
Abstract
Chronic overexposure to cobalt (Co) may result in neurotoxic effects, but the mechanism of Co-induced neurotoxicity is not yet well established. Our study was conducted to determine whether Co is associated to the induction of central nervous system damage in pregnant rats and their progeny. Twelve pregnant female rats were randomly divided into 2 groups: group I served as controls and group II received Co (350 mg/L, orally). Treatments started from the 14th day of pregnancy until day 14 after delivery. Co concentration in plasma was higher in the treated groups than in the controls. Exposure to Co also increased the levels of MDA, PCO, H2O2, and AOPP, while Na+K+-ATPase and Mg2+-ATPase, AChE, and BuChE activities decreased in the cerebrum and cerebellum of suckling pups. A smear without ladder formation on agarose gel was also shown in the cerebrum and cerebellum, indicating random DNA degradation. A reduction in GPx, SOD, CAT, GSH, NPSH, and vitamin C values was observed. The changes were confirmed by histological results. In conclusion, these data showed that the exposure of pregnant and lactating rats to Co resulted in the development of oxidative stress and the impairment of defense systems in the cerebrum and cerebellum of their suckling pups.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cobalt (Co) emission into the atmospheric, aquatic, and terrestrial environments increased dramatically during the twentieth century. Stable Co may enter the environment from both natural sources and human activities. Generally, human Co poisoning can result from occupational, accidental, and intentional exposures [1]. Indeed, increased levels of Co were found in the urine and blood of occupationally exposed workers [2]. This heavy metal is suspected to have neurotoxic effects, as indicated by reports on memory deficit among workers exposed to hard metal both as dust powder and in mist form [3]. On the other hand, gestation impairment and its complications during pregnancy and childbirth have been reported in humans and animals exposed to Co [4]. Indeed, Co crosses the placenta and may also be found in milk, causing many disorders in newborns [5]. This heavy metal is recognized as being genotoxic and carcinogenic [6]. The action of Co ions is mediated, at least in part, by their ability to enter Fenton-type reactions [7] and trigger the production of reactive oxygen species (ROS). The latter are constantly produced and eliminated by living organisms, which results in a certain ROS steady-state level. An imbalance between ROS production and elimination in favor of the former and disturbing core and regulatory processes is called oxidative stress [8]. High ROS levels cause damage via the oxidative modification of lipids, proteins, and DNA of many cell types, including neural brain cells [9, 10]. The latter tissue is especially vulnerable to oxidative damage for a number of reasons: (1) it has a high concentration of polyunsaturated fatty acids (PUFA) compared to any other tissue in the body [11]; (2) the brain has a very high oxygen content and it consumes 20 % of the blood’s oxygen, even though it makes up only 2–4 % of the body weight [12]; and (3) it undergoes a progressive accumulation of iron levels with aging [13]. Moreover, the brain uses a relatively large amount of oxygen at rather low activities of antioxidative enzymes [14]. Consequently, there is evidence of oxidative stress as an important mechanism of neuropathological disorders such as Alzheimer’s disease, schizophrenia, and Parkinson’s disease [15].
The brain, being a very sensitive tissue to oxidative stress, is vulnerable to damage induced by several chemical products, including metals. It exhibits distinct variations in the cellular as well as regional distribution of antioxidant defenses [16]. Thus, neural cells and/or brain regions respond differentially to metabolic rate changes associated with ROS generation [17]. Indeed, there is evidence invoking regional sensitivity to oxidative stress that is dependent on cellular and regional redox status [18]. According to some authors, the cerebellum is more susceptible to oxidative damage relative to other brain regions [19]. Although the effects of Co on the olfactory system have been investigated in adult rats [20], there are, to our knowledge, no reports about the effects of this metal on the cerebrum and cerebellum during late pregnancy and early postnatal periods. Thus, the present study was conducted to evaluate the redox state imbalance and disruption of membrane-bound ATPases and oxidative stress induced by Co in the cerebrum and cerebellum of suckling rats.
