
Abstract
Poly(tetrafluoroethylene) (PTFE) is well known for its very low friction coefficient and thus widely used for antifriction applications, e.g., sliding lacquers, for many years. Because of the high wear rate and cold flow of the original polymer, PTFE is usually added in higher amounts to other polymer matrices as a lubricant. However, the incompatibility of PTFE requires lots of efforts to perform a homogeneous breaking down of agglomerates and particle distribution as well as dispersion stabilization of the physical mixtures during application and curing. PTFE can be functionalized by high energy irradiation and subsequently compatibilized with a polymer matrix by reactive extrusion resulting in chemical bonding of both polymers via suitable reactive groups. In this manner, poly(amide imide) (PAI) was coupled to γ-radiation modified PTFE micro-powder prior to the dispersion procedure. This study deals with the thermal and crystallization behavior of the PTFE lubricant in chemical bonded (cb) PAI-PTFEcb compound thin layers in comparison with those of the plain radiation modified PTFE depending on the cooling rate by DSC, Flash-DSC and WAXS. A morphological characterization of the extrudate as well as the coating gives information about the fragmentation and distribution of the PTFE phase in the PAI matrix. Furthermore, mechanical and surface properties of sliding lacquer films were analyzed after application on steel substrates by varying dry film thickness and curing.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
PTFE itself is a semi-crystalline high-performance plastic and offers a lot of outstanding properties that makes the material very interesting and unique for lots of applications. Because of its chemical structure and the high C-F bonding strength, it is chemically inert and anti-adhesive. Therefore, it is a most advantageous material for the coating of chemical devices and the sealing technology especially in the chemical plant construction when very aggressive media have to be handled. Furthermore, the polymer exhibits a high thermal stability and can be used in a wide temperature range from approximately − 200°C up to 260°C. Additionally, PTFE offers a very low friction coefficient of ~ 0.15 when it is used under dry conditions and even ~ 0.04 in the presence of oil. For that reason, the polymer is widely utilized as a sliding material and solid lubricant in many technical devices to save energy and avoid damage during the operation.
However, there are also some drawbacks. Although it is a thermoplastic material, it cannot be processed by conventional thermoplastic processing machines. Besides, the cold flow prevents the use of plain PTFE for construction elements in the machinery and plant engineering on one hand. On the other hand, the very high hydrophobicity of the material resulting from a contact angle of ~ 126° makes it incompatible with almost all other polymers. So, processing stable mixtures of other polymers with original PTFE in melt or dispersions with PTFE for anti-friction coatings are not available easily and require additional stabilization approaches. During the last decades, lots of efforts were done to overcome these drawbacks. For that, the instability of PTFE against high energy irradiation was used to develop strategies and technologies for a defined functionalization of the polymer. Thus, intensive investigations, also in our institute, have shown in the past that PTFE can be functionalized in a defined way by electron or γ-irradiation.1,2,3,4,5,6,7,8,9,10,11 When the irradiation is carried out in air atmosphere, C-C and C-F scission is accompanied by the generation of reactive functional groups (COF, free and associated carboxylic acid groups) and very persistent radicals. The overall mechanism was described in detail by Lunkwitz and Lappan.12,13,14,15,16 Those formed reactive groups can be used for a chemical compatibilization of PTFE with other polymers. In addition, the irradiation results in a certain reduction of molecular weight that lowers the melting point and the melting viscosity as well. However, those changes neither limit the temperature range of application nor lead to alterations of the low coefficient of friction. Hoffmann et al. investigated the γ-irradiation of PTFE powder in an industrially like pilot plant scale.17 As expected, they found that the general reaction mechanism in principle is comparable to that what was found for the electron treatment in a lab scale. Additional findings were that the overall functionalization degree not only depends on the type of irradiation and the irradiation dose but also on the PTFE type, the added HF absorber, the used atmosphere or the addition of other reactants. Moreover, via all these parameters the ratio between generation of functional groups and radicals can be controlled too.
Based on various radiation modified PTFE types during the last two decades, several new anti-friction materials in the fields of plastics, lubricants (oils and greases) and sliding lacquers have been developed and characterized intensively.18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41 Some of them were already brought to the markets.
Extended studies on the chemical compatibilization of functionalized PTFE with various poly(amide)s (e.g., PA6, PA66) via reactive extrusion in melt have demonstrated, that the resulting physical, mechanical and tribological properties are mainly influenced by the breaking down behavior and the PTFE particle size distribution in the poly(amide) matrix and the chemical bonding as well.21 Depending on the bonding degree between the PTFE particles and the PA chains, the mechanical properties of the polyamide matrix are nearly maintained for most parameters even at higher PTFE contents of e.g., 20 wt.%.22,23,41 Tribological experiments on optimized PA-PTFEcb compounds in various tribological systems revealed the same low friction coefficients that expected from original PTFE. Furthermore, for the chemical bonded compounds a much higher wear resistivity was determined by contrast with that what was known from comparable physical mixtures of PA with PTFE.23,25,26 In this context intensive DSC studies on PA-PTFEcb compounds with varied radiation modified PTFE-grades, amounts and extrusion parameters have shown that an optimal fine dispersed particle distribution and chemical bonding results in a fractional crystallization of the PTFE phase from the melt exclusively.23,27,28,29,41 Opposite to this, in the case of low chemical bonding also bulk crystallization at higher temperatures is observed beside that. Only a bulk crystallization that corresponds to the original PTFE was detected when no chemical compatibilization is achieved. The conclusion from those outcomes was that DSC measurements are a suitable analysis method to get information about the dispersing and coupling state in PA-PTFEcb compounds very easily.
In addition to PA-PTFEcb compounds also high-performance polymers and elastomers have been investigated by Lehmann et al. initially.18,20,30,31,32,33 In all cases, a chemical compatibilization was proved. In this manner, bulk phase materials for ball bearing cages based on PAI-PTFEcb were developed.34,35,36 Similar compounds were also prepared to investigate sliding lacquers for several tribological loads, initially.37,38,39,40 Due to a similar molecule structure compared to the poly(amide) for those PAI-PTFEcb compounds, a chemical coupling between the PTFE and the polyamide can be reached too.
The aim of the current study was to investigate the crystallization behavior of PTFE in chemical bonded PAI-PTFEcb compounds in comparison with original and γ-modified PTFE. Therefore, DSC measurements were performed under defined slow cooling rates. It was of interest, if the fractional crystallization also happens and can be used as an indication for the chemical bonding state in PAI-PTFEcb compounds. In addition, it has never been tested before, if the fractional crystallization happens and can be detected under very high cooling rates via Fast Scanning Calorimetry (FSC) too. Furthermore, the crystallinity and the crystal quality were characterized on the chip sensor subsequently to each cooling ramp. Therefore, a PAI-PTFEcb extrudate grade was chosen that was prepared via reactive extrusion conditions without any optimization concerning the chemical compatibilization. In order to be able to clearly assign the various effects in thermal and X-ray investigations, TGA, DSC and X-ray studies were also carried out on the base polymers for comparison. More comprehensive details with regard to the thermal behavior are provided in the supplements. Finally, the compound was utilized to prepare a sliding lacquer dispersion and sliding lacquer films that were tested with regard to their general technical properties depending on the film thickness.
Experimental methods and materials
Materials and material treatment
Commercial chemicals
PTFE: For the experiments, an emulsion polymerized poly(tetrafluoroethylene) (PTFE) type TF 2025 (Dyneon/3M, Burgkirchen an der Alz, Germany) was purchased. In this paper, the abbreviation PTFE will be used as name for all samples without any further pre-treatment concerning subsequent experiments (e.g., γ-irradiation).
PAI: As PAI, an industrially available poly(amide imide) type Torlon®4000T (Solvay Speciality Polymers, Brussels, Belgium) was chosen. In this paper, the abbreviation PAI will be used as name for all samples without any further pre-treatment.
NMP: For dispersion preparation as well as a solvent for extraction procedures, N-methyl-2-pyrrolidone (NMP) with purity > 99.5 vol% (Emplura® series; Merck KGaA, Darmstadt, Germany) was utilized.
Ethanol: The dispersion extracts were washed by ethanol 99 vol% denounced with 1 vol% methyl ethyl ketone (MEK) (Th. Geyer GmbH & Co. KG, Renningen, Germany) for two times.
Methanol: The extrusion extracts were washed by methanol 98.5 vol% (VWR, Darmstadt, Germany).
Radiation modification of PTFE
The emulsion polymer PTFE TF 2025 was radiation modified by γ-irradiation with a radiation dose of 700 kGy at ambient temperature in air atmosphere at Beta-Gamma-Service GmbH & Co. KG (Wiehl, Germany) by a 5 MCi60Co source. The modification was carried out under normal pressure and at room temperature according to Hoffmann et al.17 PTFE samples under investigation, which were modified by this procedure, will be named as PTFEγ.
Reactive extrusion of PAI with radiation modified PTFE
In this treatment procedure, a polymer compound named as PAI-PTFEcb will be made of PAI and PTFEγ in several steps. Initially, the powdery PAI Torlon® 4000T was premixed with irradiated TF 2025 powder (20 wt.%) in a Laboratory Universal Mixer LM 15 FU (MTI, Friedrichshafen, Germany) at 550 rpm for 5 min to get a homogenized powder. Afterwards, PAI-PTFEcb-compounds were prepared by reactive extrusion in melt by using a Haake Rheomex PTW16/25 twin screw extruder [maximum housing temperature: 320°C, screw speed: 250 rpm (Thermo Fisher Scientific, Waltham, USA)]. The melt was cooled down by water bath and the resulting solid PAI-PTFEcb was then granulated by a CSG 171/1 granulator (COLLIN Lab & Pilot Solutions GmbH, Maitenbeth, Germany).
Sliding lacquer dispersion preparation
For solvent-based sliding lacquers, 50 g of PAI-PTFEcb extrudate was dispersed in 150 mL NMP at 80°C by using a Dispermat®LC30 (VMA Getzmann GmbH, Reichshof, Germany) with 20 mm tooth disc (10.000 rpm, 4 h). For the reference sample, 50 g of original PAI was used. By the same procedure as described before PAI solutions were kept.
Application of PAI-PTFEcb dispersions
Free films. To prepare free films for use in standard DSC and SEM imaging, substrate free coating foils were prepared by hand. To do this, steel sheets (steel DC 01, thickness 1 mm) were initially cleaned with a washing solution of acetone: xylene: Petroleum benzine (bp: 100–140°C) in a 1:1:1 volume ratio. Subsequently, the release agent Frekote 800NC was applied by means of a saturated kitchen paper to form a visually observable layer, which was oven-baked in a laboratory convection oven (Function Line UT12, Thermo Electron Corporation–Heraeus, Langenselbold, Germany) at 190°C for 1 h. Then, application of the antifriction coating dispersion was carried out using a 200 µm doctor blade (BYK-Gardner, Geretsried, Germany), which, according to preliminary tests, results in target dry film thicknesses between 20 and 25 µm.
The curing process was also realized in the previously specified laboratory convection oven by the following temperature-time procedure:
80°C (60 min) → 120°C (60 min) → 160°C (60 min) → 200°C (60 min) → 225°C (60 min).
Finally, the film thickness of the cured film was determined using the Byko-test 8500 film thickness tester (BYK-Gardner, Geretsried, Germany) before the paint film was peeled off.
Coated steel sheets for studying the film quality properties. The sliding lacquer application on gradient oven sheets (steel ST 37-2, thickness 1 mm) was performed by Simex automatic doctor blade (Simex GmbH, Haan, Germany) and an application speed of 20 mm s−1. For that, the sheets were degreased to remove oily surface protection before the application as described above.
The wet film thickness was varied between 50 and 450 µm. Subsequently the curing procedure was carried out by a stepwise curing with a maximum curing temperature Tcuring,max of either 200°C or 225°C in a laboratory convection oven Function Line UT 12 (Thermo electron corporation–Heraeus, Langenselbold, Germany) according to the following curing procedures:
-
(I)
80°C (60 min) → 120°C (60 min) → 160°C (60 min) → 200°C (60 min)
-
(II)
80°C (60 min) → 120°C (60 min) → 160°C (60 min) → 200°C (60 min) → 225°C (60 min)
Characterization methods
Fourier transform infrared spectroscopy (FTIR)
FTIR spectroscopy was performed on the insoluble extraction residues subsequent to extrudate and dispersion extraction (130°C/45 min by NMP for seven times each, and washing with ethanol for two times at room temperature and drying at 60°C in vacuo until constant weight as a final step).
