
Abstract
Every year, seasonal epidemics of influenza viruses are causing considerable morbidity and mortality worldwide. Also infrequent novel and rearranged strains of influenza viruses have caused quick, acute universal pandemics resulting in millions of mortalities. The usage of efficient and accurate detection is superior for infection control, effective treatment, and epidemiological supervision. Therefore, evaluation of useful real-time PCR molecular tests for the detection of pandemic viruses is important before the next wave of the pandemic. A novel quantitative real-time reverse-transcription polymerase chain reaction (qRT-PCR) assay with specific primers was used successfully for detection and monitoring of the influenza A, B, and swine influenza. The newly designed primers target highly conserved regions in influenza viruses. Our qRT-PCR assay is highly specific for detecting influenza A, B, and swine influenza viruses. The cutoff CT value was determined <38 for domestic human diagnostic test, under conditions of FDA emergency, and the reaction efficiency of the InfA, swInfA, and InfB assays were thereby estimated to be 97.9 % (R2 = 0.998), 98.3 % (R2 = 0.986), and 99.5 % (R2 = 0.995), respectively. Interestingly, based on our finding, there is no cross reactivity of detecting other viruses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza A and B viruses cause the majority of viral lower respiratory tract infections; elderly and compromised individuals are especially at risk of developing severe illness and complications [1, 2].
In April 2009, novel swine-lineage influenza A (H1N1 swl) virus entered the human population and spread rapidly throughout the world [1]. Subsequent phylogenetic characterization of this swine origin influenza virus A (H1N1 swl) from the US index case indicated that the virus had a unique genome composition that had not been previously reported [1, 2].
For virus isolation such as cell culture, shell vial culturing, serologic analysis, and antigen detection are the conventional methods used for the laboratory diagnosis of influenza viruses. Although these methods have their limitations such as virus isolation via cell culturing that is a labor-intensive and time-consuming method despite its sensitivity [3]. On the other hand, other diagnostic techniques, for example antigen detection and shell vial culturing, provide results more quickly but typically are less sensitive than conventional cell culturing or PCR [4–6].
To obtain far more rapid diagnostic results and to overcome this lack of sensitivity, PCR techniques have been developed for the specific detection and subtyping of influenza viruses [7].
The emergence of the SWL H1N1 has ramifications for existing typing and diagnostic PCR methods and may result in a failure to detect and/or type this new virus because of the genetic differences mentioned above [2].
As all influenza virus subtypes causes similar clinical symptoms, differential diagnosis of the virus is required in one sample. Mono-specific PCR tests require separate amplification of each amplicon and are therefore time consuming and resource intensive. Multiplex PCR for clinical diagnosis has a major advantage, as it permits simultaneous amplification of several pathogens in a single reaction tube, providing cost-effective diagnosis [8–10]. However, so far, these multiplex PCR assays distinguish the target by PCR amplicon size on electrophoresis or hybridization with probes post-PCR.
Real-time PCR with specific detection of the product by fluorophore probes increases the specificity of tests and significantly reduces processing time. In addition, another feature of the real-time PCR method is the ability to perform multiplex amplification and detection [11, 12].
This study describes the development and use of a multiplex real-time RT-PCR assay as a diagnostic tool for the cost effective, simultaneous, and fast diagnosis of influenza A, B, and swine origin influenza A (H1N1) viruses specifically to diagnose influenza virus strains in Iran.
Material and Methods
Virus Strains and Viral RNA Preparation
In this study, influenza viruses were grown to high titers in either Madin–Darby canine kidney cells or embryonated chicken eggs. Infectious viruses in culture supernatants or allantoic fluids was measured by determining the number of 50 % tissue culture infectious doses per milliliter or the number of 50 % egg infectious doses per milliliter, respectively. Clinical specimens (nasal washes, nasal swabs, nasopharyngeal swabs, throat swabs, and lower respiratory tract specimens) included in this study were received from Tehran, different cities in Iran, and other local public health laboratories as well as foreign laboratories between April 2011 and June 2012.