Material and Methods
Animals and Experimental Design
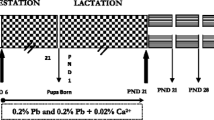
Wistar female rats weighing 170 ± 10 g, obtained from the Central Pharmacy (SIPHAT, Tunisia), were housed in plastic cages in a climate-controlled facility with a constant light–dark cycle at a temperature of 22 ± 2 °C and humidity of 40 %. Pregnant female rats were inspected daily by the presence of the vaginal plug, which indicated day 0 of pregnancy. Twelve pregnant female rats were randomly divided into 2 groups of 6 each: rats of group 1 (control group) received distilled water and served as negative controls. Those of group 2 (Co group) received, by drinking water, 350 ppm of CoCl2. The Co dose represented one ninth of the LD50 [21]. In a pre-study, we tested different doses of Co: no toxic effects and no oxidative stress were observed in rats treated with Co at doses under 350 ppm. With this dose, oxidative stress was identified in female rats without lethal effects. But with doses over 350 mg/L, Co provoked severe signs of toxicity and mortality.
Pregnant female rats were allowed to deliver spontaneously 3 weeks after coitus. The day of birth was considered as postnatal day 0. Within 24 h after delivery, litters in both control and treated groups were culled to eight pups (four males and four females if possible) in order to maintain a similar lactation performance and to ensure maternal care [21]. Water consumption and food intake by dams were recorded daily during a lactating period.
All treatments started from day 14 of pregnancy until day 14 after delivery. CoCl2 quantities ingested daily by adult rats were calculated after measuring drinking water. The experimental procedures were carried out according to the Natural Health Institute of Health Guidelines for Animal Care and approved by the Ethical Committee of Sfax Science Faculty. All animal procedures were conducted in strict conformity with the “Institute Ethical Committee Guidelines” for the care and use of laboratory animals [22]. At the end of the experimental period, mothers (n = 12) and pups (n = 96) were euthanized.
Blood and Brain Preparation
At the end of experiment, blood samples were collected in heparinized tubes. Plasma was separated from blood by centrifugation (3.500 rpm for 10 min). The cerebrum and cerebellum were quickly removed and cleaned from the adhering tissue. Some were used for oxidative stress markers analysis and others were minced, homogenized (10 % w/v) in an appropriate phosphate-buffered saline (100 mM Na2HPO4/NaH2PO4, pH 7.4) with an Ultra Turrax homogenizer in ice-cold buffer, and centrifuged at 10.000×g for 15 min at 4 °C. The resulting supernatants were used for various biochemical assays. Some cerebrums and cerebellums were immediately fixed in 10 % formalin solution for histological studies.
Biochemical Assays
Co Content
Co concentration in plasma was measured after acid digestion with nitric acid (65 %) using a microwave digestion system (CEM, USA, model MDS-2100). Co was estimated using a hydride vapor generation system (Perkin-Elmer model MHS-10) fitted with an atomic absorption spectrophotometer (Perkin-Elmer model Analyst100).
Protein Quantification
Cerebrum and cerebellum protein contents were measured according to the method of Lowry et al. [23] using bovine serum albumin as standard.
MDA Measurement
Malondialdehyde (MDA) concentrations, an index of lipid peroxidation, were determined spectrophotometrically according to Draper and Hadley [24]. The MDA values were calculated using 1,1,3,3-tetraethoxypropane as standard and expressed as nanomoles of MDA per milligram of protein.
Determination of AOPP Levels
Advanced oxidation protein product (AOPP) levels were determined according to the method of Kayali et al. [25]. The concentration of AOPP for each sample was calculated using the extinction coefficient of 261 cm−1 mM−1 and the results were expressed as micromoles per milligram of protein.
Measurement of H2O2
Measurement of hydrogen peroxide (H2O2) was carried out by the ferrous ion oxidation xylenol orange (FOX1) method [26]. The FOX1 reagent consisted of 25 mM sulfuric acid, 250 μM ferrous ammonium sulfate, 100 μM xylenol orange, and 0.1 M sorbitol. Briefly, 100 μl of extract was added to 900 μl of FOX1 reagent vortexed and incubated for 30 min at room temperature. Solutions were then centrifuged at 12.000×g for 10 min, and the amount of H2O2 in the supernatant was determined using a spectrophotometer at 560 nm.
Determination of PCO Content
Protein carbonyl (PCO) was measured using the method of Reznick and Packer [27]. PCO was calculated based on the molar extinction coefficient of DNPH (ε = 2.2 × 104 cm−1 M−1) and expressed as micromoles per milligram of protein.
DNA Fragmentation Analysis
The extent of DNA fragmentation in the cerebrum and cerebellum was determined by the method described by Kanno et al. [28]. Briefly, brain tissue was homogenized in lysis buffer. The gel was observed under an ultraviolet lamp and photographed.