Infrared spectra of insoluble content as well as of original PAI were acquired by evacuated FTIR spectrometer Vertex 80v (Bruker, Ettlingen, Germany) equipped with both liquid nitrogen cooled and room temperature detectors. Extrudate dispersion residues as well as PTFEγ were measured in vacuo as cold pressed films in the region 4,000–400 cm−1, with 2 cm−1 resolution, using room temperature detector. For this, 32 scans were co-added to every spectrum.
It was impossible to prepare the original PAI (in powdered state after milling in an A11 basic mill [IKA, Staufen, Germany)] as a cold pressed film; therefore a transmission measurement was replaced by one in the air in ATR (attenuated total reflection) mode using single reflection diamond ATR Golden Gate unit (Specac, Orpington, UK). In this case, liquid nitrogen cooled detector was used to improve signal-to-noise ratio, obtaining the ATR spectrum within 4,000–600 cm−1 with 4 cm−1 spectral resolution, consisting of 100 averaged scans.
As mathematical treatment for the further spectra comparison, base line correction and normalization were used. Baseline correction was applied to every spectrum. Normalization was performed for transmission spectra only, utilizing the combination band around 2,365 cm−1 as the internal reference band (i.e., a combination band of stretching vibrations bands of both CF2 and C-C groups with the CF2 deformation vibration band).21 The spectral processing was implemented using OPUS 7.5 software (Bruker, Ettlingen, Germany).
TGA measurements
The TGA measurements were carried out by using a TGA-DSC 3+ (Mettler Toledo, Columbus, Ohio, USA) with intracooler Julabo F250 and sample robot in a nitrogen atmosphere (40 mL min−1). For the measurements, sample material of ~ 5 mg was placed in a 70 µL platinum pan. The samples were hold isothermally (30°C, 5 min) and then were heated from 30°C to 800°C with 10 K min−1. The evaluation was carried out by means of METTLER TOLEDO STARe Software (V16.20).
DSC measurements
Standard DSC. For the standard DSC measurements, a Discovery DSC 2500 (TA Instruments, New Castle, Delaware, USA) with intracooler and sample robot was used. Sample material of about 5 mg was placed in a pierced Tzero aluminum crucible. Under nitrogen as flushing gas (50 mL min−1), the samples were heated 2 times with 10 K min−1 from − 30°C–350°C and cooled down to − 30°C in between by using the following measuring program:
Equilibrate − 30°C | Isothermal for 2 min |
Ramp 10 K min−1 to 350°C | Isothermal for 2 min |
Ramp 10 K min−1 to − 30°C | Isothermal for 2 min |
Ramp 10 K min−1 to 350°C |
For the analysis, the software TRIOS (4.4.0.41651) (TA Instruments, New Castle, Delaware, USA) was utilized.
Modulated DSC. The modulated DSC of PAI-PTFEcb was carried out by a Discovery DSC 2500 (TA Instruments, New Castle, Delaware, USA) with Tzero aluminum pans (pierced lid) as described before and a sample weight of ~ 7 mg.
The following measuring conditions were used for the separate steps:
(a) Crystallization the extrudate after melting
Equilibrate 350°C | Isothermal for 5 min |
Cooling to − 30°C by 2 K min−1, modulate temperature 0.32 K for 60.0 s | |
Equilibrate at − 30°C | Isothermal for 5 min |
(b) Melting of crystallized sample from step (a)
Heating to 350°C by 2 K min−1, modulate temperature 0.32 K for 60.0 s
The peak separation calculation was carried out by using the NETZSCH Peak Separation 3 software.
Sample mass estimation for Fast Scanning Calorimetry. For a realistic interpretation of analyzed data the estimation of sample masses is required. The following procedure was applied:
(a) Determination of the normalized PTFE/compound PTFEcb phase melting heat by Discovery DSC 2500 (TA Instruments):
Ramp 30 K min−1 to − 30°C | Isothermal for 5 min |
1st Heating 20 K min−1 from − 30°C to 350°C | Isothermal for 2 min |
Cooling 30 K min−1 from 350°C to − 30°C | Isothermal for 5 min |
2nd Heating 20 K min−1 from − 30°C to 350°C | Isothermal for 2 min |
(b) Determination of the PTFE/compound PTFEcb phase melting heat by Flash DSC 1 (Mettler Toledo):
Ramp to − 30°C | Isothermal for 0.1 s |
1st Heating 100 K s−1 from − 30°C to 350°C | Isothermal for 0.1 s |
Cooling − 0.5 K s−1 from 350°C to − 30°C | Isothermal for 0.1 s |
2nd Heating 100 K s−1 to 350°C | Isothermal for 0.1 s |
The sample mass was obtained on the basis of the 2nd heating scan each. In this way, influences of the sample history can be excluded.
Fast Scanning Calorimetry (FSC, also named as Flash DSC). The measurements were performed by using a Flash DSC 1 (Mettler Toledo, Columbus, Ohio, USA) with UFS-1 sensor. The PAI-PTFEcb extrudate samples are prepared as microtome cuts of 10 µm thickness each that were deposited directly on the sensor by using a big area around 0.2 mm2. For PTFE sample preparation, this approach based on a pressed tablet or foil was not successful. Therefore, a tablet was pressed at room temperature. A section was then placed in a DSC pan, heated to 360°C at 10 K min−1, and then held isothermal for 2 min. After cooling the sample, sections of 10 µm thickness were made by microtome. These sections were then used for the Flash DSC measurements. Nitrogen was used as a flushing gas (55 mL min−1), and the surrounding temperature was kept at − 80°C.
For the crystallization experiments, the samples were initially hold at 25°C for 0.1 s and subsequently heated from 25°C to 360°C with 1,000 K s−1. There they were kept for 30 s isothermally to melt all crystals completely and then cooled down from the melt to − 50°C by varying the cooling rates CR between 0.1 and 4,000 K s−1. Finally, the material was kept at − 50°C for 0.1 s.
For the DSC investigation a low sample size was advantageous to achieve highly resolved signals. For this sample the variation of the crystallization speed was directly done without any break after the end of the first measuring. However, for the X-ray scattering the sample size was too low for a good resolution. For this reason, a larger sample with the same sample thickness was prepared and studied by DSC too. The evaluation was done with the METTLER TOLEDO STARe software (V16.20).
SEM-EDX measurements
Cross sections of the extrudate and a cured free film sample were prepared at − 170°C using an ultramicrotome EM UC7 (Leica, Wetzlar, Germany). In addition, SEM pictures were taken on the same type samples that were prepared by cryofracture. Prior SEM and EDX analyses, samples were coated with thin layer of 3 nm Pt. SEM and EDX images were achieved by utilizing a Gemini ULTRA Plus (Carl Zeiss Microscopy GmbH, Jena, Germany) equipped with a Flat Quad XFlash 5060F EDX detector (Bruker, Berlin Germany) using a magnification of 5,000 × for cross section samples and 20,000 × for cryofracture samples. The applied excitation voltage of 3 kV results in an information depth up to 200 nm.
Rheological measurements
Rotational measurement on the PAI-PTFEcb dispersion was done by an Anton Paar MCR 301 (Anton Paar Germany GmbH, Ostfildern-Scharnhausen, Germany) under increasing shear ramp between 0.1 and 1,000 s−1 and by using plate-plate PP 50 geometry at 24°C.
X-ray scattering measurements
Wide-angle X-ray scattering (WAXS) experiments were executed by means of the multi-range device Ganesha 300 XL + (SAXSLAB ApS, Copenhagen, Denmark/Santa Barbara, USA). For the experiments, Cu-Kα radiation was utilized (µ-focus tube 50 kV, 600 µA; monochromatized with bifocal Göbel mirror). Scattering intensities were accumulated by 2D-detector Pilatus 300K (pixel size 172 × 172 µm2). Path of rays, sample and detector are completely under vacuum (p < 5·10−2 mbar).
For thin sample measurements, the UFS-1 sensor with the large sample (see Flash DSC) was fixed to the sample holder with double-sided adhesive tape. Transmission scans were executed to determine the horizontal and vertical center of the thin support membrane with the sample for 2D-measurements. The experiments were performed using asymmetric transmission (perpendicular beam to the sample surface), a 2-slit configuration (0.9 x 0.9 mm2 and 0.4 x 0.4 mm2) and a beam stop with 2 mm diameter. The measurements were carried out for 3,600 s and 110 mm sample-detector distance.
The primary data (intensity I) were presented as 2D-scattering patterns lg I(qx, qy) and radial profiles lg I(q) (calculated via azimuthal averaging of the 2D-frames; q–scattering vector). The Bragg’s law,42 is the basic equation (1) for scattering experiments:
with d—lattice plane distance, θ—half scattering angle, n—order of reflection, and λ—wavelength.
The crystallinity of the samples was determined according to the peak area method,43 with the crystallinity defined as the ratio of the integral intensity of the crystalline scattering to the total scattering relating to PTFE alone (crystalline + amorphous). The calculations were performed using underground- WAXS curves, in which all available scattering maxima (reflections and amorphous halos inside an appropriate scattering range) will be fitted by means of pseudo-Voigt functions. For an appropriate fitting of the scattering area near the left tail of the (100) reflection of PTFE an additional weak scattering maximum was inserted, which corresponds to a meso-phase of PTFE associating with short-range ordered states.44 A strong reflection of the sensor material served as internal standard to control the calibration.
The full width at half maximum (FWHM) of a scattering reflection include information about the crystallite size and the quality of the crystalline structure (e.g., lattice distortions) with respect to the crystallographic direction (i.e., perpendicular to the lattice plane). Thus, the obtained FWHM of (100) PTFE reflections served as basis to calculate average crystallite sizes \(\it \left\langle L \right\rangle_{100}\) perpendicular to PTFE chain axis using the Scherrer equation (2):45
with K-factor (~0.9, depending on the kind of FWHM determination), λ—wavelength, \(\beta\)—radian of Δ2θ, and θ—half scattering angle. The values of Δ2θ and θ were taken from partial scattering curves of the fitting of experimental scattering data and corrected by slit smearing. Due to the presence of lattice imperfections, which also contribute to the reflection broadening, \(\it \left\langle L \right\rangle_{100}\) has account as minimum of average crystallite size.
Sliding lacquer film characterization
Film thickness. The dry film thickness after complete curing was measured by a Byko-test 8500 b (BYK-Gardner, Geretsried, Germany) according to the standards DIN 50981:1979-05 and ASTM B499. The values are intermediates of ten points that were measured each.
Gloss. The film gloss was determined by a Multi Gloss 268 (Konica Minolta, München, Germany) using 60° and 85° angle according to the standards DIN 67530, ISO 2813 and 7668, ASTM D523 and 2457. The values are the intermediates of 5 points each.
Surface roughness. Ra and Rz values were determined by a Hommel–ETAMIC W5 (Jenoptik, Villingen-Schwenningen, Germany) according to the standard DIN EN ISO 11562. The measuring conditions were:
Measuring length lt | 15.0 mm |
Measuring speed vt | 0.5 mm s−1 |
Cut off length lc | 0.25 mm |
The values are the intermediates of 3 points each on two coated sheets.
Mechanical properties
Impact/Reverse Impact. Film flexibility and film adhesion against impact was studied by an impact tester (BYK-Gardner, Geretsried, Germany) according to the standard ASTM 2794.
Erichsen Cupping. For the characterization of film flexibility under quasi-static loads, an Erichsen Cupping tester (Erichsen GmbH & Co. KG, Hemer, Germany) was utilized. The test was done according to the standard DIN EN ISO 1520.
Results and discussion
PAI itself is a high-performance polymer with outstanding mechanical, thermal and chemical properties. It can be used for applications up to 275°C and is one of the most thermally stable polymers together with poly(imide). Additionally, it offers a high resistance to chemicals and a high abrasion resistance. As a thermoplastic polymer it can be processed in melt, e.g., via melt extrusion or injection molding. Usual applications are heat resistant coatings, e.g., coatings for electrical insulation or overcoats for wires. Other applications are high-performance structures, e.g., ball bearings for high-performance turbine drills or fibers for heat and flame protective clothing. For the coated items in many cases, an anti-friction behavior is required. However, the friction coefficient of various PAI grades varies between ~ 0.25 and ~ 0.4 during dynamic sliding against steel surfaces. Therefore, solid lubricants, such as MoS2 and graphite or PTFE in colorless coatings, are added to improve the antifriction properties of PAI sliding lacquers.46,47,48,49 However, in industrially applications these lubricants are distributed physically in the PAI matrix. In this manner, no compatibilization and only low or no adhesion of the PTFE particles are kept in the PAI matrix polymer that results in a bad and not stable distribution together with lowering the mechanical matrix properties and wear resistance. To overcome these drawbacks, chemical coupling of the PTFE molecules to the PAI matrix was aimed and successfully implemented in the past. It was furthermore demonstrated that the bonding between the two polymers improved the wear resistance of the chemically bonded compounds in ball bearings as well as sliding lacquers remarkably.34,35,36,37,38,39,40 In hydrodynamic sliding bearings, the new materials were confirmed to be a competitive alternative for the expensive and problematic white metal.39
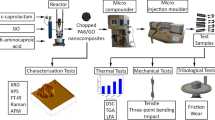
In this study, a PAI-PTFEcb compound with 20 wt.% PTFEγ was prepared by reactive extrusion in melt. This amount was confirmed to enable optimal sliding and wear properties for certain tribological applications before. The process of sample preparation and characterization is displayed in the scheme in Fig. 1. At the beginning, the extrudate was characterized by FTIR spectroscopy and SEM to prove the chemical bonding and to characterize the compound morphology. Next, the characterization will be continued by thermal analyses like TGA and DSC including a detailed examination by Flash DSC at thin samples. These samples are the objects of investigation in WAXS experiments depending on cooling rates in a broad range. Finally, dispersions and cured films will undergo tests for properties of potential applications.