RNA Extraction
Viral RNA was extracted from 100 ul virus isolates or clinical specimens and eluted in 100 ul volumes by using viral RNA isolation kit (Bioneer, Korea), according to the manufacturer’s instructions.
rRT-PCR Flu Panel Primers and Probes
Oligonucleotide primers and probes were designed based on available nucleotide sequence data from the GenBank database of the National Center for Biotechnology Information (NCBI), NIH, the influenza sequence database of Los Alamos National Laboratories, and the Global Initiative on Sharing Avian Influenza Data. Nucleotide BLAST search (NCBI) analysis was used to verify primer and probe sequence specificity and avoid potential nonspecific reactivity. The primers and probes were designed to have annealing temperatures of approximately 60 and 62 °C, respectively, using Beacon designer software ver. 8 (Primer Biosoft, USA). Dual-labeled hydrolysis probes were labeled at the 5' end with the reporter dye (FAM, Hex, Texasred, cyc5) and quenched with Black Hole Quencher 1 either at the 3' end or internally at a modified “T” residue with Spacer3 (3'Sp3) at the 3' end to prevent probe extension by Taq polymerase (Metabion international AG, Germany). Primers and dually labeled TaqMan hydrolysis probes were synthesized by Metabion international AG, Inc. (Germany, Munich). The rRT-PCR flu panel includes four sets of oligonucleotide primer and probe (universal influenza A (InfA), swine influenza A (swInfA), influenza B, and RNase P (RP)) designed for the detection and characterization of influenza A, swine influenza A, and influenza B viruses in human specimens. The InfA assay was designed for universal detection of the matrix (M) gene of all influenza A viruses. The InfB assay was designed for universal detection of the nucleoprotein gene of influenza B gene of all influenza B viruses.
The RP assay detects the human RNase P gene and used for human clinical specimens to measure the quality of nucleic acid extraction to indicate that the gene was adequately extracted from the clinical specimen (Table 1).
rRT-PCR Conditions
Reaction’s conditions for rRT-PCR were based on the Food and Drug Administration (FDA)-cleared CDC human influenza virus real-time RT-PCR detection and characterization panel (CDC rRT-PCR Flu Panel). The CDC rRT-PCR Flu Panel contains the same set of primer and probe for detection of the influenza A virus matrix gene similar to the CDC rRT-PCR Swine Flu Panel.
In order to determine the optimal annealing and extension temperatures, thermal gradient analysis was performed in triplicate using the Rotor-Gene 6000 system (Corbett research,Australia) and the RNA extracted from 2008 (H1N1) influenza A virus strain A/Tehran/2008/H1N1, A/Iran/14068/2009/H1N1, A/shiraz/15/2005/H1N1 for detection influenza A/SW and influenza B/Tehran/12/2007 virus for influenza B. Thermocycling rRT-PCR conditions were 42 °C for 30 min, Taq activation for 7 min at 95 °C, and 45 cycles of 95 °C for 15 s and an annealing extension step with temperatures ranging from 50 to 65 °C for 30 s. All three assays showed comparable levels of performance, with annealing temperatures ranging from 58 to 60 °C. The final reaction annealing temperature was set to 60 °C, which is 5 °C below the maximum optimal annealing temperature (65 °C) used to accommodate potential nucleotide mismatches in the primer–probe regions due to virus evolution (Table 2).
PCR parameters of the rRT-PCR flu panel were optimized using Real Master mix RT-PCR probe kits (5 Prime Technologies) and Rotor-Gene 6000 system (Corbett research, Australia) real-time PCR system. All rRT-PCRs were performed at total reaction volume of 25 ul. Final primer and probe reaction concentrations were 0.5 and 0.2 uM, respectively. All analytical performance data and clinical specimen data were collected using a Rotor Gene 6000 software real-time PCR instrument.
