
Abstract
Cardiotoxic side-effects of doxorubicin limit the clinical use of this anti-cancer agent. Iron chelators have been studied as protectors against doxorubicin-induced cardiotoxicity. These iron chelators do not provide optimal protection and have certain drawbacks. We therefore looked for new protectors and decided that these new compounds should combine iron chelating and antioxidant activity. Flavonoids appeared to possess those combined iron chelating and antioxidant properties. Quantum chemical evaluation of radical stabilization and determination of physico-chemical properties of a series of flavonoids brought our attention to the semi-synthetic flavonoid 7-monohydroxyetylrutoside (monoHER). Both in vitro (using an electrically paced mouse left atrium model) and in vivo (using a mouse ECG telemetry model) experiments corroborated the protective effect of monoHER. MonoHER also showed anti-inflammatory properties. A subsequent clinical phase I study showed that an i.v. dose of 1,500 mg/m2 is a feasible and safe dose to be evaluated in a phase II study to investigate the protective properties of monoHER against doxorubicin-induced cardiotoxicity in cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Molecular mechanism of the cardiotoxicity of doxorubicin
The clinical use of doxorubicin, a widely used anthracycline is hampered by the common side-effects observed upon use of the majority of anti-cancer agents, viz. bone marrow suppression, alopecia, nausea and vomiting. Doxorubicin-induced bone marrow suppression can now be reduced by the use of haematopoietic growth factors. In addition to these rather common acute toxicities, anthracyclines contain the risk of causing a cardiomyopathy. In fact, the development of cumulative dose-related cardiotoxicity currently forms the major limitation of doxorubicin use in the clinic.
The production of free radicals as by-product of doxorubicin metabolism is considered to be the main mechanism of doxorubicin-induced cardiotoxicity. The quinone moiety of doxorubicin is converted into a semiquinone form by the acquisition of one electron. This conversion can occur either enzymatically or nonenzymatically. The enzymatic reduction of the doxorubicin quinone ring is performed by cytochrome P450 reductase, NADH dehydrogenase and cytochrome P450 [1]. The produced semiquinone form is oxidized by molecular oxygen, which yields back doxorubicin in its quinone form with concomitant production of superoxide anion radicals. This process is called redox cycling. Superoxide radicals can dismutate either enzymatically catalyzed by superoxide dismutase or, albeit with a lower rate, spontaneously. From this dismutation hydrogen peroxide is formed. This may lead to toxicity. Iron ions have been suggested to play a crucial role in this process. Not only because iron reacts with hydrogen peroxide to form the reactive hydroxyl radical but also because Fe3+ may form complexes with doxorubicin. The theory on the iron-dependent formation of free radicals by doxorubicin semiquinones is supposed to be the most prevailing one to explain the cardiotoxicity. An internal electron shift may give reduction of iron, which subsequently delivers this electron to molecular oxygen yielding superoxide anion radicals.
The need for new protectors
Iron chelators have been studied as protectors against doxorubicin-induced cardiotoxicity. The bispiperazinedione ICRF 187 has been studied extensively. ICRF 187 is also known as dexrazoxane and is the more water soluble (+)enantiomorph of the racemic mixture ICRF-159, which was originally developed as a conventional antineoplastic agent [2]. In phase I trials using a variety of schedules in both adults and children, dose-limiting toxicities on most schedules were myelosuppression, except in children, in whom the major dose-limiting toxicity was hepatotoxicity [3]. In addition, research conducted at the same time as the initial phase I studies showed that the divalent chelating properties of ICRF-159 and 187 could protect animals from the cardiotoxicity induced by anthracyclines [4, 5]. This further corroborated the catalytic toxic role of iron ions in the doxorubicin-induced cardiotoxicity. There are several disadvantages associated with the use of ICRF-187. ICRF-187 exerts a dose-dependent myelosuppression, which may limit its addition to chemotherapy. Moreover, controversy exists as to whether ICRF-187 decreases the antitumour activity of doxorubicin. ICRF-187 is a prodrug which requires hydrolysis to achieve the active form. Finally, ICRF-198, the hydrolyzed form of ICRF-187 is an EDTA-like chelator. This implies that because of stoichiometrical restrictions the drug lacks the ability to occupy all six coordination sites of iron. Consequently, iron remains catalytically active to generate oxygen radicals. Besides iron chelators as ICRF-187 and for example deferoxamine, many other compounds have been evaluated for their potential to reduce doxorubicin-induced cardiotoxicity, based on a perceived ability to modulate some of the biochemical alterations that accompany doxorubicin administration. The list of compounds includes adenosine, anti-histamine agents (both H1 and H2 blockers), beta-adrenoceptor antagonists, amrinone, milrinone, fructose-1,6-diphosphate, inosine, ibuprofen, dextran, methylprednisone, salts of bismuth, zinc and calcium, tetracycline antibiotics and a number of iron chelators as deferoxamine, ICRF-187. In addition, various antioxidants have been applied as vitamin E, curcumin, lipoic acid and venoruton.