ATPase Assay
For the determination of ATPase activities, cerebrum and cerebellum were homogenized in Tris–HCl buffer, pH 7.4, according the method of Kawamoto et al. [29]. The resulting supernatants were immediately used for ATPase determination. Total ATPase activity was determined by Pi assay released from hydrolyzed adenosine triphosphate (ATP) forming a complex with molybdate. Enzyme activity was expressed as micromoles of Pi liberated per hour per milligram of protein.
Determination of Antioxidant Enzyme Activities
Catalase (CAT) activity was assayed by the method of Aebi [30]. Enzymatic reaction was initiated by adding an aliquot of 20 μl of the homogenized tissue and the substrate (H2O2) to a concentration of 0.5 M in a medium containing 100 mM phosphate buffer (pH 7.4). Changes in absorbance were recorded at 240 nm. CAT activity was calculated in terms of micromoles of H2O2 consumed per minute per milligram of protein
Superoxide dismutase (SOD) activity was estimated according to Beauchamp and Fridovich [31]. The reaction mixture contained tissue homogenates in potassium phosphate buffer (50 mM, pH 7.8), 0.1 mM EDTA, 13 mM l-methionine, 2 μM riboflavin, and 75 mM nitro blue tetrazolium (NBT). The developed blue color in the reaction was measured at 560 nm. Units of SOD activity were expressed as the amount of enzyme required to inhibit the reduction of NBT by 50 % and the activity was expressed as units per milligram of protein.
Glutathione peroxidase (GPx) activity was measured according to Flohe and Gunzler [32]. GPx catalyzes the oxidation of reduced glutathione (GSH) by cumene hydroperoxide. In the presence of reduced GSH reductase and nicotinamide adenine dinucleotide phosphate reduced form (NADPH), the oxidized reduced GSH is immediately converted to the reduced form with a concomitant oxidation of NADPH–NADP+. The decrease in absorbance at 340 nm was measured. The enzyme activity was expressed as nanomoles of GSH oxidized per minute per milligram of protein.
Cerebrum and Cerebellum Total GSH Levels
Total GSH was determined by the method of Ellman [33] modified by Jollow et al. [34]. The method is based on the development of a yellow color when DTNB is added to compounds containing sulfhydryl groups. The absorbance was measured at 412 nm after 10 min. Total GSH content was expressed as micrograms per milligram of protein.
Vitamin C Determination
Vitamin C determination was performed as described by Jacques-Silva et al. [35]. Protein was precipitated in 10 volumes of a cold 4 % trichloroacetic acid solution. An aliquot (300 μl) of supernatant adjusted with distilled H2O to a final volume of 1 ml was incubated at 38 °C for 3 h, then 1 ml H2SO4 65 % (v/v) was added to the medium. The reaction product was determined using a color reagent containing 4.5 mg/ml dinitrophenylhydrazine and CuSO4 (0.075 mg/ml). The data were expressed as micromoles of ascorbic acid per milligram of protein.
Cerebrum and Cerebellum NPSH Levels
Nonprotein thiol (NPSH) levels were determined by the method of Ellman [33]. Absorbance of colorimetric reaction was measured at 412 nm. Total NPSH content was expressed as micromoles per milligram of protein.
Determination of AChE Activity
Acetylcholinesterase (AChE) activity was measured immediately in homogenates according to the method of Ellman et al. [36], using acetylthiocholine iodide as a substrate. The reaction mixture was composed as follows: phosphate buffer (0.1 M; pH 8) and 0.01 M DTNB. The hydrolysis rate of acetylthiocholine iodide is measured at 412 nm through the release of the thiol compound which, when reacted with DTNB, produces the color-forming compound TNB. The reaction was initiated by adding 0.075 M acetylthiocholine iodide. Activities were expressed as micromoles of substrate per minute per milligram of protein.
Determination of BuChE Activity
Butylcholinesterase (BuChE) activity was determined by the method of Ellman et al. [36], with some modifications. Hydrolysis rate was measured at acetylthiocholine concentrations of 0.8 mM in 1 ml assay solutions with 100 mM phosphate buffer, pH 7.5 and 1 mM DTNB. Fifty microliters of supernatant was added to the reaction mixture and preincubated for 3 min. The hydrolysis was monitored by the formation of the thiolate dianion of DTNB at 412 nm for 2–3 min (intervals of 30 s) at 25 °C. All samples were run in duplicate. Activities were expressed as micromoles of substrate per minute per milligram of protein.