Process of sample preparation and characterization
Investigation of chemical bonding and PAI-PTFEcb morphology
Although the extrudates comprise a relatively large amount of PTFEγ (20 wt.%) a direct confirmation of the chemical bonding, e.g., via NMR or FTIR analysis of the extrudate, or the film, respectively, were not successful. It was obvious that the total amount of covalent bonds is too low and so is the sensitivity of the various analysis methods not sufficient for the evidence. For that reason, a suitable extraction method was investigated based on a physical mixture of PAI and PTFE (80:20 wt.%) that enables to remove the soluble polymer content completely. This mixture was kept by dry blending the powders in an analysis mill. Then this mixture was extracted with NMP solvent at 130°C up to so much times that the PAI was removed completely at the end. For that 7 (the same for dispersion and extrudate) extraction steps were necessary. The details for both procedures are given in the experimental part. Afterwards the solid extraction residue was proved to be the plain PTFE by FTIR on a cold-pressed film. In this manner the PAI-PTFEcb extrudate and the dispersion as well were treated according to those validated procedures to remove the soluble and not bonded PAI content. In Fig. 2 the FTIR spectra of the resulting extraction residues are compared to the original PAI and the PTFEγ (700 kGy). In contrast to the PTFEγ containing samples the PAI was measured by using the powdered sample. A cold-pressed film based on this polymer cannot be prepared at all. Fort that reason the intensities of the PAI absorption bands are not comparable to those of the PAI-PTFEcb extraction residue but band positions are correct. With the exception of the original PAI all spectra were normalized to the CF2 absorption combination band at 2,365 cm−1 for a quantitative comparison (see Experimental part).16 The absorption curves of the insoluble extrudate as well as the dispersion residue beside the typical PTFE absorption bands also exhibit those assigned typically for the imide [at 1,778 and 1,710 cm−1 (Imide I) and 1,367 cm−1 (Imide II)], for the amide (at 1,605 and 1,494 cm−1—Amide I and Amide II, respectively) functional groups as well as the stretching vibrations of aromatic ring (at 1,605 and 1,494 cm−1) in PAI (see Fig. 2).50 So, it was proved that a certain amount of PAI is chemically bonded to the PTFEγ, how it has been already shown for PA before.21,29