In view of the suggested molecular mechanism involved in doxorubicin-induced cardiotoxicity we searched for protectors which combine iron-chelating and oxygen radical scavenging properties. Moreover, the novel protector should not interfere with the cytostatic activity of doxorubicin.
The choice for flavonoids
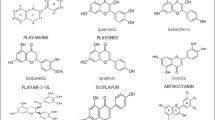
A large series of flavonoids from several subclasses were screened by quantum chemical calculation (Fig. 1) and physico-chemical characterization to find antioxidants with both iron chelating and oxygen radical scavenging properties [6–8]. To study the direct antioxidant action of the flavonoids, the compounds were tested in the iron-independent liver microsomal lipid peroxidation, which was induced with azobisamidinopropane (ABAP), a hydrophilic azo-initiator. ABAP generates a constant radical flow in the water phase of the lipid peroxidation system. An excess of EDTA was added to inactivate all traces of iron. A good correlation was found between the inhibition (pIC50-value) of the ABAP-induced lipid peroxidation by the flavonoids and their half-peak oxidation potential (Ep/2) (Fig. 2). In contrast to the ABAP-induced lipid peroxidation where iron does not play a role in the initiation of lipid peroxidation, iron is strongly involved in the lipid peroxidation induced with Fe2+/ascorbic acid. Also in the enzymatic doxorubicin induced lipid peroxidation iron is suggested to play a role. The correlations of the inhibition of the lipid peroxidation processes in which iron is involved are weaker than the correlation obtained in the iron-independent lipid peroxidation process (Fig. 2). This suggests that besides the direct radical scavenging effect also the iron chelating effect of the flavonoids is involved in their antioxidant action.

Spin distribution of the quercetine and taxifoline radical. Small changes in the molecule (in this case a 2–3 double bond) lead to variation in the spin distribution and to changes in the antioxidant activity of the molecule

Correlation between inhibition (pIC50-value) of ABAP-induced lipid peroxidation by several flavonoids and their half-peak oxidation potential. Lipid peroxidation was induced in mouse liver microsomes with 10 mmol/l ABAP in the presence of 100 μmol/l Na2EDTA at 37°C for 1.5 h
Protection against negative inotropic action ex vivo and in vitro
We subsequently tested a selection of promising flavonoids which possess combined antioxidant and iron-chelating properties in a series of in vitro and ex vivo organ bath studies [9–11]. The flavonoid 7-monohydroxyethylrutoside was able to protect almost completely (93%) against the negative inotropic effect of 30 μM doxorubicin. This concentration of doxorubicin gives 50% inhibition of the inotropy of the isolated electrically paced mouse left atrium (Fig. 3).