Histological Studies
Some cerebrum and cerebellum samples, intended for histological examination by light microscopy, were immediately fixed in 10 % of formalin and processed in a series of graded ethanol solutions. They were then embedded in paraffin, serially sectioned at 3 μm. Some sections were stained with hematoxylin–eosin. The other ones were stained with toluidine blue. Six slides were prepared from each cerebrum or cerebellum.
Statistical Analysis
The data were analyzed using the statistical package program StatView 5 Software for Windows (SAS Institute, Berkley, CA, USA). Statistical analysis was performed using one-way analysis of variance followed by Fisher’s protected least significant difference test as a post hoc test for comparison between groups. All values were expressed as the means ± standard deviation (SD). Differences were considered significant if P < 0.05.
Results
The Effects of Co on the General Health of Rats
Death and abortion were not observed in Co-treated dams during the experimental period (21 days). In their suckling pups, few clinical signs such as ataxia, reduced activity, as well as tremors and nasal discharge were observed.
Concentrations of Co in Plasma
In our experimental conditions, Co concentration in plasma was higher in suckling rats of the treated groups (P < 0.01) than in the controls (Table 1).
Cerebrum and Cerebellum AChE and BuChE Activities
CoCl2 treatment of pregnant and lactating rats produced a significant reduction of AChE and BuChE activities in both cerebrum (P < 0.001 and P < 0.05) and cerebellum (P < 0.001) (Table 1).
CoCl2 Effects on Lipid Peroxidation and H2O2 Production
Our results showed that MDA contents, the major product of lipid peroxidation, increased by 55 % in the cerebrum (P < 0.001) and by 25 % in the cerebellum (P < 0.01) of CoCl2-treated rats when compared to the controls (Table 2). In addition, the H2O2 levels generated in the brain of suckling rats significantly increased, indicating oxidative stress (P < 0.001).
CoCl2 Effects on the Activity of ATPases
Table 2 showed that Co treatment impaired Na+K+-ATPase and Mg2+-ATPase enzyme activities which significantly decreased in the cerebrum (P < 0.001) and cerebellum (P < 0.01 and P < 0.001, respectively).
Markers of Protein Oxidative Damage
AOPP and PCO levels are usually used as markers of protein oxidative damage. In the Co group, a significant increase of PCO and AOPP levels in the cerebrum and cerebellum (P < 0.001) of suckling rats occurred (Table 2).
Effects of Co on DNA Fragmentation
Agarose gel electrophoresis showed undetectable DNA laddering in the cerebrum and cerebellum of the control rats. The DNA intact band appeared to be condensed near the application point with no DNA smearing, suggesting no DNA fragmentation, while a smear (hallmark of necrosis) without ladder formation on agarose gels was observed in the cerebrum and cerebellum of the Co-treated rats, indicating random DNA degradation (Fig. 1).

Agarose gel electrophoresis of DNA fragmentation. M marker (phage lambda, HindIII (23, 9, 6, 4, 2, and 0.5 kbp), lane 1 cerebrum of the control group, lane 2 cerebrum of the Co-treated group, lane 3 cerebellum of the control group, lane 4 cerebellum of the Co-treated group
Antioxidant Activities in the Cerebrum and Cerebellum
In CoCl2-treated rats, SOD, CAT, and GPx activities decreased by 53 % (P < 0.001), 35 % (P < 0.05), and 21 % in the cerebrum and by 52 % (P < 0.001), 71 % (P < 0.001), and 39 % in the cerebellum, respectively, compared to those of the controls (Table 3). Our results also revealed a significant decrease in GSH and NPSH levels by 23 % (P < 0.05) and 50 % (P < 0.001) in the cerebrum and by 16 % (P < 0.05) and 25 % (P < 0.01) in the cerebellum, respectively (Table 4).
Vitamin C Levels
The data presented in Table 4 showed the levels of vitamin C in the cerebrum and cerebellum of the control and tested groups. The exposure of pregnant and lactating rats to Co caused a significant decrease of vitamin C levels in the cerebrum (P < 0.001) and in the cerebellum (P < 0.001) of suckling pups.
Cerebrum and Cerebellum Histopathological Findings
Light microscopic examination indicated a normal structure in the cerebrum of suckling treated rats (Fig. 2b) when compared with the controls (Fig. 2a). However, there were changes on histological sections performed on the cerebellum of 14-day-old rats whose mothers were treated with CoCl2. Normal histoarchitecture of the cerebellum showed four layers: an external granular layer (EGL), a molecular layer (ML), a PCL, and an internal granular layer (IGL). Purkinje cells in the PCL were well differentiated, showing elongated cellular bodies and dendritic arborization (Fig. 3a). Those of Co-treated rats were poorly differentiated with frequent pyknotic cells, and their number was reduced (Fig. 3b). Furthermore, the EGL became markedly developed (Fig. 3b).

