycosylation [19]. If a cancer is responding to treatment and has not spread, the altered glycosylation pattern should return to normal. Saliva has clinical advantage over other body fluids such as blood serum and urine since it provides an easy, abundant, noninvasive means to collect samples for diagnostic testing. Biomarkers from saliva have been successfully used to aid in the diagnosis of a number of diseases such as sarcoidosis, rheumatoid arthritis, breast cancer, tuberculosis, lymphoma, and Sjögren’s syndrome [1]. Our group is using a wide variety of proteome techniques [2, 8, 28, 29] to discover disease-related biomarkers from saliva in the hopes of developing a routine saliva diagnostic test to detect, diagnose, and monitor the progression of disease. This research provides a visible 2-D gel glycosylation reference map, which puts in place one more tool for the discovery of biomarkers in saliva. Ultimately, any glycosylation biomarker identified through 2-D lectin blot analysis must be studied in detail through MS/MS analysis to locate the glycosylation site(s), to completely identify the entire glycan structure(s) attached, and to understand all the post-translational modifications associated with a particular protein of interest.
Supporting Information Available
A complete spreadsheet listing the proteins identified for each spots, the reactivity of each of the spots to the lectin panel, and other information such as the experimental and theoretical weights for each of the proteins identified are available in the supplemental material along with the blot images for the entire lectin panel.
References
Slavkin HC. Toward molecularly based diagnostics for the oral cavity. J Am Dent Assoc 1998;129:1138–43.
Denny P, Hagen FK, Hardt M, et al. The proteomes of human parotid and submandibular/sublingual gland salivas collected as the ductal secretions. J Proteome Res 2008;7:1994–2006.
Oppenheim FG, Salih E, Siqueira WL, Zhang W, Helmerhorst EJ. The salivary proteome and its genetic polymorphisms. Ann N Y Acad Sci 2007;1098:22–50.
Guo T, Rudnick PA, Wang W, Lee CS, Devoe DL, Balgley BM. Characterization of the human salivary proteome by capillary isoelectric focusing/nanoreversed-phase liquid chromatography coupled with ESI-tandem MS. J Proteome Res 2006;5:1469–78.
Hirtz C, Chevalier F, Centeno D, et al. Complexity of the human whole saliva proteome. J Physiol Biochem 2005;61:469–80.
Walz A, Stuhler K, Wattenberg A, et al. Proteome analysis of glandular parotid and submandibular-sublingual saliva in comparison to whole human saliva by two-dimensional gel electrophoresis. Proteomics 2006;6:1631–9.
Amado FM, Vitorino RM, Domingues PM, Lobo MJ, Duarte JA. Analysis of the human saliva proteome. Expert Rev Proteomics 2005;2:521–39.
Hu S, Xie Y, Ramachandran P, et al. Large-scale identification of proteins in human salivary proteome by liquid chromatography/mass spectrometry and two-dimensional gel electrophoresis-mass spectrometry. Proteomics 2005;5:1714–28.
Hardt M, Thomas LR, Dixon SE, et al. Toward defining the human parotid gland salivary proteome and peptidome: identification and characterization using 2D SDS-PAGE, ultrafiltration, HPLC, and mass spectrometry. Biochemistry 2005;44:2885–99.
Huang CM. Comparative proteomic analysis of human whole saliva. Arch Oral Biol 2004;49:951–62.
Wilmarth PA, Riviere MA, Rustvold DL, Lauten JD, Madden TE, David LL. Two-dimensional liquid chromatography study of the human whole saliva proteome. J Proteome Res 2004;3:1017–23.
Vitorino R, Lobo MJ, Ferrer-Correira AJ, et al. Identification of human whole saliva protein components using proteomics. Proteomics 2004;4:1109–15.
Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell 2006;126:855–67.
Geyer H, Geyer R. Strategies for analysis of glycoprotein glycosylation. Biochim Biophys Acta 2006;1764:1853–69.
Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 2002;12:43R–56R.
