
Abstract
Purpose of Review
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease causing weakness, respiratory failure, and death within 3 to 5 years. Approximately, 10% of ALS cases have a genetic etiology (familial/fALS). The etiology of the remaining 90% of sporadic ALS (sALS) cases remains unknown. In this review, we provide an overview of approved and investigational therapies for fALS, as well as genetically informed therapeutic advances aimed at the larger sALS population.
Recent Findings
Antisense oligonucleotides (ASOs) are a promising strategy to treat toxic gain-of-function mutations underlying most forms of fALS. We discuss the recent approval of tofersen for ALS caused by mutation in SOD1. We also discuss progress in the development of therapies for fALS associated with C9orf72 hexanucleotide repeat expansions (C9orf72) and fused in sarcoma (FUS) mutations. Finally, we will discuss the rationale and status of molecular therapies for sALS targeting mediators of TDP-43 pathogenesis: ataxin-2 (ATXN2) and stathmin-2 (STMN2).
Summary
Advances in understanding the genetics of ALS have propelled the development of promising gene therapies. Lessons learned from tofersen continue to inform clinical trial design for a growing pipeline of therapies directed towards other fALS subtypes and sALS.
Similar content being viewed by others

References and Recommended Readings
Papers of particular interest, published recently, have been highlighted as: • Of importance
• Akçimen F, Lopez ER, Landers JE, Nath A, Chiò A, Chia R, et al. Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat Rev Genet. 2023;24(9):642–58 This review highlights recent discoveries in ALS, including new mutations, gene variability and gene-environment interactions, and provides a summary of the current status of ALS therapies developed over the past 30 years.
• Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021;29(12):3345–58 This review examines the current landscape of ALS directed gene therapy focusing on various methodologies such as ASO, RNA interference, CRISPR, AAV-mediated trophic support, and antibody-based techniques, and their applications in both genetic and sporadic cases.
Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330(9):585–91.
• Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, et al. Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919–30 This article provides evidence that sodium phenylbutyrate–taurursodiol results in slower functional decline than placebo over a period of 24 weeks as measured by the ALSFRS-R score.
Rothstein JD. Edaravone: a new drug approved for ALS. Cell. 2017;171(4):725.
• Boros BD, Schoch KM, Kreple CJ, Miller TM. Antisense oligonucleotides for the study and treatment of ALS. Neurotherapeutics. 2022;19(4):1145–58 This review provides a thorough overview of ASO therapies in ALS detailing their progression from preclinical development to early clinical trials.
• Ruf WP, Boros M, Freischmidt A, Brenner D, Grozdanov V, De Meirelles J, et al. Spectrum and frequency of genetic variants in sporadic amyotrophic lateral sclerosis. Brain Commun. 2023;5(3):fcad152 This article provides results of a cohort study done to characterize the mutational landscape of sporadic ALS to potentially broaden the eligibility for gene-specific therapies in future.
• van Daele SH, Moisse M, Van Vugt JJFA, Zwamborn RAJ, Van Der Spek R, Van Rheenen W, et al. Genetic variability in sporadic amyotrophic lateral sclerosis. Brain. 2023;146(9):3760–9 This article provides a comprehensive catalog of pathogenic variations in a large cohort of sporadic ALS patients of European ancestry.
Amado DA, Davidson BL. Gene therapy for ALS: a review. Mol Ther. 2021;29(12):3345–58.
Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14(9):544–58.
Conforti FL, Spataro R, Sproviero W, Mazzei R, Cavalcanti F, Condino F, et al. Ataxin-1 and ataxin-2 intermediate-length polyQ expansions in amyotrophic lateral sclerosis. Neurology. 2012;79(24):2315–20.
• Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, et al. Amyotrophic lateral sclerosis. The Lancet. 2022;400(10360):1363–80 This article provides a thorough overview of the evolving landscape of ALS and its current challenges, highlighting the disease’s phenotypic diversity, genetic factors, and recent developments in diagnostic criteria and therapeutic approaches.
• Hayes LR, Kalab P. Emerging therapies and novel targets for TDP-43 proteinopathy in ALS/FTD. Neurotherapeutics. 2022;19(4):1061–84 This review focuses on TDP-43-directed therapies being tested in current or upcoming clinical trials in ALS and highlights alternative pathways modulating TDP-43 toxicity for future therapeutic development.
• Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022;28(1):104–16 This study provides evidence for FUS silencing as a therapeutic strategy for ALS. ION363, a non-allele-specific FUS ASO, silences FUS, resulting in reduction of FUS levels and aggregates in both disease relevant mouse model and a patient with FUS.
• Abati E, Bresolin N, Comi G, Corti S. Silence superoxide dismutase 1 (SOD1): a promising therapeutic target for amyotrophic lateral sclerosis (ALS). Expert Opin Ther Targets. 2020;24(4):295–310 This review explores gene silencing strategies in treatment of SOD1-ALS including RNAi, ASO, and CRISPR/Cas9 related strategies.
Seddighi S, Qi YA, Brown AL, Wilkins OG, Bereda C, Belair C, et al. Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. Neuroscience. 2023. Available from: http://biorxiv.org/lookup/doi/10.1101/2023.01.23.525149
Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–71.
• Baughn MW, Melamed Z, López-Erauskin J, Beccari MS, Ling K, Zuberi A, et al. Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science. 2023;379(6637):1140–9 This study demonstrated that targeting the STMN2 cryptic exon using ASOs or dCas0Rx restored axon regeneration and lysosomal trafficking deficts in TDP-43 deficient human motor neurons.
• Giovannelli I, Higginbottom A, Kirby J, Azzouz M, Shaw PJ. Prospects for gene replacement therapies in amyotrophic lateral sclerosis. Nat Rev Neurol. 2023;19(1):39–52 This review focuses on gene replacement therapies and identifies loss of function mutations in ALS as potential targets for future therapeutic development.
Cerillo JL, Parmar M. Tofersen. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 [cited 2023 Nov 26]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK594270/
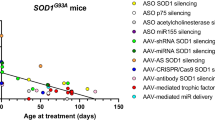
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–42.
• Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109–19 This article details the Phase I/II clinical trial demonstrating safety of tofersen in SOD1-ALS as well as key reductions in target engagement and neurodegenerative biomarkers.
McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest. 2018;128(8):3558–67.
• Ly CV, Ireland MD, Self WK, Bollinger J, Jockel-Balsarotti J, Herzog H, et al. Protein kinetics of superoxide dismutase-1 in familial and sporadic amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2023;10(6):1012–24 This study demonstrated the feasibility of using stable isotope labelling to measure SOD1 protein kinetics in vivo and demonstrated that SOD1 A5V mutant protein is less abundant and turns over faster in mutation carriers.
Self WK, Schoch KM, Alex J, Barthélemy N, Bollinger JG, Sato C, et al. Protein production is an early biomarker for RNA -targeted therapies. Ann Clin Transl Neurol. 2018;5(12):1492–504.
• Benatar M, Wuu J, Andersen PM, Bucelli RC, Andrews JA, Otto M, et al. Design of a randomized, placebo-controlled, phase 3 trial of tofersen initiated in clinically presymptomatic SOD1 variant carriers: the ATLAS study. Neurotherapeutics. 2022;19(4):1248–58 This article discusses the design considerations for the Phase III ATLAS trial of tofersen in presymptomatic SOD1-ALS.
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396–403.
Forsberg K, Andersen PM, Marklund SL, Brännström T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol (Berl). 2011;121(5):623–34.
Brotherton TE, Li Y, Cooper D, Gearing M, Julien JP, Rothstein JD, et al. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc Natl Acad Sci. 2012;109(14):5505–10.
Liu H, Sanelli T, Horne P, Pioro EP, Strong MJ, Rogaeva E, et al. Lack of evidence of monomer/misfolded superoxide dismutase-1 in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2009;66(1):75–80.
Da Cruz S, Bui A, Saberi S, Lee SK, Stauffer J, McAlonis-Downes M, et al. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol (Berl). 2017;134(1):97–111.
• Trist BG, Genoud S, Roudeau S, Rookyard A, Abdeen A, Cottam V, et al. Altered SOD1 maturation and post-translational modification in amyotrophic lateral sclerosis spinal cord. Brain. 2022;145(9):3108–30 This study use biochemical, histologic, and proteomic methods to show that structurally disordered, immature SOD1 protein abnormally accumulates in spinal cord motor neurons in SOD1-ALS and non-SOD1-linked ALS.