Cerebrum histological sections, stained with hematoxylin–eosin and blue toluidine, of suckling pups controls (a) and whose mothers were treated with Co (b) from the 14th day of pregnancy until day 14 after delivery. Optic microscopy, HE (×400).  neurons,
neurons,  glial cells
glial cells

Cerebellum histological sections, stained with hematoxylin–eosin and blue toluidine, of suckling pups controls (a) and whose mothers were treated with Co (b) from the 14th day of pregnancy until day 14 after delivery. Optic microscopy, HE (×400). EGL external granular layer, ML molecular layer, IGL internal granular layer, solid arrows Purkinje cells, dashed arrows pyknotic cells
Discussion
Co is an essential trace element required for normal growth, development, cellular homeostasis, and many enzymatic reactions [37, 38]. Although small amounts of Co are a nutritional necessity for normal brain functioning, it has been considered as neurotoxic at high doses. To our knowledge, this paper constitutes the first study evaluating cerebral and cerebellar damages induced by Co treatment in suckling rats.
Our result demonstrated that Co concentration in the plasma of pups, whose mothers were treated by Co, was higher than in the controls. This could be due to the transfer of Co2+ through placenta and/or milk from mothers to their offspring. Thus, Co, when transferred to the offspring, affected cholinergic function, as demonstrated by us. Indeed, changes in AChE and BuChE activities might be an indicator of Co-induced neurotoxicity in suckling pups. This metal, which causes a depletion of neurotransmitters [39] and inhibits synaptic transmission via the presynaptic blockade of calcium channels, can block postsynaptic responses [40] and decrease exploratory behavior [41]. Moreover, neurotoxicity could be attributed to oxidative damage caused by Co treatment. One of the main manifestations of oxidative stress in the brain is MDA. The latter has been found to play an important role in the toxicity and carcinogenicity of many xenobiotics. On the other hand, it is well known that the brain is highly vulnerable to MDA due to its high rate of oxygen utilization [42], an abundant supply of PUFA, a deficient antioxidant defense [43], and a high content of transition metals like copper and iron in several regions [44]. In our findings, Co caused an increase of MDA in the cerebrum and cerebellum, suggesting its participation in generating free radicals including H2O2. The latter can attack PUFA in the biomembrane, leading to its dysfunction and thereby destroying the special arrangement and impairing local enzyme activities. Thus, a significant decrease in the Na+K+-ATPase and Mg2+-ATPase levels was recorded after Co treatment. Additionally the inhibition of Mg2+-ATPase could lead to a reduction in ATP production which, in turn, alters Na+K+ pump activity, producing neuronal dysfunction [45]. The increased free radicals generation can also lead to protein–protein cross linkages formation; protein backbones oxidation, resulting in the protein fragmentation; and modification of amino acid side chains, including sulfhydryl moieties oxidation and PCO formation, as demonstrated by our findings. The occurrence of protein oxidative stress in the cerebrum and cerebellum of experimental rats was also confirmed by a novel marker, AOPP, which reflected an excess of free radical generation and protein oxidative damages. Free radicals attack not only proteins but also DNA bases, therefore causing mutagenic lesions. Co treatment resulted in massive DNA fragmentations with a subsequent formation of a DNA smear on agarose gel, a hallmark feature of necrosis, suggesting Co-induced brain cell damage. In fact, it has been reported that Co induces apoptosis in the neuronal PC12 cell line [46]. It triggered apoptosis in a dose-dependent and time-dependent manner as demonstrated by morphological changes and DNA fragmentation and confirmed by the decrease of the expression of Bcl-xl, a member of the Bcl-2 protein family that plays a role in the regulation of apoptosis [46].
Oxidative stress occurs when the antioxidant defense system is overwhelmed by the production of ROS. Antioxidant and other cell redox state modulating enzyme systems act as the first-line defense against ROS in all cellular and extracellular compartments [47]. SOD, CAT, and GPx, the most important antioxidant enzymes, were reduced. These results indicated that the cytotoxic actions of Co in neuronal cells were mediated, at least in part, by an oxyradical mechanism involving the overproduction of ROS and downregulation of certain key antioxidant enzymes. On the other hand, nonenzymatic antioxidants like GSH and NPSH are considered as the second line of cellular defense against Co-induced oxidative damage. In our study, these parameters were greatly impaired, as indicated by a significant decrease in GSH and NPSH levels in the cerebrum and cerebellum of the Co group, reflecting their consumption through oxidative stress. Several pathways have been proposed. First, the sulfhydryl group has a high affinity for metals, forming thermodynamically stable mercaptide complexes with several metals [48]. Second, GSH may be oxidized due to the interaction with Co-induced free radicals.