Cobb BA, Kasper DL. Coming of age: carbohydrates and immunity. Eur J Immunol 2005;35:352–6.
Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1999;1473:4–8.
Saso L, Silvestrini B, Guglielmotti A, Lahita R, Cheng CY. Abnormal glycosylation of alpha 2-macroglobulin, a non-acute-phase protein in patients with autoimmune diseases. Inflammation 1993;17:465–79.
Kobata A, Amano J. Altered glycosylation of proteins produced by malignant cells, and application for the diagnosis and immunotherapy of tumours. Immunol Cell Biol 2005;83:429–39.
Hampel DJ, Kottgen B, Dudenhausen JW, Kottgen E. Fetal fibronectin as a marker for an imminent (preterm) delivery. A new technique using the glycoprotein lectin immunosorbent assay. J Immunol Methods 1999;224:31–42.
Parekh RB, Dwek RA, Sutton BJ, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 1985;316:452–7.
Van Nieuw Amerongen A, Ligtenberg AJ, Veerman EC. Implications for diagnostics in the biochemistry and physiology of saliva. Ann N Y Acad Sci 2007;1098:1–6.
Van Nieuw Amerongen A, Bolscher JG, Veerman EC. Salivary proteins: protective and diagnostic value in cariology? Caries Res 2004;38:247–53.
Chaudhuri B, Rojek J, Vickerman MM, Tanzer JM, Scannapieco FA. Interaction of salivary alpha-amylase and amylase-binding-protein A (AbpA) of Streptococcus gordonii with glucosyltransferase of S. gordonii and Streptococcus mutans. BMC Microbiol 2007;7:60.
Van Nieuw Amerongen A, Bolscher JG, Veerman EC. Salivary mucins: protective functions in relation to their diversity. Glycobiology 1995;5:733–40.
Scannapieco FA. Saliva-bacterium interactions in oral microbial ecology. Crit Rev Oral Biol Med 1994;5:203–48.
Carpenter GH, Proctor GB. O-linked glycosylation occurs on basic parotid salivary proline-rich proteins. Oral Microbiol Immunol 1999;14:309–15.
Ramachandran P, Boontheung P, Xie Y, Sondej M, Wong DT, Loo JA. Identification of N-linked glycoproteins in human saliva by glycoprotein capture and mass spectrometry. J Proteome Res 2006;5:1493–503.
Ramachandran P, Boontheung P, Pang E, Yan W, Wong DT, Loo J. Comparison of N-linked glycoproteins in human whole saliva, parotid, submandibular, and sublingual glandular secretions identified using hydrazide chemistry and mass spectrometry. Clin Proteomics 2008;4:80–104.
Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol 2003;21:660–6.
Goldstein IJ. Lectin structure-activity: the story is never over. J Agric Food Chem 2002;50:6583–5.
Sharon N, Lis H. History of lectins: from hemagglutinins to biological recognition molecules. Glycobiology 2004;14:53R–62R.
Liener IE, Sharon N, Goldstein IJ. The Lectins: properties, functions, and applications in biology and medicine. Orlando: Academic; 1986.
Lis H, Sharon N. Lectins as molecules and as tools. Annu Rev Biochem 1986;55:35–67.
Debray H, Montreuil J. Aleuria aurantia agglutinin. A new isolation procedure and further study of its specificity towards various glycopeptides and oligosaccharides. Carbohydr Res 1989;185:15–26.
Matsumoto I, Osawa T. Purification and characterization of an anti-H(O) phytohemagglutinin of Ulex europeus. Biochim Biophys Acta 1969;194:180–9.
Pereira ME, Kisailus EC, Gruezo F, Kabat EA. Immunochemical studies on the combining site of the blood group H-specific lectin 1 from Ulex europeus seeds. Arch Biochem Biophys 1978;185:108–15.
Debray H, Decout D, Strecker G, Spik G, Montreuil J. Specificity of twelve lectins towards oligosaccharides and glycopeptides related to N-glycosylproteins. Eur J Biochem 1981;117:41–55.