Negative inotropic action of 30 μmol/l doxorubicin in the isolated mouse left atrium. Maximal protection by pre-incubation with 7-monohydroxyethyl rutoside was 93%
Protection against in vivo changes induced by doxorubicin in the ECG of mice
The promising protector 7-monohydroxyethylrutoside was further evaluated in vivo in a comparative study with ICRF-187. Balb/c mice of 20–25 g were equipped with a telemeter to measure ECG (Fig. 4). They were given 6 i.v. doses of doxorubicin (4 mg/kg) at weekly intervals. ICRF-187 (50 mg/kg) or 7-monohydroxyethylrutoside at 100, 250 or 500 mg/kg were given i.p. 1 h prior to doxorubicin administration [12, 13]. A saline and a 7-monohydroxyethylrutoside treated group served as control. At the end of the study (week 8) the ST-interval of the ECG had increased by 16.7 ± 2.7 ms (mean ± s.e. mean) in the doxorubicin-treated mice. At the same time, the ST interval had increased by only 1.8 ± 0.9 ms in ICRF-187 co-medicated mice and in 7-monohydroxyethylrutoside co-medicated mice by only 7.8 ± 1.7 ms, 4.6 ± 0.7 ms, 1.7 ± 0.8 ms for 100, 250 and 500 mg/kg, respectively. The ECG of the control animals did not change during the study. The QRS complex did not change in either group (Fig. 4).

Dose dependent protection by 7-monoHER (i.p.) on the ST-interval lengthening of the ECG of mice treated with doxorubicin. Image on the right shows the X-ray of a mouse with a transmitter to measure the ECG via telemetry
Lack of interference with the tumour growth inhibition in vitro and in vivo
Three cell lines were used i.e. the human ovarian cell lines A2780 and OVCAR-3 and the human breast cancer cell line MCF-7 to check for the effect of the selected flavonoid 7-monohydroxyethylrutoside on the IC50 values of growth inhibition by doxorubicin (Table 1). A2780 cells were approximately 10-fold more sensitive to doxorubicin than MCF-7 and OVCAR-3 cells as measured from the IC50 values of 2.05 × 10−8, 2.09 × 10−7 and 3.28 × 10−7 M, respectively. The compound 7-monohydroxyethylrutoside was not cytotoxic at concentrations of 50 and 100 μM (Table 1). More importantly, 7-monohydroxyethylrutoside did not significantly influence the IC50 of doxorubicin in the investigated cell lines (Table 1).
The antitumour activity of doxorubicin (8 mg/kg i.v. twice weekly) in A2780 xenografts and OVCAR-3 (Fig. 5) xenografts in nude mice was not decreased by 7-monohydroxyethylrutoside, administered 1 h before doxorubicin in a dose schedule of 500 mg/kg i.p. 2 or 5 days per week [13].

Growth of OVCAR-3 xenografts in nude mice and the influence of the cardioprotectors monoHER and ICRF-187 on the antitumour activity of doxorucin. Control group: no treatment, Doxorubicin group: 8 mg/kg i.v., on days 0 and 7, Doxorubicin + monoHER 5-day group: 500 mg/kg i.p., days 0–4 and 7–11 (monoHER on days 0 and 7 were given 1 h prior to doxorubicin), Doxorubicin + monoHER 2 day group: 500 mg/kg i.p., days 0 and 1 and 7 and 8 (monoHER on days 0 and 7 were given 1 h prior to doxorubicin), Doxorubicin + ICRF-187 group: 100 mg/kg ICRF i.p., days 0 and 7 (ICRF-187 was given 1 h prior to doxorubicin), MonoHER 5-day group: 500 mg/kg i.p., days 0–4 and 7–11 without doxorubicin
Involvement of iron, inflammation and superoxide anion radicals
Although the initial choice for selecting 7-monohydroxyethylrutoside was among others based on its iron-chelating properties, we recently obtained data, which question the involvement of iron in oxidative stress mediated toxicity in doxorubicin. We compared several well-known iron chelators (ICRF-187, 7-monohydroxyethylrutoside, deferoxamine and pyridoxal isonicotinoyl hydrazone) and found that only ICRF-187 and 7-monohydroxyethylrutoside protected the human lung adenocarcinoma cells A549 against doxorubicin-induced oxidative stress while other chelators did not [14].