It is also important to emphasize that GSH plays a critical role in regenerating vitamin C from its oxidized by-products. Our findings showed a significant decrease in the level of vitamin C after Co treatment, leading to the increased susceptibility of the brain tissue to free radical damage.
Enhanced oxidative stress in the brain after Co exposure was reflected in the significant histological changes, especially in the cerebellum of nursing pups. In fact, it is well known that the cerebellum is one of the brain parts that are strongly influenced during the early postnatal period, a critical period of life [49]. Because rat pups are born with a relatively undeveloped brain, especially an immature cerebellum, prenatal perturbation dramatically affects cerebellar development [49]. In the control group, the Purkinje cells were well differentiated, showing elongated cellular bodies and visible dendritic arborization. In contrast, the Purkinje cells of Co-treated rats were rounded, poorly differentiated, and frequently necrotic. There was also a loss of Purkinje cell numbers, as evidenced by a decreased cellularity in the PCL. Furthermore, the EGL became markedly developed in the treated group. This could be an indication of a delayed migration of granular cells towards the ML and IGL. According to Farwell et al. [50], granule cell migration depends on the recognition of extracellular neuronal guidance molecule(s) like laminin, a key guidance molecule in the developing brain. These data suggested that ingested Co was retained by the cerebellum, interfering with its physiology and inducing neurotoxicity, cell damage, and even cell death.
Conclusion
To our knowledge, this study may constitute the first attempt to evaluate the effects of Co on suckling rat brain. This metal, administered to pregnant and lactating rats at a dose of 350 ppm, caused free radical generation, increased MDA levels and protein oxidation products, and induced DNA fragmentation in the cerebrum and cerebellum of suckling rats. Co was considered as a pro-oxidant agent. It disturbed the oxidant–antioxidant balance and induced a depletion of cerebral and cerebellar antioxidant activities. Thus, the present study suggested that Co affected the structure and maturation of the brain, suggesting its neurotoxic effects.
Abbreviations
- AChE:
-
Acetylcholinesterase
- AOPP:
-
Advanced oxidation protein product
- Bcl-xl:
-
B cell lymphoma-extra large
- Bcl-2:
-
B cell lymphoma 2
- BuChE:
-
Butylcholinesterase
- CAT:
-
Catalase
- Co:
-
Cobalt
- DTNB:
-
5,5′-Dithiobtis-2-nitrobenzoic acid
- EGL:
-
External granular layer
- GPx:
-
Glutathione peroxidase
- GSH:
-
Glutathione
- H2O2 :
-
Hydrogen peroxide
- IGL:
-
Internal granular layer
- MDA:
-
Malondialdehyde
- ML:
-
Molecular layer
- NBT:
-
Nitro blue tetrazolium
- NPSH:
-
Nonprotein thiol
- PCL:
-
Purkinje cell layer
- PCO:
-
Protein carbonyl
- PUFA:
-
Polyunsaturated fatty acids
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
References
Gault N, Sandre C, Poncy JL, Moulin C, Lefaix JL, Bresson C (2010) Cobalt toxicity: chemical and radiological combined effects on HaCaT keratinocyte cell line. Toxicol in Vitro 24(1):92–98
Karovic O, Tonazzini I, Rebola N, Edstrom E, Lovdahl C, Fredholm BB (2006) Toxic effects of cobalt in primary cultures of mouse astrocytes. Similarities with hypoxia and role of HIF-1 alpha. Biochem Pharmacol 73:694–708
Jordan CM, Whitman RD, Harbut M (1997) Memory deficits and industrial toxicant exposure: a comparative study of hard metal, solvent and asbestos workers. Int J Neurosci 90:113–128
Agency for Toxic Substances and Disease Registry (ATSDR) (2004) Toxicological profile for cobalt. US Department of Health and Human Services, The National Academies Press, Atlanta, p 39
Kratchler M, Rossipal SLE, Irgolic KJ (1998) Changes in the concentrations of trace elements in human milk during lactation. J Trace Elem Med Biol 12:159–176