FTIR spectra of PAI-PTFEcb dispersion and extrudate extraction residue in comparison with PTFEγ and PAI powder (for PAI: the right intensity scale is valid)
Furthermore, because of the PAI containing amide structures it is supposed, that the chemical bonding during the reactive extrusion process is based on the same general principles that were observed for the PA6-PTFE compounds.26,29,50 This means that the PTFEγ carboxylic acid groups cause an in situ transamidation reaction in the molten PAI phase that might result in a block copolymer structure too. As it was expected, the chemical modification grade is similar for both extrudate and dispersion. Besides, the decrease of the carbonyl fluoride (COF) absorption intensity (COF at 1,885 cm−1,12) is another confirmation for the chemical bonding between the PAI matrix and PTFEγ.
In order to get an idea about the morphology of PAI-PTFEcb SEM-EDX measurements were performed on the extrudate as well as on cured free films subsequent to an application of the prepared dispersion to steel sheets. As can be seen in Fig. 3 for the cross section pieces, the film shows a slightly improved fragmentation of the PTFE agglomerates and a more homogeneous distribution of the F-content and the PTFE particle size compared to the extrudate. This could be a result of the high-speed shearing during the additional dispersion procedure. Furthermore, a drastically lower maximum particle size of the PTFE particles is found with values of lower than 2 µm. A large amount even exhibits particle sizes < 1 µm, which is a necessary requirement for the occurrence of so-called fractional crystallization, which will be described in more detail in the discussion of the thermal analyses. The particle size distribution is thus still not optimal in both samples, since crystallization of the PTFE exclusively via fractional crystallization cannot be expected in either case. Optimization of the process parameters during reactive extrusion and the resulting improved shear of the melt in the extruder should also allow further improvement of the PTFE breaking down and distribution for the extrudate.

SEM-EDX of PAI-PTFEcb in the extrudate (left) and in the cured film (right)—quantitative distribution of F—content (red color) in the PAI-matrix (Color figure online)
In addition, SEM figures of cryofractures (see SI-Fig. 3) have demonstrated that the PTFEγ particles are well covered by the PAI matrix material, respectively. It can be concluded that all these results are another evidence for the high quality compatibilization that is supported by chemical bonding between the both polymers. In this way, a process-stable morphology is achieved, which is a prerequisite for the recyclability of the material.
Thermal behavior of the composite original polymer components
In order to understand the thermal behavior of the chemical bonded PAI-PTFEcb the plain components were studied by TGA-DSC and standard DSC as well. PAI itself is well known as a high-performance polymer and can be used in various applications in continuous service temperatures up to 220°C. So, it is one of the most thermal stable polymers that can be processed by injection molding. However, because of its high apparent viscosity the PAI polymers are processed in industrial applications at a relatively low molecular weight resulting in weak and brittle as-molded parts with poor chemical as well as wear resistance and not optimal thermal resistance. In order to achieve maximum properties a stepwise multi-step post-curing is required by the PAI producers that has to be carried out between 149°C and 280°C and overall 4.5 to 23 days depending on the structure thickness and application needs. During this thermal treatment the molecular weight increases via chain extension and imidization by condensation reactions. Final, the polymer can no longer be brought to the melt state for processing purposes.
Thermogravimetric analysis of the used PAI grade proved the expected high thermal stability of the polymer up to almost 400°C in nitrogen and up to 375°C in air atmosphere under non-isothermal conditions (SI-Fig. 1, SI-Table 1). The observed small peaks in the 1st deviation at around 55°C (mass loss ~ 1.5%) and 270°C (mass loss ~ 0.5%) each are related to loss of adhered water and a slight amount of post-condensation and not to any polymer decomposition. More detailed description of the TGA results is available in the supplements.
Standard DSC measurements on PAI extrudate granulate and PAI film from NMP solution after curing at 225°C (curing procedure II) were carried out after drying at 80°C for 8 hours in order to eliminate the physically adhered water before the measurements. Fig. 4 demonstrates that these conditions were not sufficient. As can be seen from the 1st heating the water evaporates up to ~ 120°C for the powdered granulate and up to 150°C for the PAI film. Furthermore, the film Tg was determined at approximately 215°C and is much lower than expected. It is supposed that residual NMP solvent molecules cause the shift to lower values. The wide endothermal effect above 220°C is a strong indication for this assumption. So, the glass transition temperatures were determined in the 1st cooling and the 2nd heating scan after the materials history was levelled and the water and residual solvent were removed. In the 1st cooling and the subsequent 2nd heating, the Tg-values for granulate as well as for the film were then detected in the expected temperature range between 270 and 280°C and there is nearly no difference in between the two states of materials.

Thermal phase transitions of origin PAI granulate and PAI film (from NMP solution, Tcuring,max = 225°C) after drying at 80°C/8 h by standard DSC
PTFE itself is well known as a highly crystalline polymer with a thermal stability up to approximately 450°C,51 independently of the synthesis method and so can be utilized in a wide temperature range from − 250°C to 270°C.52 However, it cannot be processed by injection molding. The molecular weight is usually approximately 106 g mol−1.53,54 As it was already mentioned in the introduction, a high energy irradiation is the only way to modify the polymer matrix that leads to C-C and C-F scissions resulting in the generation of polar functional groups on one hand and persistent perfluoroalkyl alkoxy radicals on the other hand. In the presence of air mainly COF groups are formed as functional groups during the irradiation procedure that subsequently hydrolyze to carboxylic acid groups in the presence of humidity.15,17,55 The formation of those groups enhances the hydrophilicity and polar properties of the polymer that improves the compatibility with other polymers and, furthermore, enables a chemical bonding to other polymers if suitable reactive groups are available. A shortened overall reaction route is given in the literature.12
In this study, a PTFE emulsion polymer TF2025 was used that was subsequently radiation modified by γ-rays with a dose of 700 kGy in a commercial line as described in the experimental part. TGA-DSC 3+ curves of PTFEγ compared to the original industrial polymer PTFE grade showed almost no difference in the maximum degradation temperature between the two samples (see SI-Fig. 2 and SI-Table 2). The only decrease is in the “T1% Weight loss” value from 498°C for original PTFE to 470°C for PTFEγ. However, this change is neither relevant for the reactive extrusion nor for the DSC investigations, since in the considered combination the PAI is the less stable polymer. More details concerning the TGA-DSC-investigations are provided in the SI.
In standard DSC studies 2 transitions in the heating cycles as well as in the cooling cycle were detected, each (Fig. 5, SI-Tables 3, 4, 5). A peak maximum between 10 and 20°C that relates to a solid phase transition from a triclinic to a hexagonal crystal structure (i.e., the transition of PTFE phase II to phase IV,56 compare the discussion in the “X-ray measurements” section, below), and another peak maximum between 320 and 350°C that corresponds to the melting were found. As previously written for the PAI samples, the location of the peak maxima of the phase transitions and the peak area of the associated enthalpies in the first heating cycle (SI-Table 3) are strongly influenced by the sample treatment and history before. For this reason and the subsequent FSC investigations and WAXS experiments, further evaluation here focuses on the 1st cooling and the 2nd heating. Results for the first heating and determined values for all DSC curves are given in the SI.

Melting and cooling of original PTFE and PTFEγ (700 kGy) in DSC by 10 K min−1
Compared to the first heating in the 1st cooling run under a well-defined cooling rate, there are only small differences between the crystallization peak maxima Tc,min of non-modified PTFE and PTFEγ that was slightly shifted to lower values. However, the crystallization enthalpy ΔHc was more than twice as high at PTFEγ, resulting from more perfect crystallization as a consequence of molar weight reduction (Fig. 5, SI-Table 4).
In the 2nd heating cycle (Fig. 5), no variations of the melting peak maximum Tm,max were observed for both PTFE grades, but the melting enthalpy was more than doubled for the PTFEγ. More detailed information on the determined values of the various effects can be found in SI-Table 5. The glass transition Tg did not appear in the two heating and the cooling ramps. Even after a quick cooling from the melt with 100 K min−1 a Tg was not visible in both cases.
Thermal behavior of PAI-PTFEcb by defined slow cooling rates
TGA-DSC investigations for the chemical bonded PAI-PTFEcb compound were carried out in nitrogen and air atmosphere too (Fig. 6, Table 1). Although in both cases a mass loss of 1 wt.% was already detected at approximately 190°C and of approximately 2 wt.% at 410°C, the 1st derivative does not show a peak minimum in this temperature range. The reason for this should be the extrudate grain used, from which the water contained is transported only very slowly to the surface of the grain. The actual polymer degradation, on the other hand, only starts at temperatures above 400°C and independently of the sample gas used. In the presence of oxygen, the chemically coupled compound even exhibits a somewhat higher thermostability than was found for the original PAI matrix material. The DSC signal of the TGA-DSC shows a melting peak with a maximum at 323°C for both measurements.

Degradation behavior and thermal phase transitions of PAI-PTFEcb in N2 and air atmosphere by TGA-DSC 3+ (heating rate 10 K min−1)
Subsequently, the specific standard DSC analyses were carried out in the same temperature range as used for the plain PAI. In contrast to the extrudate a vaporization of adhered water (cured at 225°C for 1 h at the end of the curing procedure) occurred between 10 and 140°C for the PAI-PTFEcb film and the Tg was detected at a significantly lower temperature in the first heating cycle (Fig. 7, Table 2). On the other hand, the PAI glass transition was determined at a much higher temperature than it was determined for the virgin PAI film after curing under the same curing procedure. So, it can be concluded that the NMP residue in the PAI-PTFEcb film was much lower. The melting of the PTFE phase was detected with a peak maximum at almost the same value each.

Melting and cooling of PAI-PTFEcb extrudate and cured film (Tcuring,max = 225°C) after drying at 80°C/8 h
In the first cooling, two crystallization peaks are visible in both samples that are obviously overlapped by the glass transition of the PAI phase at lower temperatures. The first peak at ~ 310°C relates to the PTFE bulk crystallization Tc,bulk and occurred at the same temperature than it was detected in the PTFEγ sample (Table 3).
Besides, another crystallization peak was found at approximately 275°C. From the literature it is well known that this crystallization peak results from the so-called fractionated crystallization as it was described for chemical bonded PA-PTFEcb block copolymers earlier.21,28,29 The occurrence of this peak (Tc,frac) is another evidence for the chemical bonding between PTFEγ and PAI molecules as well as an indication for a good breaking down behavior of PTFEγ agglomerates into small primary particles (particle size required ≤ 1 µm) by high shear rates during reactive extrusion in melt. Furthermore, the crystallization peak temperatures are independent of the PAI-PTFEcb preparation. The PTFE amorphous phase Tg that is expected at approximately 137°C was not detectable in the DSC curve for both materials as well.
In the 2nd heating run the PAI phase glass transition temperature as well as the PTFE crystal melting were analyzed (see Table 2). The film Tg that is due to the amorphous PAI phase is almost the same as detected for the extrudate but significantly higher than in the 1st heating run. This means that no solvent or adhered water residue was remaining in the amorphous PAI phase neither in the extrudate nor inside the film after the 1st heating. It can be concluded that the thermal properties of 2nd heating run only result from the molecule structure of the polymer in the copolymer itself. In addition, the melting behavior of the PTFE phase was found with a melting peak in the same temperature range as observed in the 1st heating cycle. So, it can be concluded that comparable to PA-PTFEcb compounds as described by Pompe and Häußler et al., real copolymers were kept by the reactive extrusion of PAI and PTFEγ in melt.21
In order to separate the PAI glass transition from the PTFE crystallization process to get more accurate values modulated DSC measurements were carried out. As can be seen from Fig. 8 the PAI phase Tg-value was detected in the reversing heat flow at 284°C and was successfully removed from the complex PTFE crystallization in the non-reversing cooling scan. Subsequently, for the calculation of bulk and fractional crystallization a peak separation was performed by using the non-reversing heat flow. Via a simulation with two peaks (bulk and fractional crystallization), the PTFEγ crystallization peak maxima were determined at 308°C (bulk) and 282°C (fractional) (SI-Fig. 4) (correlation coefficient: 0.996). However, the simulated sum curve does not adequately model the measured curve in the temperature range between ~ 275–285°C and 290–300°C and intended further simulation with 3 and 4 peaks, respectively.