Sastry MV, Banarjee P, Patanjali SR, Swamy MJ, Swarnalatha GV, Surolia A. Analysis of saccharide binding to Artocarpus integrifolia lectin reveals specific recognition of T-antigen (beta-D-Gal(1–3)D-GalNAc). J Biol Chem 1986;261:11726–33.
Mahanta SK, Sastry MV, Surolia A. Topography of the combining region of a Thomsen-Friedenreich-antigen-specific lectin jacalin (Artocarpus integrifolia agglutinin). A thermodynamic and circular-dichroism spectroscopic study. Biochem J 1990;265:831–40.
Ahmed H, Chatterjee BP. Further characterization and immunochemical studies on the carbohydrate specificity of jackfruit (Artocarpus integrifolia) lectin. J Biol Chem 1989;264:9365–72.
Knibbs RN, Goldstein IJ, Ratcliffe RM, Shibuya N. Characterization of the carbohydrate binding specificity of the leukoagglutinating lectin from Maackia amurensis. Comparison with other sialic acid-specific lectins. J Biol Chem 1991;266:83–8.
Wang WC, Cummings RD. The immobilized leukoagglutinin from the seeds of Maackia amurensis binds with high affinity to complex-type Asn-linked oligosaccharides containing terminal sialic acid-linked alpha-2,3 to penultimate galactose residues. J Biol Chem 1988;263:4576–85.
Matsumoto I, Uehara Y, Jimbo A, Seno N. Immunochemical and spectral studies on Vicia faba agglutinin. J Biochem (Tokyo) 1983;93:763–9.
Kornfeld K, Reitman ML, Kornfeld R. The carbohydrate-binding specificity of pea and lentil lectins. Fucose is an important determinant. J Biol Chem 1981;256:6633–40.
Yamamoto K, Tsuji T, Osawa T. Requirement of the core structure of a complex-type glycopeptide for the binding to immobilized lentil- and pea-lectins. Carbohydr Res 1982;110:283–9.
Kornfeld R, Ferris C. Interaction of immunoglobulin glycopeptides with concanavalin A. J Biol Chem 1975;250:2614–9.
Baenziger JU, Fiete D. Structural determinants of concanavalin A specificity for oligosaccharides. J Biol Chem 1979;254:2400–7.
Shibuya N, Goldstein IJ, Van Damme EJ, Peumans WJ. Binding properties of a mannose-specific lectin from the snowdrop (Galanthus nivalis) bulb. J Biol Chem 1988;263:728–34.
Kaku H, Van Damme EJ, Peumans WJ, Goldstein IJ. Carbohydrate-binding specificity of the daffodil (Narcissus pseudonarcissus) and amaryllis (Hippeastrum hybr.) bulb lectins. Arch Biochem Biophys 1990;279:298–304.
Osawa T, Irimura T, Kawaguchi T. Bauhinia purpurea agglutinin. Methods Enzymol 1978;50:367–72.
Wu AM, Kabat EA, Gruezo FG, Allen HJ. Immunochemical studies on the combining site of the D-galactopyranose and 2-acetamido-2-deoxy-D-galactopyranose specific lectin isolated from Bauhinia purpurea alba seeds. Arch Biochem Biophys 1980;204:622–39.
Kaladas PM, Kabat EA, Kimura A, Ersson B. The specificity of the combining site of the lectin from Vicia villosa seeds which react with cytotoxic T-lymphoblasts. Mol Immunol 1981;18:969–77.
Tollefsen SE, Kornfeld R. Isolation and characterization of lectins from Vicia villosa. Two distinct carbohydrate binding activities are present in seed extracts. J Biol Chem 1983;258:5165–71.
Desai NN, Allen AK, Neuberger A. Some properties of the lectin from Datura stramonium (thorn-apple) and the nature of its glycoprotein linkages. Biochem J 1981;197:345–53.
Ashford D, Desai NN, Allen AK, Neuberger A, O'Neill MA, Selvendran RR. Structural studies of the carbohydrate moieties of lectins from potato (Solanum tuberosum) tubers and thorn-apple (Datura stramonium) seeds. Biochem J 1982;201:199–208.