Lison D, De Boeck M, Verougstraete V, Kirsch-Volders M (2001) Update on the genotoxicity and carcinogenicity of cobalt compounds. Occup Environ Med 58(10):619–625
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39:44–48
Nazıroglu M (2009) Role of selenium on calcium signaling and oxidative stress-induced molecular pathways in epilepsy. Neurochem Res 34:2181–2191
Naziroğlu M, Kutluhan S, Uğuz AC, Celik O, Bal R, Butterworth PJ (2009) Topiramate and vitamin E modulate the electroencephalographic records, brain microsomal and blood antioxidant redox system in pentylentetrazol-induced seizure of rats. J Membr Biol 229:131–140
Hassoun EA, Li F, Abushaban A, Stohs SJ (2001) Production of superoxide anion, lipid peroxidation and DNA damage in the hepatic and brain tissues of rats after subchronic exposure to mixture of TCDD and its congeners. J Appl Toxicol 21:211–219
Cotman CW, Peterson C (1989) Aging in the nervous system. In: Fleischer S, Packer L (eds) Basic neurochemistry. Academic, New York, pp 382–389
Hoyt KR, Gallagher AJ, Hastings TG, Reynolds IJ (1997) Characterization of hydrogen peroxide toxicity in cultured rat forebrain neurons. Neurochem Res 22:333–340
Dawson VL, Dawson TM (1996) Free radicals and neuronal cell death. Cell Death Differ 3:71–78
Dringen R, Gutterer JM, Hirrlinger J (2000) Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem 267:4912–4916
Abdollahi M, Rainba A, Shadnia S, Nikfar S, Rezaie A (2004) Pesticide and oxidative stress: a review. Med Sci Monitor 10:RA141–RA147
Santamaria A, Vazquez-Roman B, La Cruz VP, Gonzalez-Cortes C, Trejo-Solis MC, Galvan-Arzate S, Jara-Prado A, Guevara-Fonseca JB, Ali SF (2005) Selenium reduces the proapoptotic signaling associated to NF-kappaB pathway and stimulates glutathione peroxidase activity during excitotoxic damage produced by quinolinate in rat corpus striatum. Synapse 58:258–266
Yeo JE, Kang SK (2007) Selenium effectively inhibits ROS-mediated apoptotic neural precursor cell death in vitro and in vivo in traumatic brain injury. Biochim Biophys Acta 1772:1199–1210
Yousuf S, Atif F, Ahmad M, Hoda MN, Khan MB, Ishrat T, Islam F (2007) Selenium plays a modulatory role against cerebral ischemia-induced neuronal damage in rat hippocampus. Brain Res 1147:218–225
Atif F, Yousuf S, Agrawal SK (2008) Restraint stress-induced oxidative damage and its amelioration with selenium. Eur J Pharmacol 600:59–63
Eva P, Jörgen H, Hans T (2003) Uptake of cobalt from the nasal mucosa into the brain via olfactory pathways in rats. Toxicol Lett 145:19–27
Fishbeck KL, Rasmussen KM (1987) Effect of repeated cycles on maternal nutritional status, lactational performance and litter growth in ad libitum-fed and chronically food restricted rats. J Nutr 117:1967–1975
Maryland RC (1996) Guide for the care and use of laboratory animals. Institute of Laboratory Animal Resources Commission on Life Sciences, National Research Council National Academy Press, Washington, DC
Lowry OH, Rosenbrough NJ, Farr AL (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Draper HH, Hadley M (1990) Malondialdehyde determination as an index of lipid peroxidation. Meth Enzymol 86:421–431
Kayali R, Cakatay U, Akcay T, Altug T (2006) Effect of alpha-lipoic acid supplementation on markers of protein oxidation in post-mitotic tissues of ageing rat. Cell Biochem Funct 24:79–85
Ou P, Wolff SP (1996) A discontinuous method for catalase determination at near physiological concentrations of H2O2 and its application to the study of H2O2 fluxes within cells. J Biochem Biophys Methods 31:59–67
Reznick AZ, Packer L (1994) Oxidative damage to proteins: spectrophotometric method for carbonyl. Methods Enzymol 233:357–363
Kanno S, Shouji A, Hirata R, Asou K, Ishikawa M (2004) Effects of naringin on cytosine arabinoside (Ara-C)-induced cytotoxicity and apoptosis in P388 cells. Life Sci 75:353–365
Kawamoto EM, Munhoz CD, Glezer I, Bahia VS, Caramelli P, Nitrini R, Gorjao R, Curi R, Scavone C, Marcourakis T (2005) Oxidative state in platelets and erythrocytes in aging and Alzheimer’s disease. Neurobiol Aging 26:857–864