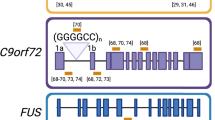
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, et al. Stress granule assembly disrupts nucleocytoplasmic transport. Cell. 2018;173(4):958-971.e17.
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci. 2013;110(47). Available from: https://pnas.org/doi/full/10.1073/pnas.1318835110
Bertrand A, Wen J, Rinaldi D, Houot M, Sayah S, Camuzat A, et al. Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol. 2018;75(2):236.
Lee SE, Sias AC, Mandelli ML, Brown JA, Brown AB, Khazenzon AM, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. NeuroImage Clin. 2017;14:286–97.
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313–25.
• Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23(5):615–24 This study showed that reduced expression of endogenous C9orf72 exacerbated autophagic, neuronal loss, and cognitive deficits in a mouse model expressing the C9 HRE suggesting that C9orf72 haploinsufficiency contributes to pathogenesis.
• Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright CL, Pinto C, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2022;28(1):117–24 This study provided pre-clinical evidence that afinersen, a mixed backbone ASO to the C9orf72 repeat expansion, reduced RNA foci and poly(GP) in C9orf72 model mice and a single participant with C9-ALS.
• Sattler R, Traynor BJ, Robertson J, Van Den Bosch L, Barmada SJ, Svendsen CN, et al. Roadmap for C9ORF72 in frontotemporal dementia and amyotrophic lateral sclerosis: report on the C9ORF72 FTD/ALS summit. Neurol Ther. 2023;12(6):1821–43 This article summarizes recommendations from the 2023 C9orf72 FTD/ALS Summit to harmonize biomarkers and clinical trial design for C9orf72-related ALS-FTD.
• Hung ST, Linares GR, Chang WH, Eoh Y, Krishnan G, Mendonca S, et al. PIKFYVE inhibition mitigates disease in models of diverse forms of ALS. Cell. 2023;186(4):786–802.e28 This study showed the pharmacologic and ASO-mediated PIKfyve inhibition in human-derived motor neurons representing diverse forms of ALS and animal models led to expulsion of aggregate-prone protein through a novel exosome-based protein clearance mechanism.
Scotter EL, Chen HJ, Shaw CE. TDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics. 2015;12(2):352–63.
Shang Y, Huang EJ. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016;1647:65–78.
Sharma A, Cao EY, Kumar V, Zhang X, Leong HS, Wong AML, et al. Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy. Nat Commun. 2018;9(1):4931.
• Tanemoto M, Hisahara S, Ikeda K, Yokokawa K, Manabe T, Tsuda R, et al. Sporadic amyotrophic lateral sclerosis due to a FUS P525L mutation with asymmetric muscle weakness and anti-ganglioside antibodies. Intern Med. 2021;60(12):1949–53 This case report details the early onset and rapid progression characteristic of FUS-ALS due to P525L mutations.
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–75.
Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM. Amyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol. 2014;71(12):1529.
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22(2):167–79.
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–68.
Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349(6248):650–5.
Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180–90.
• López-Erauskin J, Bravo-Hernandez M, Presa M, Baughn MW, Melamed Z, Beccari MS, et al. Stathmin-2 loss leads to neurofilament-dependent axonal collapse driving motor and sensory denervation. Nat Neurosci. 2023. https://doi.org/10.1038/s41593-023-01496-0. This study showed that STMN2 depletion in aging mice causes pronounced collapse of axonal caliber, reduced interneurofilament spacing, and muscle denervation providing insight into the consequences of TDP-43 nuclear depletion and STMN2 cryptic exon inclusion.
Willemse SW, Harley P, Van Eijk RPA, Demaegd KC, Zelina P, Pasterkamp RJ, et al. UNC13A in amyotrophic lateral sclerosis: from genetic association to therapeutic target. J Neurol Neurosurg Psychiatry. 2023;94(8):649–56.
Funding
This work is supported by Target ALS and NIH NINDS K08 NS107621 to C.V.L.
Author information
Authors and Affiliations
Contributions
C.M, C.V.L, N.R, and W.F. wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Meadows, C., Rau, N.A., Faridi, W. et al. Translating the ALS Genetic Revolution into Therapies: A Review. Curr Treat Options Neurol 26, 35–49 (2024). https://doi.org/10.1007/s11940-024-00781-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11940-024-00781-y