Nachbar MS, Oppenheim JD. Tomato (Lycopersicon esculentum) lectin. Methods Enzymol 1982;83:363–8.
Kilpatrick DC. Purification and some properties of a lectin from the fruit juice of the tomato (Lycopersicon esculentum). Biochem J 1980;185:269–72.
Allen AK, Neuberger A, Sharon N. The purification, composition and specificity of wheat-germ agglutinin. Biochem J 1973;131:155–62.
Bhavanandan VP, Katlic AW. The interaction of wheat germ agglutinin with sialoglycoproteins. The role of sialic acid. J Biol Chem 1979;254:4000–8.
Goldstein IJ, Hammarstrom S, Sundblad G. Precipitation and carbohydrate-binding specificity studies on wheat germ agglutinin. Biochim Biophys Acta 1975;405:53–61.
Konami Y, Yamamoto K, Osawa T, Irimura T. Strong affinity of Maackia amurensis hemagglutinin (MAH) for sialic acid-containing Ser/Thr-linked carbohydrate chains of N-terminal octapeptides from human glycophorin A. FEBS Lett 1994;342:334–8.
Shibuya N, Goldstein IJ, Broekaert WF, Nsimba-Lubaki M, Peeters B, Peumans WJ. The elderberry (Sambucus nigra L.) bark lectin recognizes the Neu5Ac(alpha 2–6)Gal/GalNAc sequence. J Biol Chem 1987;262:1596–601.
Dai Z, Liu YK, Cui JF, et al. Identification and analysis of altered alpha1,6-fucosylated glycoproteins associated with hepatocellular carcinoma metastasis. Proteomics 2006;6:5857–67.
Ohtsu I, Nakanisi T, Furuta M, Ando E, Nishimura O. Direct matrix-assisted laser desorption/ionization time-of-flight mass spectrometric identification of proteins on membrane detected by Western blotting and lectin blotting. J Proteome Res 2005;4:1391–6.
Kim YS, Hwang SY, Oh S, et al. Identification of target proteins of N-acetylglucosaminyl-transferase V and fucosyltransferase 8 in human gastric tissues by glycomic approach. Proteomics 2004;4:3353–8.
Ekuni A, Miyoshi E, Ko JH, et al. A glycomic approach to hepatic tumors in N-acetylglucosaminyltransferase III (GnT-III) transgenic mice induced by diethylnitrosamine (DEN): identification of haptoglobin as a target molecule of GnT-III. Free Radic Res 2002;36:827–33.
Taniguchi N, Ekuni A, Ko JH, et al. A glycomic approach to the identification and characterization of glycoprotein function in cells transfected with glycosyltransferase genes. Proteomics 2001;1:239–47.
Birrell GW, Earl S, Masci PP, et al. Molecular diversity in venom from the Australian Brown snake, Pseudonaja textilis. Mol Cell Proteomics 2006;5:379–89.
Ramagli LS. Quantifying protein in 2-D PAGE solubilization buffers. Methods Mol Biol 1999;112:99–103.
Denny PC, Denny PA, Takashima J, Galligan J, Navazesh M. A novel caries risk test. Ann N Y Acad Sci 2007;1098:204–15.
Smejkal GB, Robinson MH, Lazarev A. Comparison of fluorescent stains: relative photostability and differential staining of proteins in two-dimensional gels. Electrophoresis 2004;25:2511–9.
Lopez MF, Berggren K, Chernokalskaya E, Lazarev A, Robinson M, Patton WF. A comparison of silver stain and SYPRO Ruby Protein Gel Stain with respect to protein detection in two-dimensional gels and identification by peptide mass profiling. Electrophoresis 2000;21:3673–83.
Denny PC, Denny PA, Takashima J, Si Y, Navazesh M, Galligan JM. A novel saliva test for caries risk assessment. J Calif Dent Assoc 2006;34:287–290, 292–4.