Crystallization behavior of PAI-PTFEcb extrudate between 350 and − 30°C (2 K min−1) subsequent to melting at 350°C /5 min by modulated DSC
As demonstrated in Fig. 9 the choice of 4 peaks improved the correlation coefficient to 0.999 and lead to an optimal agreement between the simulation and the measured curve. While the crystallization peak at Tc,bulk = 309°C should refer to the bulk crystallization the peak maxima Tc,frac1 to Tc,frac3 are assumed to relate to various qualities of the fractional crystallization (Table 4). A similar behavior with a two-step crystallization was described for PA-PTFEcb compounds with a special grade of PA6 (Miramid SH3) in the literature found by Pompe et al.28 The authors stated in their hypothesis that the first part of the fractionated crystallization step starts from the surface caused by the nucleating effect of the molten PA6 Miramid SH3 matrix. Then, crystallization occurs within the PTFE particles, initiating from heterogeneous nuclei in the PTFE phase. Based on this hypothesis, for PAI-PTFEcb it is assumed that Tc,frac1 is nucleated from the PAI melt and crystallization starts from the surface of the PTFE phase as well. In contrast to PA6-PTFEcb, the temperature maximum for the first part of the fractional crystallization is shifted to higher temperatures. One possible reason is that the PAI melt already has a relatively high melt viscosity at 295°C only slightly above the solidification temperature. The second part of the fractional crystallization (Tc,frac2) is then already nucleated by the solidified PAI phase, but should also lead to crystallization starting from the surface of the PTFE phase. The third part of the fractional crystallization is assumed to be crystallization within the PTFE particles, as in the case of PA6-PTFEcb with PA6 Miramid SH3 grade.

Peak separation of PTFE crystallization peak in PAI-PTFEcb extrudate non-reversing heat flow cooling curve (integration limits: 263–325°C) − 4 peaks
By the peak area calculation, it was found that the copolymers still exhibit a relatively large content of bulk crystallization of about 50% (2 peak simulation, SI-Table 6) and about 35% (4 peak simulation, Table 4). In other words, the amount of fractional crystallization, which is a measure for the fragmentation and distribution quality of PTFEγ micro powders, is also about 50% (2 peaks) and 65% (4 peaks), respectively. In the optimum case, up to almost 100% fractional crystallization can be achieved if very finely dispersed breaking down and distribution and intense interactions of the incompatible PTFEγ phase with the matrix are obtained.28 So, high shear during reactive processing,22 a high irradiation dose of the PTFE28,29 and, the degree of chemical coupling have so far been identified as the main influencing factors. Obviously, the stabilization of the very finely dispersed morphology is only possible via chemical bonding. Despite of a high coupling proportion the interaction as well as self-organization tendency of the CF2 segments results in crystallization in the PTFE-phase.
It can be concluded that the reactive extrusion process and thus the breakdown behavior and distribution of the PTFEγ particles in the PAI matrix, as well as the chemical bonding between the two polymers, are not yet optimal.
The 2nd heating run of the modulated DSC shows the PAI phase glass transition as well as a smaller melting peak of the PTFE melting in the reversing heat curve (Fig. 10). That the last still occurred is supposed by a fast melting of the PTFE phase so that the peak could not be leveled under the used modulation conditions. A variation of the modulation conditions to overcome this drawback was not done yet. The small endothermic peak at ~ 285°C in the non-reversing heat is supposed to be the Tg relaxation Trelax,max of the PAI glass transition. The determined data are collected in Table 5.

Melting behavior of PAI-PTFEcb extrudate subsequent to the crystallization between − 30°C and 350°C by modulated DSC
Crystallization behavior by fast cooling
In order to analyze the crystallization behavior of PAI-PTFEcb in comparison with original PTFEγ small as well as larger samples with a defined thickness of 10 µm each were studied by fast cooling from the melt via FSC. In preliminary measurements were found, that for the validation of the thermal transitions small samples as shown in Fig. 11 (left) are more suitable. However, they were not sufficient to get a good resolution via X-ray investigation too. For that reason, approximately 10 times larger samples were prepared for those studies (Fig. 11, right) and treated by a comparable heating-cooling procedure afterwards. Therefore, those samples were not suitable for a quantitative DSC parameter calculation. For that purpose, a specific sample investigation strategy by FSC and WAXS was established that enables reliable results for each method (see scheme in Fig. 12). In addition, it makes sense that FSC studies of selected samples are carried out on small as well as large samples for comparison.

PAI-PTFEcb extrudate sample sizes on the FSC chip for subsequent DSC parameter analysis (small, left) and WAXS measurements (large, right). The sample thickness is 10 µm each (extrudate microtome cut)

Strategy of sample investigation by FSC and WAXS
In the case of PAI-PTFEcb the samples were cut from an extrudate granulate. In contrast, a PTFEγ film made of γ-modified micro-powder was too brittle to be cut by microtome section. So, a PTFE film was pressed at room temperature first, then heated in the DSC up to 360°C by 10 K min−1 and kept there for 2 min to achieve a complete melting of all crystals. After cooling down the sample was cut into the aimed thickness via microtome section. The masses m of the FSC samples were estimated based on the DSC peak areas of the PTFE melting peak using the following tailored equation:
It was found that the large samples are approximately nine times larger in the case of PAI-PTFEcb and even 18 times larger for PTFEγ (Table 6).
The crystallization curves of PTFEγ from the melt by varying the cooling rate between 0.1 and 4,000 K s−1 are collected in Fig. 13. In the temperature range between 180 and 330°C a single crystallization peak with a maximum between ~ 305°C (4 K s−1) and ~ 270°C (4,000 K s−1) depending on the cooling rate was observed. This peak relates to the PTFE bulk phase crystallization. It is well known from the literature that PTFE crystallizes very quickly.57 For that reason, only a slight drop of ΔHc was calculated in the observed cooling rate range (Table 7). In addition, in the literature is described that a leveling of the crystallization peak can be only reached at CR > 106 K s−1.58 This state was not achieved. A glass transition was not visible in all cases as already observed for the standard DSC measurement at CR = 10 K min−1.

Fast crystallization behavior of PTFEγ (small sample, microtome cut 10 µm thick) depending on the cooling rate CR by FSC
Figure 14 shows the melting peaks after melting with a heating rate of 1,000 K s−1 subsequent to each cooling rate variation. After cooling with CR between 0.1 and 3,000 K s−1 a single peak with a slight shift of the peak maximum temperature Tm,max from 320 to 331°C with decreasing CR was detected. The ΔHm-values raise with decreasing cooling rates (Table 8). Furthermore, the shifts of the peak maximum temperature as well as the melting enthalpy are stronger in the range of cooling rates below 500 K s−1.

Melting behavior of PTFEγ (small sample) by 1,000 K s−1 subsequent to the crystallization at varied rates CR by FSC
As it is obvious for the PTFEγ samples in Fig. 14 (Table 8), the value of CR has a strong influence on the subsequent melting behavior with a fast heating rate. There are two ranges of CR, which merge into each other at CR ~ 100…500 K s−1. The sample, which was crystallized by the lowest CR = 0.1 K s−1 (“quasi-isotherm”), has the highest melting temperature Tm (= 331°C) and a broader melting range. The DSC curve should reflect the melting of crystallites with comparatively best quality (i.e., lattice perfection, crystallite size, crystallite size distribution). But, the broadened melting range suggested an enlarged crystallite size distribution. As the CR increases, these parameters become more stringent. In consequence, Tm becomes smaller significantly and the melting range becomes narrower. Beginning with CR ~ 500 K s−1, the Tm-values change only slightly. That means, during the cooling procedure in the range with higher CR only crystallites with quite similar parameters (especially here with the lowest Tm ~ 320°C each) could be generated due to the freezing process of PTFEγ.
The crystallization maxima of large samples Tc,max offered the same tendency that was already observed for the small ones (see SI-Table 7). However, the crystallization enthalpy values ΔHc do not follow a unique tendency. Besides, the subsequent heating cycle showed that the melting was not completed in the non-isothermal step. For that reason, it was not possible to quantify the ΔHm-values with accuracy and a detailed analysis is not given. Nevertheless, it can be supposed that the remaining crystallite portion is completely melted in the subsequent isothermal step at 360°C for 30 s leaving no crystallization nuclei.
Crystallization curves of Flash-DSC measurements on small PAI-PTFEcb samples depending on the cooling rate CR are collected in Fig. 15. Two exothermic crystallization peaks were detected in the temperature range between 200 and 300°C, both arising from CR ≥ 100 K s−1.

Crystallization behavior of PAI-PTFEcb extrudate (small sample) depending on the cooling rate CR by FSC
The crystallization peak Tc,max2 determined at higher temperatures corresponds to PTFEγ bulk crystallization, while the peak Tc,max1 with a maximum between 264 and 248°C can be assigned to fractional crystallization of the PTFEγ phase (Table 9). Thus, it can be concluded that in chemically coupled PAI-PTFEcb, fractional crystallization also occurs under very high CR, as previously demonstrated in the standard DSC measurements with CR = 10 K min−1. The ratio peak-area/peak-height between bulk crystallization and fractional crystallization is largely independent of CR. Thus, it can be assumed that the crystallization kinetics of both processes is similar. In contrast, the glass transition of the PAI phase is not visible in the FSC at all CR.
After the defined crystallization at different cooling rates, a simple peak was found in the melt scans of the PAI-PTFE samples (each with 1,000 K s−1, Fig. 16) in the range of cooling rate from 3,000 to 500 K s−1. The peak maxima Tm,max1 were determined at approximately 320°C (Table 10) except for cooling with CR between 100 and 4 K s−1, where a shoulder Tm,max2 occurs at continuously increasing temperatures. At CR = 0.1 K s−1 the shoulder changes to the main peak and the former main peak still exists as a shoulder. Additionally, when two melting peaks occurred, a significant increase in the total enthalpy of melting was observed with decreasing cooling rate. In contrast to the Flash DSC (here 1,000 K s−1) such a behavior was not observed at slow heating rates of 10 K min−1 in standard DSC (0.16 K s−1), where a single peak at 224°C was measured.

Melting behavior of PAI-PTFEcb extrudate (small sample) by 1,000 K s−1 subsequent to the crystallization at varied rates CR by FSC
For the PAI-PTFEcb samples (compare Fig. 16 and Table 10), a very similar behavior to PTFEγ can be found in parts. There are also two ranges of CR, which merge into each other at CR ~ 100…500 K s−1, too. The sample, which was crystallized by the lowest CR = 0.1 K s−1, has its main melting peak (Tm,max2) at 328°C and a shoulder at 321°C. The main peaks for the following CR are related to the previous shoulder, whereas the former main peak appears only as a slowly disappearing shoulder. Following the interpretation for PTFEγ, it is assumed that here only during the “quasi-isotherm” cooling well-ordered crystallites were generated, which have the highest melting temperature [328°C, which is significantly lower (3 K) compared to that in PTFEγ due to the presence of PAI]. For higher cooling rates this ability quickly disappears again. On the other site, the new main peak Tm,max1 is nearby unchanged at ~ 320°C (beginning with CR = 4 K s−1). This means that almost the entire set of crystallites is formed independently of CR with similar quality parameters due to freezing process in PAI-PTFEcb as well as additionally by the chemical bonds in between the polymer chains. In other words, the deteriorating quality of the crystallites is determined more by the presence of PAI coupled to PTFE than by an increased cooling rate.
The results of the evaluation of the crystallization behavior of the large PAI-PTFEcb sample under variation of the cooling rate CR are summarized in Table 11. Both bulk crystallization and fractional crystallization of the PTFE phase are generally observed in the large sample as well, depending on CR. However, bulk crystallization of the large sample was detectable only at CR = 500 K s−1, while it was visible in the small sample at CR = 100 K s−1. In contrast, fractional crystallization was detected as earlier CR = 20 K s−1 as in the small sample.
The evaluation of the melting behavior of the previously defined crystallized samples showed a simple melting peak with a peak maximum at almost identical temperatures for all cooling rates (Table 12). However, with increasing CR, a significant trend towards drastically lower melting heats ΔHm was observed, indicating a significantly lower degree of crystallization after very high CR.
X-ray measurements
Scattering experiments (2D-WAXS) were executed by subsequent treatment of the same samples of PTFEγ and of PAI-PTFEcb: heating up to melt followed by cooling with defined (increasing) cooling rate CR. To get an impression of the basic 2D-WAXS patterns selected ones with different cooling rates were presented in Fig. 17 for PTFEγ and in Fig. 18 for PAI-PTFEcb. Already, first information could be drawn to the structural behavior: all samples were isotropic and show partial crystallinity caused by the content of PTFEγ. PAI is an amorphous component.

2D-WAXS pattern log(I) vs (qx, qx) of PTFEγ with different cooling rates CR, left: 0.1 K s−1, central: 4 K s−1, right: 4,000 K s−1 (miscolored pattern: low \(\to\) high intensity: blue-green-yellow-red)

2D-WAXS pattern log(I) vs (qx, qx) of PAI-PTFEcb extrudate with different cooling rates CR, left: 0.1 K s−1, central: 4 K s−1, right: 4,000 K s−1 (miscolored pattern: low \(\to\) high intensity: blue-green-yellow-red) (Color figure online)
Due to the isotropic scattering behavior more detailed (metric) data were calculable by azimuthal integration of the intensity I(q, φ) over the whole φ-range = (0 … 360)° with φ—azimuth angle. As examples, a zoomed q-region of radial scattering curves log(I) vs q of PTFEγ and PAI-PTFEcb with different cooling rates CR were shown in Figs. 19 and 20, respectively, containing all CR under investigation (scattering curves are vertically shifted, q-axis has a break). The left q-range indicate the (100) reflection of PTFE (in hexagonal crystal structure), the right part represents the invariant reflection of the high crystalline sensor material and the rather weak (110) reflection of PTFE. Both PTFE reflections are influenced by the cooling rates.