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Beauchamps C (1971) Superoxide dismutase: improved assays and assay applicable to acrylamide gels. Anal Biochem 44:276–287
Flohe L, Gunzler WA (1984) Assays of glutathione peroxidase. Methods Enzymol 105:114–121
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem 82:70–77
Jollow DJ, Mitchell JR, Zampaglione N, Gillette JR (1974) Bromobenzene-induced liver necrosis. Protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. Pharmacology 11:151–169
Jacques-Silva MC, Nogueira CW, Broc LC (2001) Diphenyl diselenide and ascorbic acid changes deposition of selenium and ascorbic acid in liver and brain of mice. Pharm Toxicol 88:119–125
Ellman GL, Courtney KD, Andres V, Feather-Stone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95
Schrauzer GN, Deutsch E (1969) Reactions of cobalt(I) supernucleophiles. The alkylation of vitamin B12s, cobaloximes(I), and related compounds. J Am Chem Soc 91:3341–3350
Schrauzer GN (1984) The discovery of the essential trace elements. An outline of the history of biological trace element research. In: Frienden E (ed) Biochemistry of the essential ultratrace elements. Plenum, New York, pp 17–31
Hasan M, Ali S, Anwar J (1980) Cobalt-induced depletion of dopamine, norepinephrine & 5-hydroxytryptamine concentration in different regions of the rat brain. Indian J Exp Biol 18(9):1051–1053
Gerber U, Gahwiler BH (1991) Cobalt blocks postsynaptic responses induced by neurotransmitters in the hippocampus in vitro. Neurosci Lett 134:53–56
Czarnota M, Whitman D, Berman R (1998) Activity and passive avoidance learning in cobalt-injected rats. Int J Neurosci 93:29–33
Travacio M, Polo JM, Liesuy S (2000) Chromium (VI) induces oxidative stress in the mouse brain. Toxicology 150:137–146
Mates JM (2001) Effects of antioxidant enzymes in the molecular control of reactive oxygen species. Toxicology 163(2–3):219
Calabrese V, Bates TE, Stella AMG (2000) NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders; the role of oxidant/antioxidant balance. Neurochem Res 25:1315–1341
Siraj Mohiyuddin S, Rajeswara Reddy S, Ananda Kumar L, Jacob Doss P (2010) Acephate induced alterations in Mg2+ and Na+K+ ATPases of different brain regions of albino rats. Biasean 5:153–156
Zou W, Yan M, Xu W, Huo H, Sun I, Zheng Z, Liu X (2001) Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP-1 activation. J Neurosci Res 67(6):646–653
Karihtala P, Soini Y (2007) Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. APMIS 115:81–103
El-Sharaky AS, Newairy AA, Badreldeen MM, Eweda SM, Sheweita SA (2007) Protective role of selenium against renal toxicity induced by cadmium in rats. Toxicology 235:185–193
Hasebe M, Matsumoto I, Imagawa T, Uehara M (2008) Effects of an anti-thyroid drug, methimazole, administration to rat dams on the cerebellar cortex development in their pups. Int J Dev Neurosci 26:409–414
Farwell AP, Dubord-Tomasettia SA, Pietrzykowskia AZ, Stachelek SJ, Leonard JL (2005) Regulation of cerebellar neuronal migration and neurite outgrowth by thyroxine and 3,3′,5′-triiodothyronine. Dev Brain Res 154:121–135
Acknowledgments
The authors thank Mrs. Dalinda Kchaou for her assistance in the histological techniques. The present work was supported by DGRST grants (Direction Générale de la Recherche Scientifque et Technique-Tunisie, Appui à la Recherche Universitaire de base 99/UR/08-73). We also wish to extend our thanks to Mr. Bejaoui Hafedh, teacher of English at the Sfax Faculty of Science, who proofread the English of this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ibtissem Ben Amara and Elmouldi Garoui contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Garoui, E., Amara, I.B., Driss, D. et al. Effects of Cobalt on Membrane ATPases, Oxidant, and Antioxidant Values in the Cerebrum and Cerebellum of Suckling Rats. Biol Trace Elem Res 154, 387–395 (2013). https://doi.org/10.1007/s12011-013-9746-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-013-9746-0