Results
rRT-PCR Flu Panel Reaction Efficiency
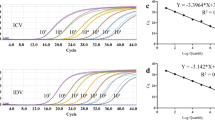
Reaction’s efficiency for primer and probe contained sets of InfA, swInfA, and InfB assays which were determined by testing a fivefold serial dilution series of viral RNA of 2008 A (H1N1) influenza virus strain A/Tehran/2008, A/Iran/14068/2009/H1N1, A/shiraz/15/2005/H1N1, and influenza B/Tehran/12/2007 virus for InfB duplicated. The resulting threshold cycle (CT) values were plotted versus relative RNA concentration values, and linear regression analysis was applied to determine the slopes. The reaction efficiency of the InfA, swInfA, and InfB assays were thereby estimated as 97.9 % (R2 = 0.998), 98.3 % (R2 = 0.986), and 99.5 % (R2 = 0.995), respectively (data not shown).
Analytical Sensitivity
The limits of detection (LoD) of the InfA, swInfA, and InfB assays were determined by analyzing a tenfold dilution series of grown influenza viruses (influenza A virus strain A/Tehran/2008/H1N1, A/Iran/14068/2009/H1N1, A/shiraz/15/2005/H1N1 for detection influenza A/SW, and influenza B/Tehran/12/2007 virus) (Influenza Lab center, Tehran, Iran). A tenfold dilution series were tested in triplicate for each extracted virus strain (Table 3). The LoD of the rRT-PCR flu panel was determined according to the lowest concentration at which all three assays (InfA, swInfA, InfB) gave positive results. The LoD was determined between 101 and 101.5 ID50/ml virus concentrations to test three viruses. Similarly, quantified synthetic RNA of viruses was tested in duplicate with a commercial real-time PCR kit (ILS, Inter Lab Service, Russia) to determine the detectable minimum RNA copy number assay. The LoD of the InfA, swInfA, and InfB assays were found in 12 copies of RNA per reaction. The cutoff CT value for the flu panel was determined <38 for domestic human diagnostic test, under conditions of FDA Emergency Use Authorization in the US. This cutoff value is based upon the data from LoD analysis that are consistent with the cutoff value of <38 previously established for the rRT-PCR flu panel (Table 4).
Analytical Specificity and Inclusivity
Assay specificity was showed by testing four strains of influenza A (H1N1) viruses from Iranian population. Viruses were diluted approximately tenfold above the limit of detection of the assay, and the extracted RNA was tested in triplicate. Average Ct values for commercial kit and the rRT-PCR flu panel was compromised. As expected, all four viruses tested positive in the InfA, swInfA, and InfB assays at low virus concentrations (Table 5). All test results were 100 % concordant with the expected results.
Analytical Specificity and Exclusivity
In order to demonstrate the absence of cross-reactivity with other common human respiratory pathogens, exclusivity testing was performed by examining 34 non-influenza virus strains and bacterial organisms commonly present in the nasopharynx region of the human respiratory tract. Cross-reactivity was not observed in any of the non-influenza organisms which were tested at high titers. Extracted RNA from non-SW animal influenza viruses were also tested at high virus titers. As expected, all non-SW animal influenza viruses were InfA positive and swH1 negative, although some viruses were positive in the swInfA assay. Interestingly, not all three assays were positive for any non-SW animal influenza virus, so these results would be considered inconclusive and require further testing.
Discussion
We report the development of a multiplex RT-PCR to rapidly detect and subtype all currently circulating strains of human influenza virus in Iran, including influenza A, B, and swine origin influenza A (H1N1) virus.
Only a few assays that are capable of subtyping human and animal influenza viruses have been developed. One of these assays focuses specifically on swine influenza viruses and can detect and differentiate the H1, H3, N1, and N2 genes from currently circulating strains [13]. The primers from this multiplex assay appear to be capable of detecting swine origin influenza (S-OIV) H1 and N1 genes but are not likely to be capable of detecting the H1 gene of human viruses. Additionally, this study was designed to test swine specimens and may not be proper for human samples. A group of similar methods are capable of detecting H1 to H12 and N1 to N9 subtypes of the virus. These systems consist of five multiplex reactions (three of which are capable of detecting four different HA subtypes and two of them can each detect two different NA subtypes). The authors presenting these assays describe that they are capable of detecting human, avian, and swine viruses [14]. All of these methods subtype viruses by product size as visualized on an agarose gel [9, 10, 13].