Radial scattering curves log(I) vs q of PTFEγ with different cooling rates CR, calculated from 2D-pattern by azimuthal integration over the whole φ-range = (0 … 360)°

Radial scattering curves log(I) vs q of PAI-PTFEcb with different cooling rates CR, calculated from 2D-pattern by azimuthal integration over the whole φ-range = (0 … 360)°
For PTFE as a partially crystalline polymer a number of crystalline as well amorphous phases are known, which are dependent on temperature and pressure as well.44,59,60,61 The different amorphous phases are generated from varied molecular weight polymer amounts. Phase diagrams provide a fast-informative overview about the phase behavior with its possible phase transitions.60,62,63,64
In the case of ambient air pressure, the crystalline PTFE phases II, IV, and I have to consider:
< ~ 19°C : | PTFE phase II has a 136 helix conformation (six turns per 13 CF2-groups), with a twisted zig-zag arrangement and triclinic crystal structure. |
~ 19…. ~ 30°C : | PTFE phase IV (after crystal-to-crystal phase transformation), which is characterized by a 157 helix conformation (seven turns per 15 CF2-groups) and hexagonal crystal structure. |
> ~ 30°C : | PTFE phase I (after crystal-to-crystal phase transformation), which is characterized by a helical conformation (up to about 150°C ) and pseudo-hexagonal crystal structure. |
Under pressure (at least ~ 4.5 kg cm−2) the phases II and I undergo a crystal-to-crystal phase transition to PTFE phase III, which has an orthorhombic structure (planar zig-zag arrangement). | |
Considering the experimentally found positions of the PTFE reflections, the crystalline structure could be assigned with phase IV of PTFE (stabile around room temperature), which is hexagonal in conformity with unit cell parameters ahex = a = b = 0.5665 nm, c = 1.9559 nm (space group P31 [144]), ICDD: PDF Card 00-060-1504,65 and 7 turns per 15 CF2-groups (along c-axis).59
In contrast to a number of publications, in which the influence of any irradiation on structure and properties of polymers is discussed after an appropriate sample preparation, here the investigations are directed on the characterization of a chemical bonded compound, in which one component is radiation modified from the beginning. It is well known that despite irradiation degradation in polymers, which is often stronger in amorphous polymer phases (mainly at low irradiation doses as result of chain scissions allowing strain relaxation and creating further crystalline areas). This leads to an increase of crystallinity. On the other hand, any irradiation with high-energy electromagnetic rays causes more or less strong lattice distortions in crystalline polymer phases (lattice damage, especially at higher doses and in presence of high initial crystallinity). In general, the former well-ordered domains cannot longer contribute to the crystallinity which thus becomes smaller.57,66 Also for polymers, WAXS experiments supply information about important parameters of the solid-state structure (e.g., crystal structure, crystallinity, crystallite sizes).67,68,69,70 In comparison with other crystallizable polymers, PTFE is distinguished by an extreme fast crystallization under cooling conditions up to several 105 K s−1, investigated by Bosq et al.58 The scientists also found that it is impossible to suppress crystallization and to generate a metastable glassy state of PTFE. Crystallites formed under these extreme conditions are not so stable than those arisen under moderate cooling conditions, what causes a shift of Tm to lower values.
By peak fitting of the WAXS curves the behavior of the crystallinity Xα in dependence on the cooling rate CR were calculated. In Fig. 22 the PTFE crystallinity Xα of PTFE, PTFEγ and PAI-PTFEcb vs cooling rate lg(CR) was presented with error bars. PTFE crystallinity is defined as the crystallinity in the PTFE phase, only. The amorphous scattering amount of the PAI phase was subtracted in advance. It was expected that Xα goes down with increasing CR. Inside of the error bars the crystallinity follows to an exponential function (decreasing linear slopes in Fig. 21). With two small deviations: first, for PTFEγ the slope can be fitted with two partial straights having a kink at CR = 100 K s−1, and second, for PTFE a similar, but considerably weaker kink into the other direction will be found too. Nevertheless, in comparison of the three samples the expectations were met generally. Increased cooling rates reduce the crystallinity. Non-modified PTFE shows the largest values for the crystallinity, but PAI-PTFEcb the lowest (based on the PTFE content). The minimal difference at CR = 0.1 K s−1 in comparison between PTFE and PTFEγ could be a hint on the stronger radiation degradation in the amorphous PTFE phase (but the error bar does not allow real proof). An increase of CR by 4.5 decades yields in loss of crystallinity ΔXα ~ 0.9 for PTFE, ~ 0.26 for PTFEγ, and ~ 0.10 for PAI-PTFEcb, respectively. In relation to the start values of Xα at CR = 0.1 K s−1 there are the corresponding values of Xα decreasing: ~ 12.5%, ~ 23.5%, and ~ 40.0%, respectively. As result of γ-irradiation it can be assumed that Tg and Tm of PTFE becomes lower with respect to the related values assumed of the non-irradiated PTFE.71,72 Especially at higher irradiation doses, the reasons are main chain scissions (tantamount to reduction of molecular weight), which change strongly the long-range orientation of chains in the intermediate and high melting crystalline blocks of PTFE. The generated structure is distinguished by a short range orientation of cluster association structures and leads ultimately to transformation into the amorphous phase.66,72 Besides, it has to be considered that the actual effect of irradiation onto structural and thermal properties is enormously influenced by the initial crystallinity. In the case of Tg the amorphous PTFE phase shows peculiarities found with dynamic rheometry and mechanical tests by Calleja et al.,73 which were discussed as two different regions: first, a “mobile amorphous fraction” (MAF) with a relaxation area at quite lower temperature (T ≈ − 103°C), and second, a “rigid amorphous fraction” (RAF) specific for PTFE chain segments having lower mobility near the crystalline phase. Thus, their relaxation area is located at a higher temperature (T ≈ 116°C), which is accepted as Tg of PTFE generally. Via the previously described DSC measurements in the temperature range between − 30°C and 360°C, the PTFE-associated Tg could neither be detected by standard and modulated DSC nor by FSC. This is in agreement with the explanation that was given by Henri et al..74

PTFE crystallinity Xα of PTFE, PTFEγ and PAI-PTFEcb vs cooling rate lg(CR). Errors ΔXα are ~ 1.5% (for PTFE and PTFEγ) and ~ 2.0% (for PAI-PTFEcb)
Besides of the crystallinity several parameters basing on the partial scattering curves of the fitting procedure (peak deconvolution) could be determined. The position of the strong (100) reflection of PTFE qmax is a measure of the lateral distance of the hexagonally ordered PTFE chains concerning d100 = 1/2√3·ahex. The full width at half maximum (FWHM) include information about the crystallite size and the quality of the crystalline structure (e.g., lattice distortions). These parameters as function vs cooling rate CR are presented in Figs. 22 and 23.

Maximum position as qmax and 2θmax of the (100) PTFE reflection of PTFE, PTFEγ and PAI-PTFEcb vs cooling rate lg(CR)

Full width at half maximum (FWHM) as Δq and Δ2θ of the (100) PTFE reflection of PTFE, PTFEγ and PAI-PTFEcb vs cooling rate lg(CR)
For PTFEγ (only one chemical phase) a continuous decrease of qmax vs CR was found with steady reduction of the slope at low CR. From CR ≥ 2,000 K s−1, a nearby linear behavior is visible. These changes correlate to a coefficient named as “freezing coefficient” (κfreez) for d100. That is to say the lateral distance dmax of the PTFEγ chains become larger with increasing of CR and so κfreez is positive (compare also Table 13). On the other site, PAI-PTFEcb (two chemical phases with covalent bonds in between) depicted a quite other behavior. It aroses two ranges of CR. Beginning with CR ~ 0 up to 100 K s−1 an increasing of qmax vs CR and reaching a maximum (i.e., a minimum in dmax) are shown. Thereafter, the behavior turned over to a decreasing function, but weaker compared to PTFEγ. Here, a nearby linear behavior is visible from CR ≥ 1,000 K s−1. In this connection, the cooling rate CR ~ 100 K s−1 occupies a special position, were κfreez changes the sign. Non-modified PTFE showed a peculiar behavior. At first glance, its freezing behavior vs CR seems to be mirror-image in comparison with PTFEγ (see Fig. 22). This finding seems to match the behavior of the crystallinity with the special point at CR ~ 100 K s−1. A continuous increase of qmax vs CR was found with steady enhancement of the slope at low CR. From CR ≥ 1,000 K s−1, a nearby linear behavior is visible, which correlates to a negative κfreez for d100. So far, there is no explanation for this unexpected behavior. Besides, a comparison of PAI-PTFEcb in detail with both PTFE and PTFEγ shows that the behavior of qmax up to CR ~ 100 K s−1 is similar to that of PTFE, whereas for CR > 100 K s−1 it is much more similar to PTFEγ. That suggests that the freezing behavior of PAI-PTFEcb initially resembles that of PTFE and later that of PTFEγ. Again, the change point is CR ~ 100 K s−1.
In Fig. 23 the full width at half maximum (FWHM) is illustrated as scattering parameters Δq and Δ2θ, respectively, characterizing the quality of ordering. The related crystallite sizes confirm the gradually different behavior in dependence of the applied cooling rate as well. It was expected that FWHM increases with rising CR. But, up to cooling rates of ~ 100 K s−1 a different behavior was detected. For PTFE a nearby unchanged FWHM was found, whereas for PTFEγ and PAI-PTFEcb a distinct increasing occurred. Between ~ 100 K s−1 and ~ 400 K s−1 a changeover is visible. Then, with still higher cooling rates a linear slope was reached for all samples. The slopes of PTFEγ and PAI-PTFEcb (i.e., samples, which contain radiation modified PTFE) are akin to each other and steeper in comparison with PTFE. As discussed above for qmax, here the cooling rate CR ~ 100 K s−1 occupies a special position, too. Compared to PTFEγ, for PAI-PTFEcb could be found additionally a clear lower level of \(\it \left\langle L \right\rangle_{100}\), which was caused by the presence of the PAI phase in the whole and particularly by its interplay with the PTFEγ phase because of chemical linkages in between. For selected FWHM the calculated average crystallite sizes \(\it \left\langle L \right\rangle_{100}\) are listed in Table 13 as a quick overview, in which the tendencies of the parameters change, are noted in a simplified manner as well. The values confirm the knowledge that the generation of large crystallites is preferred at low cooling rates and in crystallizable polymers having a comparatively lower molecular weight (here caused by irradiation).75
Clark76 has discussed two peculiarities of the crystal structure of PTFE: 1st - the narrow region of existence of PTFE IV in the phase diagram (at approximately room temperature) and 2nd - the phase transition to PTFE I adjoining to higher temperature. PTFE IV seems to be compatible with the restrictions on molecular untwisting resulting from the favorable packing for the space group P31. This model required substantial angular motion (librations) of linear segments within a well-defined lateral chain array (lattice), based on left/right-defects and clustering of like-handed segments. PTFE I exhibits increased motion of defects accompanied by further untwisting of the chains, without achieving the planarity of polyethylene chains. The planar alignment of CF2 groups gradually dissipates with increasing temperature above 30°C, but the parallel hexagonal alignment of the chain axes remains up to the melting point. The experiments discussed here follow the reverse temperature curve (starting at T > Tm) with successively increasing cooling rates CR. With detailed examination of structure parameters (e.g., with WAXS and DSC) could be discovered that it is possible to subdivide CR in three ranges roughly: 0 < CR ≤ 100 K s−1, ~ 80…100 K s−1 < CR < ~ 400…500 K s−1 (with 100 K s−1 as peculiarity), and ~ 400…500 K s−1 < CR (with 4,000 K s−1 as experimental maximum). Considering the Clark’s scientific findings, it can be concluded that at low CR the librations are able to follow the cooling process and a phase transition PTFE I → PTFE IV could take place. At significantly higher CR the librations can no longer follow and freeze up and as a result a supercooled phase I should be created. Therefore, CR ~ 100 K s−1 could be considered as the "freezing cooling rate" of PTFE.
Based on general experiences relating to polymers the perfectness of crystalline structures drops drastically with increasing cooling rates. Heterogeneities and nucleation processes influence the crystallization processes significantly. So that increasing amounts of the copolymer PAI-PTFEcb are transferred into a more or less non-ordered state. The prominent values found in the WAXS experiments (i.e., qmax, d100, and FWHM [or rather: Δq, Δ2θ], \(\it \left\langle L \right\rangle_{100}\)) at the special cooling rate CR ~ 100 K s−1 can be attached to a peculiar behavior in DSC investigations, too.
Dispersion and film characterization
For the subsequent film formation and characterization, the PAI-PTFEcb extrudate was dispersed in NMP, where the PAI-phase is completely soluble. Because of the insolubility of the PTFE phase in NMP it can be assumed that the dispersion procedure itself does not lead to any change of the crystallization state compared to the state of the material after the reactive extrusion processing. Resulting from the partial chemical bonding of the PTFEγ on one hand and the better compatibilization of the functionalized PTFE particles with the PAI matrix on the other hand the dispersions are very stable and no sedimentation was observed while storing for month. Rheological measurements of the viscosity η depending on the shear rate \(\dot{\gamma }\) after the preparation gave an intention about the needed minimum shear rate to get shear-thinning behavior what is an absolute precondition for a well-defined film thickness after curing.
Figure 24 shows the viscosity η (left ordinate) and the shear stress τ (right ordinate) of the chemically coupled PAI-PTFEcb dispersion and pure PAI coating lacquer as a reference as a function of the shear rate in double logarithmic scale. The measurement was performed in rotation. A shear rate ramp between \(\dot{\gamma }\) = 0.01 s−1 and 1,000 s−1 was performed. With the plot of the axes used in the diagram, one can determine a yield point by means of the shear stress - shear rate curve. For the specific measurement mode used here, the yield point for the PAI-PTFEcb dispersion is η ≈ 8.0 Pa·s (viscosity) at a shear rate \(\dot{\gamma }\) = 0.01 s−1. In comparison, the yield point for the PAI dispersion is significantly higher with η ≈ 12.0 Pa·s.