Human umbilical cord vascular endothelial cells (HUVECs) were incubated with increasing concentrations of doxorubicin (up to 25 μM) to investigate whether doxorubicin could induce an inflammatory response in vitro. It appeared that proliferating HUVECs were more sensitive to doxorubicin (IC50 value of 60 ± 21 nM) than resting cells (IC50 value of 4.0 ± 0.3 μM) [15]. Doxorubicin also increased the adhesion of neutrophils, which was accompanied by overexpression of VCAM and E-selectin. Treatment with 1 mM of 7-monohydroxyethylrutoside prevented this overexpression. The increased levels of adhesion molecules probably explain the stimulation of neutrophil adhesion to the endothelial cells by 0.6 μM doxorubicin. The flavonoid 7-monohydroxyethylrutoside (1 mM) prevents the doxorubicin-stimulated neutrophil adhesion [15].
This protective effect of the flavonoid against inflammation was also observed recently in a study on ischaemia-reperfusion in mice [16]. In this study, heart ischaemia was induced for 30 min by ligating the left anterior descending coronary artery. Afterwards, the ligature was removed and reperfusion was allowed for 6 or 24 h. The protector 7-monohydroxyethylrutoside was given i.p. (500 mg/kg) 1 h before reperfusion and strongly attenuated myocardial neutrophil influx at both 6 and 24 h after reperfusion by 58% and 49%, respectively.
Clinical studies
A phase I human volunteer i.v. dose of monoHER escalation study was performed in order to find the maximal plasma concentration (C max) and the area under the plasma concentration time curve (AUC∞) as observed in mice after administration of the protecting dose of 500 mg/kg monoHER i.p. The study was performed as a single blind, randomized trial in healthy volunteers (age between 19 and 56 years). At each dose level, six subjects received monoHER and three placebo. MonoHER was solubilized in 100 ml dextrose 5% and administered as an i.v. infusion in 10 min. The placebo consisted of 100 ml dextrose 5%. The i.v. starting dose in human volunteers of monoHER was 100 mg/m2. Dose escalation by 100% of the preceding dose took place after finishing each dose level until the protecting pharmacokinetic values for C max and AUC∞ (as observed in mice after 500 mg/kg monoHER i.p.) were reached and/or serious side effects were observed. The dose was escalated up to 1500 mg/m2. The mean values of C max and AUC∞ were 360 ± 69.3 μM and 6.8 ± 2.1 μmol min/ml, respectively. These values were comparable to the C max and AUC∞ observed under the protecting conditions in mice. No serious side effects occurred during the entire study. From this phase I study it could be concluded that 1,500 mg/m2 is a feasible and safe dose to be evaluated in a phase II study to investigate the protective properties of monoHER against doxorubicin-induced cardiotoxicity in cancer patients [17]. This phase II study is currently conducted.
References
Goeptar, A. R., Te Koppele, J. M., Lamme, E. K., Piqué, J. M., & Vermeulen, N. P. E. (1993). Cytochrome P450 2B1-mediated one-electron reduction of adriamycin: A study with rat liver microsomes and purified enzymes. Molecular Pharmacology, 44, 1267–1277.
Creighton, A. M., Hellmann, K., & Whitecross, S. (1969). Antitumour activity in a series of bisketopiperazines. Nature, 222, 384–385.
Holcenberg, J. S., Tutsch, K. D., Earhart, R. H., Ungerleider, R. S., Kamen, B. A., Pratt, C. B., Gribble, T. J., & Glaubiger, D. L. (1986). Phase I study of ICRF-187 in paediatric cancer patients and comparison of its pharmacokinetics in children and adults. Cancer Treatment Reports, 70, 703–709.
Decorti, G., Bartoli Klugmann, F., Mallardi, F., Klugmann, S., Benussi, B., Grill, V., & Baldini, L. (1983). Effects of ICRF 159 on adriamycin-induced cardiomyopathy in rats. Cancer Letters, 19, 77–83.