In comparison, conventional PCR assays use gel electrophoresis, blotting, or hybridization to detect the amplified targets and take significantly more hands-on time [9]. The multiplex real-time PCR provides us a diagnostic result within one working day and eliminates post-PCR processing besides reducing the risk of cross-contamination.
Multiplex real-time PCR was found to be more sensitive than cell culture on a range of different respiratory samples. These findings are consistent with those of other studies, which employed RT-PCR for the detection of viral infections [15–17].
Our findings demonstrate that the multiplex TaqMan PCR is a sensitive and specific method for the simultaneous rapid detection of influenza viruses A, B, and swine-lineage influenza A. In the design of these real-time PCRs, an alignment of conserved regions of the target viruses was made with publicly available GenBank sequences. The detection limit of the multiplex real-time PCR method was measured by using tenfold serial dilution series of viral RNA of three different flu panel. The limit of detection of InfA, swInfA, and InfB assays were found in 12 copies of RNA per reaction. The cutoff CT value for the flu panel was determined <38, and regardless of the method chosen for sensitivity determination, it is difficult to ascertain the minimum number of target viruses needed for a detectable PCR product.
The analytical specificity of our multiplex real-time PCR assay was tested, and we detected no cross-reactivity between influenza viruses A/Tehran/2008/H1N1, A/shiraz/15/2005/H1N1, and B/Tehran/12/2007 or B/Tehran/8/02.
As demonstrated in this study, it is often difficult to achieve both high sensitivity and high specificity for viral nucleic acid detection. The primer–probe sets SwH1 (Table 2) are highly sensitive (ten copies/ml) but react nonspecifically with other influenza A subtypes (Table 3).
In summary, we have developed a rapid multiplex real-time PCR assay with the ability to accurately detect and differentiate influenza A, influenza B, and swine origin influenza A (H1N1) virus and this multiplex real-time PCR assay constitutes a specific and sensitive alternative to conventional culture and IF methods, and use of this assay would aid in the diagnosis of respiratory disease. Specific and sensitive results within 6 h are important in a clinical setting, and therefore, this assay could improve patient management by appropriate therapy following rapid diagnosis of a viral infection.
References
Esghaei, M., Monavari, S. H., Tavassoti-Kheiri, M., Shamsi-Shahrabadi, M., Heydarchi, B., Farahmand, B., Saleh, M., & Fotouhi, F. (2012). Expression of the influenza M2 protein in three different eukaryotic cell lines. Journal of Virological Methods, 179, 161–165.
(2009) Update: infections with a swine-origin influenza A (H1N1) virus--United States and other countries, April 28, 2009. MMWR. Morbidity and mortality weekly report 58, 431–433.
Hayden, F. G., Osterhaus, A. D., Treanor, J. J., Fleming, D. M., Aoki, F. Y., Nicholson, K. G., Bohnen, A. M., Hirst, H. M., Keene, O., & Wightman, K. (1997). Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenza virus infections. GG167 Influenza Study Group. New England Journal of Medicine, 337, 874–880.
Steed, L. L., Salmon, V. C., & Overall, J. C., Jr. (1994). Identification of influenza A virus by shell vial culture and two commercially available antigen detection methods. Clinical and Diagnostic Virology, 2, 181–189.
Vasoo, S., Stevens, J., & Singh, K. (2009). Rapid antigen tests for diagnosis of pandemic (Swine) influenza A/H1N1. Clinical Infectious Diseases, 49, 1090–1093.
Ziegler, T., Hall, H., Sanchez-Fauquier, A., Gamble, W. C., & Cox, N. J. (1995). Type- and subtype-specific detection of influenza viruses in clinical specimens by rapid culture assay. Journal of Clinical Microbiology, 33, 318–321.