Dispersion viscosity η and shear stress τ depending on the shear rate \(\dot{\gamma }\) of PAI and PAI-PTFEcb
Noticeable in the viscosity curve of PAI-PTFEcb are two changes, which are due to a changing viscosity behavior of the sample as a function of the shear rate. At very low shear rates between 0.1 and about 4 s−1, a largely Newtonian behavior of the viscosity was obtained (blue tangent). That is, the viscosity value is independent of the shear rate in this range. Above \(\dot{\gamma }\) ~ 4 s−1 a shear thinning behavior is observed. Initially, there is a linear drop (red tangent) in viscosity, which then continues to drop drastically in a curve-like progression from \(\dot{\gamma }\) ~ 160 s−1. Three tangents were created and their intersections determined. The first intersection point was identified at a shear rate \(\dot{\gamma }\) = 4.0 s−1 and a viscosity η = 8.1 Pa·s. The second crossover point has been detected at a shear rate \(\dot{\gamma }\) ~ 270 s−1 and η = 6.9 Pa·s. Thus, above the shear rates found in the crossover point, a rheological change in the sample takes place in each case. Since the sample consists of completely dissolved material (PAI) and finely dispersed undissolved PTFE particles with chemically bonded PAI molecules, these changes can be explained as follows. It is known from the literature,77 that shear thinning can occur during the alignment of physically dispersed particles. This requires relatively low shear rates. Therefore, it is assumed that the 1st transition is due to the alignment of chemical bonded and undissolved PAI-PTFEcb particles and unbonded PTFEγ in the dispersion. Furthermore, dissolved polymer nodules may disentangle when a certain shear rate is reached. In this process, the viscosity drops further. It is supposed that this process occurs in the PAI-PTFEcb dispersion under consideration above the second transition, since higher shear rates are required for this. For comparison, the viscosity curve of a PAI solution with the same mass concentration of solids was measured. In contrast to PAI-PTFEcb, this shows only one crossover point at \(\dot{\gamma }\) = 245 s−1, and η = 12.1 Pa·s which was determined approximately in the region of the 2nd transition for PAI-PTFEcb. This can be considered as a confirmation of the previously established hypothesis. The viscosity value of the PAI solution was detected to be ~ 1/3 higher in the shear rate independent range compared to the PAI-PTFEcb dispersion. A calculation of the shear rate during the used doctor blade application led to values between \(\dot{\gamma }\) = 44 s−1 (50 µm wet film thickness) and 400 s−1 (450 µm wet film thickness). This means that the required condition of shear thinning during application to get defined film thicknesses was fulfilled in every case.
The shear stress increases linearly with the shear rate and showed no deviation from an ideal viscous behavior up to a shear rate of \(\dot{\gamma }\) ~ 250 s−1. Above this value a shear thinning was detected. The difference between the viscosity and flow curve with regard to the shear thinning behavior is supposed to result from the two different polymer states in the dispersion: insoluble PAI-PTFEcb and PTFEγ molecules on one hand and soluble PAI polymer molecules on the other hand. For a more detailed description of the viscosity and flow behavior, further experiments have to be done with a maximum PAI-PTFEcb bonding degree (no bulk crystallization) and also up to higher shear rates of at least 104, nay, 105 s−1.
In order to get an idea of the film properties depending on the dry film thickness the dispersions were applied to degreased steel sheets. Therefore, the wet film thickness was varied between 50 and 450 µm. The curing procedure was carried out stepwise in a convection oven via curing procedure I with a maximum curing temperature Tcuring,max of 200°C. This is slightly below the boiling temperature of NMP (202°C) and corresponds to industrially usual curing temperatures of PAI lacquers (see detailed results in SI-Table 8). In addition, another curing procedure II with an elevated maximum temperature of 225°C was utilized that has been found by our own experimental work earlier to perform the maximal available mechanical film performance with regard to the film flexibility for a final dry film thickness between 15 and 30 µm. The film flexibility was tested according to DIN EN ISO 1520 for Erichsen cupping and ASTM 2794 for direct (dir) or reverse (rev) Impact after cooling down and storing the applied sheets for at least 2 or 3 days in laboratory climate. Results depending on the wet Dwet and dry film thickness Ddry by using curing procedure II were collected in Table 14. The maximum possible resistance to direct and reverse impact loading was found up to Ddry ≈ 25 µm. That is almost the same, what was achieved for curing procedure I.
A comparison of the Erichsen film flexibility that was measured under quasi stationary conditions (Fig. 25) showed the expected slight decrease of film flexibility with the increase of dry film thickness. Furthermore, the flexibility of the film with a maximum curing temperature of 225°C seems to be slightly lower at higher dry film thicknesses than of those with a maximum curing temperature of 200°C (Detailed information based on the measurement points cannot be derived.). TGA measurements on both films with a dry film thickness of ~ 20 µm demonstrated that a smaller content of residual NMP in the film with the higher maximum curing temperature (curing procedure II) should have caused this behavior.

Film flexibility of PAI-PTFEcb films on degreased steel depending on the dry film thickness after curing at a temperature maximum of 200°C (curing procedure I) or 225°C (curing procedure II) by Erichsen cupping
Furthermore, a comparison with plain PAI films with an approximately film thickness of 25 µm (values see Table 15) demonstrated that the relatively high PTFE content of 20 wt.% does not reduce the overall film flexibility. This is another indication for the good compatibilization, breaking down and distribution of PTFE primary particles in the PAI matrix.
For the characterization of the overall surface film appearance gloss and roughness measurements were carried out. Independent of the dry film thickness high gloss was observed in all cases for curing procedure II (Table 16) as well as curing procedure I (SI-Table 9). So those values that are measured via the 85° angle geometry delivered a more accurate picture for comparison.78 It was determined that almost independent of the curing procedure the gloss increases up to ~ 30 µm film thickness (Fig. 26). At higher film thicknesses the gloss values reach a plateau each and were found in the same range. In contrast, the mean roughness values Ra and the roughness depths Rz drop nearly continuously with increasing Ddry-values. It cannot be excluded that the changes in the surface smoothness affect the gloss values. Especially at low film thicknesses a deviation of the reflected angle during the gloss measurement can be supposed that leads to a reduction of the surface gloss.

Surface appearance (gloss, surface roughness) of PAI-PTFEcb films on degreased steel depending on the dry film thickness after curing at a temperature maximum of 200°C (curing procedure I) or 225°C (curing procedure II)
In contrast, the reference films made from pure PAI matrix show significantly higher 60° but surprisingly lower 85° gloss values than the films made from the PAI-PTFEcb compounds (Table 17). The latter should be due to the higher surface roughness values determined for the PAI films. However, because of the very low primary particles size and the very homogeneous particle size distribution of the PTFEγ particles that were already found by SEM images a very high gloss level and low surface roughness were achieved for all PAI-PTFEcb compound films.
Conclusions
The subject of the studies in this work was the characterization of the morphology, melting and crystallization behavior of chemically compatibilized and chemically coupled PAI-PTFEcb by means of thermal analyses (TGA, DSC, FSC) in combination with X-ray diffraction analysis of the crystallization state. The results were discussed in comparison with radiation-modified (PTFEγ) and untreated PTFE. For this purpose, a compound of PAI and 20 wt.% modified PTFEγ was prepared via reactive melt extrusion. A lacquer dispersion was then formulated on the basis of the extrudate under high shear and slightly elevated temperatures. The dispersion was characterized with respect to rheological properties and the cured films with respect to application-relevant film properties.
In order to investigate the coupling state of the PTFEγ to the PAI matrix after melt extrusion or after dispersion of the resulting extrudate, an extraction method was developed to remove non-bonded matrix polymer. The occurrence of characteristic PAI imide and amide absorption bands, as well as the decrease in the intensity of the COF band in the FTIR spectra of solid residues, indirectly demonstrated that PAI molecules are coupled to PTFEγ via covalent bonds in both cases. Subsequent high shear dispersion of the extrudate did not result in any further increase in the amount of bonded PAI and thus in no further increase in the degree of coupling. Since PAI, like poly(amide), contains amide structures in the polymer chain, it can be assumed that transamidation reactions are responsible for the chemical coupling. The fine breakdown and distribution of PTFEγ particles, as found in SEM-EDX studies, and the matrix coverage are evidence of good compatibility between matrix and solid lubricant, and of chemical coupling, respectively. A high amount of PTFEγ primary particles in the submicrometer range are observed in the cured film. The absence of aggregates in the film shows that a largely process-stable morphology has been achieved. In addition, the high shear in the dispersion process led to a further slight improvement in the breaking down and distribution of the PTFEγ particles in the PAI-matrix.
TGA and DSC preliminary tests on the neat matrix components provided suitable temperature limits and served as a reference for elucidating and assigning the thermal effects in the chemically coupled compound. The PAI grade and PTFEγ used are thermostable up to at least 400°C. Standard DSC measurements with heating and cooling rates of 10 K min−1 have shown Tg-values of ~ 270°C (1st cooling) and ~ 280°C (2nd heating) after leveling the thermal history on both granules and films for the amorphous matrix polymer. For the crystalline PTFE, in addition to the melting peak, the known solid phase transitions between 18 and 30°C were also detected. For the melting peak maximum Tm,max, drastic differences of 20 K were found between PTFE and PTFEγ in the 1st heating scan. PTFEγ exhibits the lower value at 326°C, while the melting enthalpies differ by only ~ 3 J g−1. The reason for this is the radiation-chemical degradation of the PTFE chains during γ-modification, which thus largely took place in the amorphous regions. When cooling from the melt, the position of the crystallization peak minima Tc,min differs only by 4 K and Tc,min is slightly shifted to lower values for PTFEγ. However, the crystallization enthalpy ΔHc for PTFEγ is more than twice as high, which is due to the improved crystallization ability of the now shorter PTFE chains. In the 2nd heating, the temperatures of the melting peak maxima of both PTFE types do not deviate. The melt enthalpies essentially correspond to the previously determined crystallization enthalpies, which is an indication that no further increase in crystallinity occurs after defined cooling at 10 K min−1. No glass transition temperatures were found for either PTFE or PTFEγ.
TGA-DSC measurements on PAI-PTFEcb granules revealed differences in the degradation behavior between N2 and air atmosphere. The curves, as well as the determined values for mass loss, illustrate that the compound is thermostable in the range within the temperature limits. Standard DSC measurements (1st heating) on the extrudate and cured films demonstrated that residual solvent components are still present in the film after curing, resulting in a significantly lower glass transition temperature Tg of the PAI phase of 253°C in the 1st compared to 275°C in the 2nd heating. For the PAI-PTFEcb-extrudate, differences of 8 K were observed between 1st and 2nd heating, which are supposed to be due to absorbed water traces and resulting H-bridge formation with amide structures in the compound. In the 2nd heating run the Tg-values for the film are only a little lower than in the extrudate. The melting peak maximum of the PTFE phase was determined at ~ 324°C for both the extrudate and the film, independent of the sample history. The same applies to the values of ΔHm with ~ 10 J g−1. In the cooling curves of both PAI-PTFEcb specifications, a second minimum at ~ 275°C was found in addition to the crystallization peak with a minimum at ~ 310°C, which can be attributed to PTFEγ bulk crystallization. This is fractional crystallization, which only happens in very finely dispersed phases.28 Fractional crystallization is another indication of chemical covalent coupling between the two matrix components. It is assumed that true copolymers, or more precisely block copolymers, were formed by the transamidation reaction. The fractional crystallization as well as partly the bulk crystallization of the PTFE phase of the compound are affected by the glass transition of the PAI phase. Modulated DSC measurements were performed to separate the reversible part from the irreversible phase transitions and to obtain a more relevant picture of the crystallization processes. Based on the non-reversing cooling scan, peak separation and simulation were performed to quantify the fractional crystallization fraction. The evaluation with 2 peaks provided a nearly 1:1 ratio between the two processes as shown in the supplements in detail. A peak separation with 4 peaks led to a more exact description of the processes occurring in parallel to the vitrification of the PAI phase. A total of 3 peaks were assigned to fractional crystallization, resulting in a ratio between bulk and fractional crystallization of 1:3 from this evaluation.
Since the results of the standard DSC measurements for PAI-PTFEcb did not differ significantly for extrudate and film, the crystallization investigations were carried out under high cooling rates (4 and 4,000 K s−1) using FSC on 10 µm extrudate microtome sections. PTFEγ was used as a comparison. Sample sizes in ng are sufficient for reliable FSC evaluation. In contrast, for a good resolution of the crystallization effects via X-ray diffraction, a sample mass of 10 to 20 times was required. For PTFEγ, a simple crystallization peak was identified in the utilized cooling rate range, the maximum of which shifted from 304°C to 267°C as the cooling rate CR increased. In contrast, the crystallization enthalpy decreased marginally above a CR of 100 K s−1. A leveling of the crystallization peak was not achieved with these CRs, as expected according to Bosq.58 In each subsequent heating scan with a unified heating rate of 1,000 K s−1, a shift of the melting peak maximum Tm,max from 331°C to 320°C was found. The melting peaks become narrower and the crystallite size smaller with increasing cooling rate. The normalized melting enthalpies follow the same trend as the previously determined crystallization enthalpies, but are slightly higher.
For the PAI-PTFEcb sample, two crystallization peaks were found starting at a cooling rate CR ≥ 100 K s−1 regardless of the sample mass. This is the very first evidence that fractional crystallization occurs even under the conditions of very high cooling rates. Because the ratio between peak area and peak height is approximately the same regardless of CR, it can be assumed that the crystallization kinetics do not change. In the heating scan following each cooling rate, a single peak was detected for high cooling rates ≥ 500 K s−1. The peak maximum at ~ 320°C corresponds exactly to the peak maximum for the plain PTFEγ and is independent of CR in this cooling rate range. At cooling rates between 0.1 K s−1 and 100 K s−1, a shoulder was observed for each melt, with a reversal of intensity ratios between the main peak and shoulder at 0.1 K s−1. The trend of values for Tm,max2 is very similar to that previously found for PTFEγ at these CRs. In contrast, Tm,max1 is completely independent of CR. It can be concluded that the quality of the crystallites is mainly determined by the covalent coupling between PAI and PTFE. In contrast, this effect was not detected in studies on the sample with 10-fold sample mass, which was the basis for the X-ray diffraction studies. The position of Tm,max here is almost independent of CR at cooling rates between 0.1 and 3,000 K s−1, which correlates with the behavior of neat PTFE between 500 and 3,000 K s−1.
Extensive investigations were carried out by means of X-ray diffraction to obtain detailed insights into the structural behavior of untreated PTFE and PTFEγ as well as PAI- PTFEcb. Within the chosen measurement strategy, thin samples were successively cooled from the melt with different cooling rates CR in a wide range from 0.1 to 4,000 K s−1, were each subjected to the X-ray experiment, then re-melted and cooled again with the next higher CR, and another X-ray experiment was then started. All samples are semi-crystalline, with crystallinity produced only by PTFE. Original PAI is amorphous. Under normal pressure, PTFE crystallizes in three different crystal structures, all of which can exist in the room temperature range, because the transition temperatures between the solid phases are close to each other: PTFE phase II ← ~ 19°C → PTFE phase IV ← ~ 30°C → PTFE phase I.
Since the measurement samples had to be very thin due to the high cooling rates to be achieved, the measurable scattering intensities were relatively small. In the experiment, therefore, the focus was on a scattering range q between ~ 2.5 and ~ 22.5 nm−1 (2θ: ~ 3.5 … ~ 32.0°). This allowed the following structural parameters to be determined from the scattering curves: the degree of crystallinity Xα of the PTFE phase, the position qmax of the most intense PTFE reflection (for hexagonal PTFE phase IV this is the (100) reflection) and the half-width Δq for this reflection. Δq is used to determine the average crystallite size \(\it \left\langle L \right\rangle_{100}\). All parameters could be determined within acceptable error limits for each cooling rate.
Suitable graphs were used to visualize the peculiarities in the structural behavior as a function of the cooling rate CR or lg(CR). The PTFE crystallinity follows the expectation: With higher cooling rates, the crystallinity decreases, even more so if the PTFE has been modified at the molecular level (by radiation chemistry or compounding). However, this parameter already reveals a special feature that is obviously specific to PTFE. For CR = 100 K s−1 the curve Xα = f(lg(CR)) undergoes a change of direction. The behavior of qmax = f(CR) appears even more impressive. PTFE and PTFEγ show almost opposite freezing behavior. So far, there is no explanation for this unexpected behavior. Considering the change of the (100) reflection of PTFE as a function of CR, one can define a freezing coefficient κfreez for the maximum lateral distance dmax = 2π/qmax of the PTFE chains. For PTFE, κfreez is negative, while for PTFEγ it is positive. CR = 100 K s−1 is also a prominent cooling rate here, where the shape of the curve changes significantly. For its part, the compound resembles untreated PTFE for low CR, while it behaves more like PTFEγ for high CR. For CR = 100 K s−1 the compound has the smallest chain distance dmax. The half-width Δq also shows a noticeable change in the shape of the curve for CR between 100 and ~ 400 K s−1, which is ultimately also reflected in the behavior of the average crystallite size \(\it \left\langle L \right\rangle_{100}\).
Clark76 has discussed two peculiarities of the crystal structure of PTFE: first - the narrow range of existence of PTFE phase IV in the phase diagram at about room temperature and second - the phase transition to PTFE phase I which follows at higher temperatures. PTFE phase IV appears to be compatible with respect to molecular "untwisting" constraints arising from energetically favorable packing in the P31 space group. Such a model requires substantial angular motions within a well-defined lateral arrangement of molecular chains ("lattice"), which consists of right/left defects and clustering of left-handed chain segments. During the IV → I-phase transition, the characteristic local ordering principles are lost, but the hexagonal arrangement of the PTFE chains is retained up to the melting point. Starting at T > Tm, the experiments presented here follow the reverse path with respect to the temperature response, repeating under steadily increasing cooling rate CR. The evaluation of the structural parameters obtained by DSC and WAXS allowed a consideration of the particular structural behavior of PTFE within three CR regions: 0 < CR ≤ 100 K s−1, ~ 80…100 K s−1 < CR < ~ 400…500 K s−1 (with 100 K s−1 as a special feature), and ~ 400…500 K s−1 < CR (with 4,000 K s−1 as the experimental maximum). Considering the research results found by Clark, it can be concluded that at low CR the librations are still able to follow the cooling process and the phase transition PTFE I → PTFE IV could take place. At significantly higher cooling rates, the librations can no longer comply, they freeze. As a result, a supercooled PTFE phase I should be formed. CR ~ 100 K s−1 could therefore be called the "freezing cooling rate" of PTFE.
Essential results of PAI-PTFEcb phase transitions during cooling in the temperature range between 350°C (360°C FSC + WAXS) and 100°C are summarized in Fig. 27.