Perkins, W. E., Schroeder, R. L., Carrano, R. A., & Imondi, A. R. (1982). Effect of ICRF-187 on doxorubicin-induced myocardial effects in the mouse and guinea pig. British Journal of Cancer, 46, 662–667.
van Acker, S. A. B. E., van den Berg, D.-J., Tromp, M. N. J. L., Griffioen, D. H., van Bennekom, W. P., van der Vijgh, W. J. F., & Bast, A. (1996a). Structural aspects of antioxidant activity of flavonoids. Free Radical Biology & Medicine, 20, 331–342.
van Acker, S. A. B. E., van den Berg, D.-J., Tromp, M. N. J. L., Griffioen, D. H., van Bennekom, W. P., van der Vijgh, W. J. F., & Bast, A. (1996b). A quantum chemical explanation for the antioxidant activity of flavonoids. Chemical Research in Toxicology, 9, 1305–1312.
van Acker, S. A. B. E., Plemper van Balen, G., van den Berg, D.-J., Bast, A., & van der Vijgh, W. J. F. (1998). Influence of iron chelation on the antioxidant activity of flavonoids. Biochemical Pharmacolgy, 56, 935–943.
van Acker, S. A. B. E., Voest, E. E., Beems, D. B., Madhuizen, H. T., de Jong, J., Bast, A., & van der Vijgh, W. J. F. (1993). Cardioprotective properties of O-(beta-hydroxyethyl)-rutosides in doxorubicin-pretreated BALB/c mice. Cancer Research, 53, 4603–4607.
Voest, E. E., van Acker, S. A. B. E., van der Vijgh, W. J. F., van Asbeck, B. S., & Bast, A. (1994). Comparison of different iron chelators as protective agents against acute doxorubicin-induced cardiotoxicity. Journal of Molecular and Cellular Cardiology, 26, 1179–1185.
Hüsken, B. C. P., de Jong, J., Beekman, B., Onderwater, R. C. A., van der Vijgh, W. J. F., & Bast, A. (1995). Modulation of the in vitro cardiotoxicity of doxorubicin by flavonoids. Cancer Chemotherapy and Pharmacology, 37, 55–62.
van Acker, S. A. B. E., Kramer, K., Grimbergen, J. A., van den Berg, D.-J., van der Vijgh, W. J. F., & Bast, A. (1995). Monohydroxyethylrutoside as protector against chronic doxorubicin-induced cardiotoxicity. British Journal of Pharmacology, 115, 1260–1264.
van Acker, S. A. B. E., Boven, E., Kuiper, K., van den Berg, D.-J., Grimbergen, J. A., Kramer, K., Bast, A., & van der Vijgh, W. J. F. (1997). Monohydroxyethylrutoside, a dose dependent cardioprotective agent, does not affect the antitumor activity of doxorubicin. Clinical Cancer Research, 3, 1747–1754.
Kaiserova, H., den Hartog, G. J. M., Simunek, T., Schoterova, L., Kvasnickova, E., & Bast, A. (2006). Iron is not involved in oxidative stress-mediated cytotoxicity of doxorubicin and bleomycin. British Journal of Pharmacology, 149, 920–930.
Abou El Hassan, M. A. I., Verheul, H. M. W., Jorna, A. S., Schalkwijk, C., van Bezu, J., van der Vijgh, W. J. F., & Bast, A. (2003). The new cardioprotector monohydroxyethylrutoside protects against doxorubicin-induced inflammatory effects in vitro. British Journal Cancer, 89, 357–362.
de Celle, T., Heeringa, P., Strzelecka, A. E., Bast, A., Smits, J. F., & Janssen, B. J. (2004). Sustained protective effects of 7-monohydroxyethylrutoside in an in vivo model of cardiac ischemia-reperfusion. European Journal of Pharmacology, 494, 205–212.
Willems, A. M., Bruynzeel, A. M., Kedde, M. A., van Groeningen, C. J., Bast, A., & van der Vijgh, W. J. F. (2006). A phase I study of monohydroxyethylrutoside in healthy volunteers. Cancer Chemotherapy and Pharmacology, 57, 678–684.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bast, A., Haenen, G.R.M.M., Bruynzeel, A.M.E. et al. Protection by flavonoids against anthracycline cardiotoxicity: from chemistry to clinical trials. Cardiovasc Toxicol 7, 154–159 (2007). https://doi.org/10.1007/s12012-007-0018-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-007-0018-0