Claas, E. C., van Milaan, A. J., Sprenger, M. J., Ruiten-Stuiver, M., Arron, G. I., Rothbarth, P. H., & Masurel, N. (1993). Prospective application of reverse transcriptase polymerase chain reaction for diagnosing influenza infections in respiratory samples from a children’s hospital. Journal of Clinical Microbiology, 31, 2218–2221.
Fan, J., Henrickson, K. J., & Savatski, L. L. (1998). Rapid simultaneous diagnosis of infections with respiratory syncytial viruses A and B, influenza viruses A and B, and human parainfluenza virus types 1, 2, and 3 by multiplex quantitative reverse transcription-polymerase chain reaction-enzyme hybridization assay (Hexaplex). Clinical Infectious Diseases, 26, 1397–1402.
Grondahl, B., Puppe, W., Hoppe, A., Kuhne, I., Weigl, J. A., & Schmitt, H. J. (1999). Rapid identification of nine microorganisms causing acute respiratory tract infections by single-tube multiplex reverse transcription-PCR: feasibility study. Journal of Clinical Microbiology, 37, 1–7.
Liolios, L., Jenney, A., Spelman, D., Kotsimbos, T., Catton, M., & Wesselingh, S. (2001). Comparison of a multiplex reverse transcription-PCR-enzyme hybridization assay with conventional viral culture and immunofluorescence techniques for the detection of seven viral respiratory pathogens. Journal of Clinical Microbiology, 39, 2779–2783.
van Elden, L. J., Nijhuis, M., Schipper, P., Schuurman, R., & van Loon, A. M. (2001). Simultaneous detection of influenza viruses A and B using real-time quantitative PCR. Journal of Clinical Microbiology, 39, 196–200.
Whiley, D. M., Syrmis, M. W., Mackay, I. M., & Sloots, T. P. (2002). Detection of human respiratory syncytial virus in respiratory samples by LightCycler reverse transcriptase PCR. Journal of Clinical Microbiology, 40, 4418–4422.
Lee, C. S., Kang, B. K., Lee, D. H., Lyou, S. H., Park, B. K., Ann, S. K., Jung, K., & Song, D. S. (2008). One-step multiplex RT-PCR for detection and subtyping of swine influenza H1, H3, N1, N2 viruses in clinical samples using a dual priming oligonucleotide (DPO) system. Journal of Virological Methods, 151, 30–34.
Kumar, S., Chusid, M. J., Willoughby, R. E., Havens, P. L., Kehl, S. C., Ledeboer, N. A., Li, S. H., & Henrickson, K. J. (2009). Introduction of a novel swine-origin Influenza A (H1N1) virus into Milwaukee, Wisconsin in 2009. Viruses, 1, 72–83.
Doller, G., Schuy, W., Tjhen, K. Y., Stekeler, B., & Gerth, H. J. (1992). Direct detection of influenza virus antigen in nasopharyngeal specimens by direct enzyme immunoassay in comparison with quantitating virus shedding. Journal of Clinical Microbiology, 30, 866–869.
van Milaan, A. J., Sprenger, M. J., Rothbarth, P. H., Brandenburg, A. H., Masurel, N., & Claas, E. C. (1994). Detection of respiratory syncytial virus by RNA-polymerase chain reaction and differentiation of subgroups with oligonucleotide probes. Journal of Medical Virology, 44, 80–87.
Weinberg, A., Zamora, M. R., Li, S., Torres, F., & Hodges, T. N. (2002). The value of polymerase chain reaction for the diagnosis of viral respiratory tract infections in lung transplant recipients. Journal of Clinical Virology, 25, 171–175.
Acknowledgments
This study was supported by a grant from Iran National Science Foundation (INSF). The authors would like to thank Dr. Keyvani.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Monavari, S.H.R., Mollaie, H.R. & Fazlalipour, M. Simultaneous Detection of Influenza Viruses A, B, and Swine Origin Influenza A Using Multiplex One-Step Real-Time RT-PCR Assay. Appl Biochem Biotechnol 172, 984–992 (2014). https://doi.org/10.1007/s12010-013-0583-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-013-0583-6