Perry GH, Dominy NJ, Claw KG, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet 2007;39:1256–60.
Loster K, Kannicht C. 2D-electrophoresis. Detection of glycosylation and influence on spot pattern. Methods Mol Biol 2002;194:301–16.
Brennan SO. Electrospray ionization mass analysis of normal and genetic variants of human serum albumin. Clin Chem 1998;44:2264–9.
Denny PA, Wu W, Denny PC. Individual variation of mucin concentration in human saliva. J Dent Res 2002;81:A–501.
Serrano SM, Shannon JD, Wang D, Camargo AC, Fox JW. A multifaceted analysis of viperid snake venoms by two-dimensional gel electrophoresis: an approach to understanding venom proteomics. Proteomics 2005;5:501–10.
Kanninen K, Goldsteins G, Auriola S, Alafuzoff I, Koistinaho J. Glycosylation changes in Alzheimer's disease as revealed by a proteomic approach. Neurosci Lett 2004;367:235–40.
Alvarez S, Goodger JQ, Marsh EL, Chen S, Asirvatham VS, Schachtman DP. Characterization of the maize xylem sap proteome. J Proteome Res 2006;5:963–72.
Van Dellen KL, Chatterjee A, Ratner DM, et al. Unique posttranslational modifications of chitin-binding lectins of Entamoeba invadens cyst walls. Eukaryot Cell 2006;5:836–48.
Larsen MR, Jensen SS, Jakobsen LA, Heegaard NH. Exploring the sialiome using titanium dioxide chromatography and mass spectrometry. Mol Cell Proteomics 2007;6:1778–87.
Kristiansen TZ, Bunkenborg J, Gronborg M, et al. A proteomic analysis of human bile. Mol Cell Proteomics 2004;3:715–28.
Koyama I, Komine S, Yakushijin M, Hokari S, Komoda T. Glycosylated salivary alpha-amylases are capable of maltotriose hydrolysis and glucose formation. Comp Biochem Physiol B Biochem Mol Biol 2000;126:553–60.
Yang Z, Hancock WS. Approach to the comprehensive analysis of glycoproteins isolated from human serum using a multi-lectin affinity column. J Chromatogr A 2004;1053:79–88.
Madera M, Mechref Y, Klouckova I, Novotny MV. High-sensitivity profiling of glycoproteins from human blood serum through multiple-lectin affinity chromatography and liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2007;845:121–37.
Plavina T, Wakshull E, Hancock WS, Hincapie M. Combination of abundant protein depletion and multi-lectin affinity chromatography (M-LAC) for plasma protein biomarker discovery. J Proteome Res 2007;6:662–71.
Madera M, Mechref Y, Klouckova I, Novotny MV. Semiautomated high-sensitivity profiling of human blood serum glycoproteins through lectin preconcentration and multidimensional chromatography/tandem mass spectrometry. J Proteome Res 2006;5:2348–63.
Qiu R, Regnier FE. Use of multidimensional lectin affinity chromatography in differential glycoproteomics. Anal Chem 2005;77:2802–9.
Field MC, Amatayakul-Chantler S, Rademacher TW, Rudd PM, Dwek RA. Structural analysis of the N-glycans from human immunoglobulin A1: comparison of normal human serum immunoglobulin A1 with that isolated from patients with rheumatoid arthritis. Biochem J 1994;299(Pt 1):261–75.
Mattu TS, Pleass RJ, Willis AC, et al. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fc alpha receptor interactions. J Biol Chem 1998;273:2260–72.
Rifai A, Fadden K, Morrison SL, Chintalacharuvu KR. The N-glycans determine the differential blood clearance and hepatic uptake of human immunoglobulin (Ig)A1 and IgA2 isotypes. J Exp Med 2000;191:2171–82.
Stockert RJ. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol Rev 1995;75:591–609.
Novak J, Julian BA, Tomana M, Mesteck J. Progress in molecular and genetic studies of IgA nephropathy. J Clin Immunol 2001;21:310–27.