As the SEM-EDX investigations showed, dispersion of the PAI-PTFEcb extrudate leads to a further improvement in the fragmentation and distribution of the PTFE primary particles in the coating dispersion. How this affects the rheological behavior was examined as a function of shear rate. While the PAI solution exhibits a shear-thinning behavior only above a shear rate \(\dot{\gamma }\) of ~ 100 s−1, the chemical compatibilization and coupling of the PTFEγ to the PAI matrix already leads to a linear shear-thinning behavior above 4 s−1. This one changes to an exponential shear-thinning at high shear rates of > 200 s−1.
For the characterization of the film properties, lacquer films with varying dry film thicknesses were realized. The flexibility of the coatings under quasi-static load varies insignificantly with the two curing regimes. Under impact loading, on the other hand, there are indications suggesting a slight decrease in flexibility at higher film thicknesses. However, it cannot be ruled out that the failure/delamination effects considered are partly due to inadequacies in substrate preparation, e.g., corrosion phenomena not yet visually visible on the substrate surface. Additionally, a comparison with pure PAI films with a solid film thickness of 25 µm has shown that the PTFE content does not lead to a significant reduction in film flexibility.
In terms of optical film appearance, glossy films were achieved regardless of film thickness and curing procedure, with gloss increasing linearly up to a film thickness of 25–30 µm and then plateauing. In parallel, the measured values for the roughness depth RZ also decrease linearly, which explains the increase in gloss values at higher film thicknesses. A matting effect, as usually occurs with PTFE incorporation into a matrix, was not identified.
In this research was shown, that fractional crystallization in chemically bonded PAI-PTFEcb compounds happens under slow (standard DSC) and even various fast CR (FSC). In combination with subsequent X-ray scattering analysis a deeper understanding of the real structure behavior of such extrudates and sliding lacquer films were kept. The property found for unmodified PTFE, contrary to expectations, to show a negative freezing coefficient κfreez with increasing cooling rate CR, could not be elucidated in the present investigations. PTFE as unique material itself revealed some specific peculiarities with regard to both structural and technical properties that are of relevance for the use as solid lubricants in anti-friction coatings as well as bulk phase materials.
References
Bro, M, Lovejoy, E, McKay, G, “Reactions of Irradiated Polytetrduoroethylene Resin.” J. Appl. Polym. Sci., 7 (6) 2121–2133 (1963)
Florin, RE, Parker, MS, Wall, LA, “The Mechanism of the Depolymerization of Polytetrafluoroethylene with Pyrolytic and Radiolytic Initiation.” J. Res. Natl. Bur. Stand. Sect. A Phys. Chem., 70 (2) 115 (1966)
Charlesby, A, “Radiation Effects in Polymers.” In: Jenkins, AD (ed.) Polymer Science, pp. 1544–1559. North-Holland Publishing Company (1972)
Florin, R, “Radiation Chemistry of Fluorocarbon Polymers.” In: Fluoropolymers, pp. 317–331. Wiley (1972)
Lunkwitz, K, “Zum Abbau von Polytetrafluoräthylen durch energiereiche Strahlung in inerter Atmosphäre.” Plaste und Kautschuk, 26 (6) 318–321 (1979)
Ferse, A, Lunkwitz, K, Grimm, H, Dietrich, P, Engler, G, Gross, U, Prescher, D, Schulze, J, “Zum Abbau von Polytetrafluoräthylen durch energiereiche Strahlung in Gegenwart von Sauerstoff.” Acta Polym., 30 (6) 348–352 (1979)
Babkin, IY, Solov’ev, S, “Present State and Prospects of Chemical Synthesis Under Irradiation.” Radiat. Phys. Chem., 18 (1–2) 371–381 (1981)
Gross, U, Dietrich, P, Engler, G, Prescher, D, Schulze, J, Lunkwitz, K, Ferse, A, “Zum Mechanismus des strahlenchemischen Abbaus von Polytetrafluorethylen.” J. Fluor. Chem., 20 (1) 33–52 (1982)
Fisher, WK, Corelli, J, “Effect of Ionizing Radiation on the Chemical Composition, Crystalline Content and Structure, and Flow Properties of Polytetrafluoroethylene.” J. Polym. Sci. Polym. Chem. Edn., 19 (10) 2465–2493 (1981)
Takenaga, M, Hamada, R, Yamagata, K, “Short-Time Annealing Behavior of Polytetrafluoroethylene Subjected to γ-Irradiation.” J. Appl. Polym. Sci., 29 (6) 2245–2246 (1984)
Lunkwitz, K, Brink, H-J, Handte, D, Ferse, A, “The Radiation Degradation of Polytetrafluoroethylene Resulting in Low-Molecular and Functionalized Perfluorinated Compounds.” Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem., 33 (6) 523–532 (1989)
Bürger, W, Lunkwitz, K, Pompe, G, Petr, A, Jehnichen, D, “Radiation Degradation of Fluoropolymers: Carboxylated Fluoropolymers from Radiation Degradation in Presence of Air.” J. Appl. Polym. Sci., 48 (11) 1973–1985 (1993)
Lunkwitz, K, Bürger, W, Lappan, U, Brink, H-J, Ferse, A, “Surface Modification of Fluoropolymers.” J. Adhes. Sci. Technol., 9 (3) 297–310 (1995)
Lunkwitz, K, Bürger, W, Geißler, U, Petr, A, Jehnichen, D, “Studies on γ-radiation Treatment of Poly(Tetrafluoroethylene) Powder.” J. Appl. Polym. Sci., 60 (11) 2017–2024 (1996)
Schierholz, K, Lappan, U, Lunkwitz, K, “Electron Beam Irradiation of Polytetrafluoroethylene in Air: Investigations on the Thermal Behaviour.” Nuclear Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms, 151 (1–4) 232–237 (1999)
Lunkwitz, K, Lappan, U, Scheler, U, “Modification of Perfluorinated Polymers by High-Energy Irradiation.” J. Fluor. Chem., 125 (6) 863–873 (2004)
Hoffmann, T, Heller, M, Jehnichen, D, Engelhardt, T, Lehmann, D, “Influence of Absorbing Materials on the Functionalization of Poly(Tetrafluoroethylene) During γ-Irradiation.” J. Appl. Polym. Sci., 130 (3) 1787–1793 (2013)
Frick, A, Sich, D, Heinrich, G, Lehmann, D, Gohs, U, Stern, C, “Properties of Melt Processable PTFE/PEEK Blends: The Effect of Reactive Compatibilization Using Electron Beam Irradiated Melt Processable PTFE.” J. Appl. Polym. Sci., 128 (3) 1815–1827 (2013)
Hoffmann, T, Lehmann, D, “Chemical Modification of Poly(Tetrafluoroethylene) Micropowder—Basis for Special Lubricant Additives.” Lubr. Sci., 25 (4) 313–327 (2013)
Khan, M, Lehmann, D, Heinrich, G, “Modification of PTFE Nanopowder by Controlled Electron Beam Irradiation: A Useful Approach for the Development of PTFE Coupled EPDM Compounds.” Express Polym. Lett., 2 (4) 284–293 (2008)
Lehmann, D, “Melt Modification of Polyamides.” In: Polymers-Opportunities and Risks II, pp. 163–192. Springer (2009)
Lehmann, D, Hupfer, B, Lappan, U, Pompe, G, Häußler, L, Jehnichen, D, Janke, A, Geißler, U, Reinhardt, R, Lunkwitz, K, Franke, R, Kunze, K, “New PTFE-Polyamide Compounds.” Des. Monomers Polym., 5 (2–3) 317–324 (2002)
Nguyen, T-D, Schmidt, T, Malanin, M, Schneider, A, Scheibner, H, Gedan-Smolka, M, Voit, B, “Chemisch gekoppelte Öl-PTFE-PA66-Verbundwerkstoffe als neuartige tribologische Materialien.” Proc. 61. Tribologie-Fachtagung, Göttingen, Germany, 28.-30.09.2020, 2020
Nguyen, T-D, Simo Kamga, L, Gedan-Smolka, M, Sauer, B, Emrich, S, Kopnarski, M, Voit, B, “Chemical Bonded PA66-PTFE-Oil Composites as Novel Tribologically Effective Materials: Part 2.” In: Proc. IOP Conference Series: Materials Science and Engineering, 2021
Simo Kamga, L, Nguyen, T-D, Gedan-Smolka, M, Oehler, M and Sauer, B, “Einfluss von strahlenmodifiziertem PTFE auf die tribologischen Eigenschaften von Polyamid 46.” In: Proc. 61. Tribologie-Fachtagung, Göttingen, Germany, 28.-30.09.2020, 2020
Franke, R, Lehmann, D, Kunze, K, “Tribological Behaviour of New Chemically Bonded PTFE Polyamide Compounds.” Wear, 262 (3–4) 242–252 (2007)
Häußler, L, Pompe, G, Lehmann, D, Lappan, U, “Fractionated Crystallization in Blends of Functionalized Poly(Tetrafluoroethylene) and Polyamide. In: Proc. Macromolecular Symposia, 2001
Pompe, G, Häußler, L, Pötschke, P, Voigt, D, Janke, A, Geißler, U, Hupfer, B, Reinhardt, G, Lehmann, D, “Reactive Polytetrafluoroethylene/Polyamide Compounds. I. Characterization of the Compound Morphology with Respect to the Functionality of the Polytetrafluoroethylene Component by Microscopic and Differential Scanning Calorimetry Studies.” J. Appl. Polym. Sci., 98 (3) 1308–1316 (2005)
Pompe, G, Häußler, L, Adam, G, Eichhorn, KJ, Janke, A, Hupfer, B, Lehmann, D, “Reactive Polytetrafluoroethylene/Polyamide 6 Compounds. II. Study of the Reactivity with Respect to the Functionality of the Polytetrafluoroethylene Component and Analysis of the Notched Impact Strength of the Polytetrafluoroethylene/Polyamide 6 Compounds.” J. Appl. Polym. Sci., 98 (3) 1317–1324 (2005)
Khan, MS, Lehmann, D, Heinrich, G, “Newly Developed Chloroprene Rubber Composites Based on Electron-Modified Polytetrafluoroethylene Powder.” Acta Mater., 57 (16) 4882–4890 (2009)
Khan, M, Lehmann, D, Heinrich, G, Gohs, U, Franke, R, “Structure-Property Effects on Mechanical, Friction and Wear Properties of Electron Modified PTFE Filled EPDM Composite.” Express Polym. Lett., 266 39–48 (2009)
Khan, MS, Franke, R, Gohs, U, Lehmann, D, Heinrich, G, “Friction and Wear Behaviour of Electron Beam Modified PTFE Filled EPDM Compounds.” Wear, 266 (1–2) 175–183 (2009)
Taeger, A, Hoffmann, T, Butwilowski, W, Heller, M, Engelhardt, T, Lehmann, D, “Evidence of Chemical Compatibilization Reaction Between Poly(Ether Ether Ketone) and Irradiation-Modified Poly(Tetrafluoroethylene).” High Perform. Polym., 26 (2) 188–196 (2014)
Edelmann, M, Lehmann, D, “New Chemically Coupled Poly(Amide-Imide) + PTFE Compounds and Their Use in High Precision Bearings - Part I.” In: Proc. 52. Tribologie-Fachtagung, Göttingen, Germany, 26.-28.09.2011, 2011
Niedermeier, H, “New Chemically Coupled Poly (Amide-Imide) + PTFE Compounds and Their Use in High Precision Bearings - Part II.” In: Proc. 52. Tribologie-Fachtagung, Göttingen, Germany, 26.-28.09.2011, 2011
Edelmann, M, Lehmann, D, Niedermeier, H, “New Chemically Coupled Poly(Amide-Imide)+ PTFE Compounds and Their Use in High Precision Bearings.” In: Proc. High Performance Plastics 2011, Cologne, Germany, 23–24 February, 2011
Gedan-Smolka, M, Lehmann, D, Marschner, A, Kunze, K, Franke, R, Haase, I, “Chemisch kompatibilisierte PAI-PTFE-cg-Gleitlacke.” In: Proc. 52. Tribologie-Fachtagung, Göttingen, Germany, 26.-28.09.2011, 2011
Gedan-Smolka, M, Marschner, A, Lehmann, D, Franke, R, Klemm, M, Leidich, E, Prase, B, “Formulation and Characterization of PAI-PTFE-cg Antifriction Coatings.” Key Eng. Mater., 721 362 (2017)
Gedan-Smolka, M, Marschner, A, Kunze, K, Franke, R, Lehmann, D, “Influence of Chemical Bonding of PTFE Lubricant to a Polyamideimide (PAI) Matrix in Antifriction Coatings. In: Proc. 6th World Tribology Congress, Beijing, China, 17.-22. 09.2017, 2017
Gedan-Smolka, M, Marschner, A, Pondicherry, KS, Großmann, H, Lehmann, D, “Physical Mixing Versus Chemical Bonding—Polyamideimide (PAI) Sliding Lacquers with PTFE as a Solid Lubricant.” In: Proc. the 18th Nordic Symposium on Tribology – NORDTRIB 2018, Uppsala, Sweden, 18.-21.06.2018, 2018
Nguyen, T-D, Gedan-Smolka, M, Simo Kamga, L, Sauer, B, Emrich, S, Kopnarski, M, Voit, B, Chemical Bonded Oil-PTFE-PA66 Composites as Novel Tribologically Effective Materials: Part 1. Solid State Phenomena. Trans Tech Publ. Ltd, Switzerland, 320 113-118 (2021)
Bragg, WH, Bragg, WL, X rays and Crystal Structure. G. Bell & Sons, London (1915)
Alexander, LE, X-ray Diffraction Methods in Polymer Science. Wiley, New York (1969)
Shen, JT, Pei, YT, De Hosson, JTM, “Structural Changes in Polytetrafluoroethylene Molecular Chains upon Sliding Against Steel.” J. Mater. Sci., 49 (4) 1484–1493 (2013)
Guinier, A, X-ray Diffraction in Crystals, Imperfect Crystals, and Amorphous Bodies. W.H Freyman and Company, San Francisco (1963)
Li, G, Ma, Y, Wan, H, Chen, L, An, Y, Ye, Y, Zhou, H, Chen, J, “Flake Aluminum Reinforced Polyamideimide-Polytetrafluoroethylene Bonded Solid Lubricating Composite Coating for Wear Resistance and Corrosion Protection.” Eur. Polym. J., 152 110485 (2021)
Rempp, A, Widmann, M, Guth, P, Killinger, A, Gadow, R, “Ceramic/Metal-Polymer Multilayered Coatings for Tribological Applications Under Dry Sliding Conditions.” Adv. Ceram. Coat. Mater. Extreme Environ. Ceram. Eng. Sci. Proc., 32 37–50 (2011)
Sheiretov, T, Van Glabbeek, W, Cusano, C, “Evaluation of the Tribological Properties of Polyimide and Poly(Amide-Imide) Polymers in a Refrigerant Environment.” Tribol. Trans., 38 (4) 914–922 (1995)
Tanaka, K, Yamada, Y, Ueda, S, “Effect of Temperature on the Friction and Wear of Heat-Resistant Polymer-Based Composites.” J. Synth. Lubr., 8 (4) 281–294 (1992)
Socrates, G, Infrared and Raman Characteristic Group Frequencies: Tables and Charts. Wiley, New York (2004)
Eichhorn, KJ, Lehmann, D, Voigt, D, “Characterization of Low Molecular Weight Carboxyl-Terminated Polyamides Obtained by Reactive Extrusion of Polyamide 6 with Trimellitic Anhydride.” J. Appl. Polym. Sci., 62 (12) 2053–2060 (1996)
Gangal, SV, “Polytetrafluoroethylene, Homopolymers of Tetrafluoroethylene.” In: Encyclopedia of Polymer Science and Engineering, pp. 577–600. Wiley, New York (1989)
Domininghaus, H, “Fluorkunststoffe.” In: Die Kunststoffe und ihre Eigenschaften, Springer, Berlin (2008)
Suwa, T, Takehisa, M, Machi, S, “Melting and Crystallization Behavior of Poly(Tetrafluoroethylene). New Method for Molecular Weight Measurement of Poly(Tetrafluoroethylene) Using a Differential Scanning Calorimeter.” J. Appl. Polym. Sci., 17 (11) 3253–3257 (1973)
Lappan, U, Geißler, U, Häußler, L, Pompe, G, Scheler, U, “The Estimation of the Molecular Weight of Polytetrafluoroethylene Based on the Heat of Crystallization. A Comment on Suwa’s Equation.” Macromol. Mater. Eng., 289 (5) 420–425 (2004)
Androsch, R, “Reversibility of the Low-Temperature Transitions of Polytetrafluoroethylene as Revealed by Temperature-Modulated Differential Scanning Calorimetry.” J. Polym. Sci. Part B Polym. Phys., 39 (7) 750–756 (2001)
Khatipov, SA, Serov, SA, Buznik, VM, “Radiation Modification of Polytetrafluoroethylene.” In: Ameduri, B, Fomin, S (eds.) Opportunities for Fluoropolymers - Synthesis, Characterization, Processing, Simulation and Recycling, pp. 137–188. Elsevier (2020)
Bosq, N, Guigo, N, Zhuravlev, E, Sbirrazzuoli, N, “Nonisothermal Crystallization of Polytetrafluoroethylene in a Wide Range of Cooling Rates.” J. Phys. Chem. B, 117 (12) 3407–3415 (2013)
Hirakawa, S, Takemura, T, “Transitions and Phases of Polytetrafluoroethylene.” Jpn. J. Appl. Phys., 8 (6) 635–641 (1969)
Resnyansky, AD, Bourne, NK, Millett, JCF, Brown, EN, “Constitutive Modeling of Shock Response of Polytetrafluoroethylene.” J. Appl. Phys, 110 (3) 033530 (2011)
Honda, Y, Nishijima, S, “Characterization of the Melting Process of PTFE Using Positron Annihilation Spectroscopy.” J. Phys. Conf. Ser., 618 012024 (2015)
Clark, ES, “The Molecular Conformations of Polytetrafluoroethylene: Forms II and IV.” Polymer, 40 (16) 4659–4665 (1999)
Rae, PJ, Dattelbaum, DM, “The Properties of Poly(Tetrafluoroethylene) (PTFE) in Compression.” Polymer, 45 (22) 7615–7625 (2004)
Conte, M, Igartua, A, “Study of PTFE Composites Tribological Behavior.” Wear, 296 (1–2) 568–574 (2012)
“Powder diffraction file PDF-2.” International Centre for Diffraction Data (ICDD®), Newtown Square, PA 19073, USA (2018)
Subrahmanyam, H, Subramanyam, S, “Thermal Expansion of Irradiated Polytetrafluoroethylene.” J. Polym. Sci. Part B Polym. Phys., 25 (7) 1549–1556 (1987)
Briskman, BA, Tlebaev, KB, “Radiation Effects on Thermal Properties of Polymers II. Polytetrafluoroethylene.” High Perform. Polym., 20 (1) 86–114 (2008)
Vaselabadi, SA, Shakarisaz, D, Ruchhoeft, P, Strzalka, J, Stein, GE, “Radiation Damage in Polymer Films from Grazing-Incidence X-ray Scattering Measurements.” J. Polym. Sci. Part B Polym. Phys., 54 (11) 1074–1086 (2016)
Saito, H, Furuta, M, Matsugaki, A, Nakano, T, Oishi, M, Okazaki, M, “Radiation-Resistant Properties of Cross-Linking PTFE for Medical Use.” Polym. Bull., 76 (12) 6111–6122 (2019)
Barylski, A, Aniołek, K, Swinarew, AS, Kaptacz, S, Gabor, J, Waśkiewicz, Z, Stanula, A, “Novel Organic Material Induced by Electron Beam Irradiation for Medical Application.” Polymers, 12 (2) 306 (2020)
Tutiya, M, “Effect of γ-Irradiation on the Crystalline Transition Temperature in Poly(tetrafluoroethylene).” Polym. J., 6 (1) 39–44 (1974)
Allayarov, S, Olkhov, YA, Dixon, D, Nikles, D, “Features of the Phase Behavior of Gamma-Irradiated Polytetrafluoroethylene Powder.” High Energy Chem., 48 (2) 104–111 (2014)
Calleja, G, Jourdan, A, Ameduri, B, Habas, J-P, “Where is the Glass Transition Temperature of Poly(Tetrafluoroethylene)? A New Approach by Dynamic Rheometry and Mechanical Tests.” Eur. Polym. J., 49 (8) 2214–2222 (2013)
Henri, V, Dantras, E, Lacabanne, C, Dieudonne, AG, Koliatene, F, “Thermal Ageing of PTFE in the Melted State: Influence of Interdiffusion on the Physicochemical Structure.” Polym. Degrad. Stab., 171 1–8 (2020)
Gall, MJ, “Effect of Molecular Weight on the Morphology of Melt-Crystallized PTFE and the Occurrence of Row-Nucleated Crystallization.” Polymer, 15 (5) 272–276 (1974)
Clark, ES, “The Crystal Structure of Polytetrafluoroethylene, Forms I and IV.” J. Macromol. Sci. Part B Phys., 45 (2) 201–213 (2006)
Mezger, TG, Das Rheologie Handbuch. Vincentz, Hannover (2012)
Kalinna, G, “Glänzende Qualität? Glanzmessung.” JOT J. Oberflächentech., 52 (6) 64–67 (2012)
Acknowledgments
The authors are grateful to Anne Garbe for dispersion preparation, application and film quality testing as well as rheological measurements and to Kornelia Schlenstedt for the SEM images. In addition, special thanks go to Prof. R. Androsch (Center for Engineering Sciences, Martin Luther University Halle-Wittenberg, Halle, Germany) for helpful discussions and recommendations regarding the FSC analyses.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
M. G.-S. and D. J. contributed to conceptualization; M. G.-S., D. J., M.M., A.S., M.G., A.G., A.M. and T. S. contributed to investigation; M. G.-S. and D. J. contributed to methodology; M. G.-S. contributed to project administration; M. G.-S. and D. J. contributed to supervision; M. G.-S., D. J. and M. M. contributed to validation; M. G.-S., D. J., A. S., A. G. and A.M. contributed to visualization; M. G.-S., D. J., M. M. and A. M. contributed to writing—original draft; M. G.-S. and D. J. contributed to writing—review & editing.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gedan-Smolka, M., Jehnichen, D., Malanin, M. et al. Relationships between crystallization behavior and application properties of chemically coupled PAI-PTFE coatings. J Coat Technol Res 21, 623–654 (2024). https://doi.org/10.1007/s11998-023-00845-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11998-023-00845-9