Leung JC, Tang SC, Chan DT, Lui SL, Lai KN. Increased sialylation of polymeric lambda-IgA1 in patients with IgA nephropathy. J Clin Lab Anal 2002;16:11–9.
Allen AC, Harper SJ, Feehally J. Galactosylation of N- and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clin Exp Immunol 1995;100:470–4.
Sharon N, Lis H. Carbohydrates in cell recognition. Sci Am 1993;268:82–9.
Karlsson KA. Microbial recognition of target-cell glycoconjugates. Curr Opin Struct Biol 1995;5:622–35.
Prakobphol A, Boren T, Ma W, Zhixiang P, Fisher SJ. Highly glycosylated human salivary molecules present oligosaccharides that mediate adhesion of leukocytes and Helicobacter pylori. Biochemistry 2005;44:2216–24.
Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 2006;19:449–90.
Ilver D, Arnqvist A, Ogren J, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998;279:373–7.
Boren T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 1993;262:1892–5.
Gillece-Castro BL, Prakobphol A, Burlingame AL, Leffler H, Fisher SJ. Structure and bacterial receptor activity of a human salivary proline-rich glycoprotein. J Biol Chem 1991;266:17358–68.
Ligtenberg AJ, Veerman EC, Nieuw Amerongen AV. A role for Lewis a antigens on salivary agglutinin in binding to Streptococcus mutans. Antonie Van Leeuwenhoek 2000;77:21–30.
Groenink J, Ligtenberg AJ, Veerman EC, Bolscher JG, Nieuw Amerongen AV. Interaction of the salivary low-molecular-weight mucin (MG2) with Actinobacillus actinomycetemcomitans. Antonie Van Leeuwenhoek 1996;70:79–87.
Prakobphol A, Tangemann K, Rosen SD, Hoover CI, Leffler H, Fisher SJ. Separate oligosaccharide determinants mediate interactions of the low-molecular-weight salivary mucin with neutrophils and bacteria. Biochemistry 1999;38:6817–25.
Thomsson KA, Prakobphol A, Leffler H, et al. The salivary mucin MG1 (MUC5B) carries a repertoire of unique oligosaccharides that is large and diverse. Glycobiology 2002;12:1–14.
Hang HC, Bertozzi CR. The chemistry and biology of mucin-type O-linked glycosylation. Bioorg Med Chem 2005;13:5021–34.
Klein A, Carnoy C, Wieruszeski JM, et al. The broad diversity of neutral and sialylated oligosaccharides derived from human salivary mucins. Biochemistry 1992;31:6152–65.
Bolscher J, Veerman E, Van Nieuw Amerongen A, Tulp A, Verwoerd D. Distinct populations of high-M(r) mucins secreted by different human salivary glands discriminated by density-gradient electrophoresis. Biochem J 1995;309(Pt 3):801–6.
Acknowledgments
We would like to thank Rachel Ogorzalek Loo for her help with the 2-D gel work and mass spectrometry data analysis, Dan Malamud for his valuable insight and recommendations, Deborah Leon-Russell for sharing her lectin blot results from Methanosarcina acetivorans, and Yang Li for collecting the whole saliva. In addition, we would like to thank Shawn Than, Weixia Yu, and Weihong Yan for construct of the Human Salivary Proteome Project database. The 2-D gel work was performed in the UCLA Mass Spectrometry and Proteomics Technology Center, which was established with a grant from the W. M. Keck Foundation, and the 2-D gel images were generated in the UCLA Biological Chemistry Imaging Facility. This work was supported by the National Institutes of Health (National Institute of Dental and Craniofacial Research; U01 DE016275 to DTW and JAL) and UCLA Fundamental Clinical Research Training Grant (T32) (DE007296 to P. R.).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary materials.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sondej, M., Denny, P.A., Xie, Y. et al. Glycoprofiling of the Human Salivary Proteome. Clin Proteom 5, 52–68 (2009). https://doi.org/10.1007/s12014-008-9021-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12014-008-9021-0