
Abstract
Purpose of the review
Cataplexy, an involuntary loss of muscle activity triggered by strong emotions is the most impressive symptom in narcolepsy. This review gives an overview of the current understanding of cataplexy and its available treatment options.
Recent findings
With the discovery of hypocretin/orexin, the understanding of the pathophysiology of cataplexy advanced in the past decades. In the recent years, with the development of new anticataplectic agents (e.g., Pitolisant) symptomatic treatment of cataplexy has further improved. Abrupt cessation of anticataplectic medication especially antidepressants increase the risk of status cataplecticus, a virtually continuous series of long-lasting cataplectic attacks.
Summary
Cataplexies still remain an under-recognized phenomenon due to missing diagnostic measures. Treatment for cataplexy still remains symptomatic but new agents with better tolerability and usability are continuously developed. New therapeutic actions either targeting the autoimmune mechanisms underlying orexin cell death or substituting orexin action are promising treatments for the near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the beginning of the twentieth century, episodes of muscle weakness caused by emotions were first described and named cataplexy [1], which is a term from the Greek and consists of the terms kata = down and plessein = beat. A “weak in the knees” in situations of high emotional intensity may be experienced in healthy people, probably representing a physiological minor expression of cataplexy. Emotionally triggered muscle weakness has even made it into a famous Beatles song (“Honey Pie,” White Album, 1968) reflecting its popularity in the normal population. An interesting neurophysiological study confirmed a reduction of the size of the H reflex in normal people when laughing [2].
The finding of an association between excessive daytime sleepiness and the occurrence of emotion-triggered cataplexies was an important step in the understanding of narcolepsy [3]. The “narcoleptic tetrade” as described by the authors refers to the four-element symptom constellation: daytime sleepiness, cataplexy, sleep paralysis, and hypnagogic hallucinations. In this first description, they also introduce methylphenidate as a possible treatment for daytime sleepiness. Around the same time with the discovery of tricyclic antidepressants, the combined therapy for narcolepsy with cataplexy consisting of stimulants against sleepiness and the antidepressant imipramine against cataplexy was established [4].
Till today, the treatment of cataplexy in the context of narcolepsy is a symptomatic therapy. The treatment response varies between patients and reflects its complex and difficult interaction between the different brain-networks involved in the narcolepsy-cataplexy complex. A compromised hypocretin/orexin signaling obviously plays one of the most important roles in the pathophysiology of cataplexy but compounds targeting specifically to hypocretin/orexin receptors are lacking so far. Available treatments enhance catecholamine availability or modulate locus coeruleus norepinephrine neuron activity to reduce excessive daytime sleepiness and cataplexy [5••]. Interestingly in a study with the purpose to characterize cataplexy clinical features with a questionnaire in 109 patients, only 56% of patients with proven cataplexy used medication to control these. When treated, 97.9% reported a reduced frequency of cataplexy (86.4%). In 12.5%, a reduction of severity was reported (cataplexy changing from complete/generalized to partial). Modification of behavioral habits reduced the frequency of cataplexy in 27.1% (e.g., regular sleep schedule, avoidance of sleep deprivation) [6••].
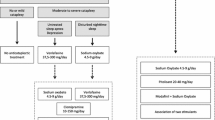
Today, pharmacotherapeutic treatment of cataplexy includes antidepressants, sodium oxybate (gamma hydroxybutyrate), and pitolisant (Table 1).
Definition and epidemiology
Cataplexy is the pathognomonic symptom of narcolepsy type 1 (narcolepsy with cataplexy, NT1)—a sleep disorder that affects 0.06% of the adult population [7, 8]. Cataplexy is defined as a not suppressible loss of muscle tone elicited by an emotional trigger. During this episode of muscle tone loss, consciousness is mostly preserved. Episodes are typically triggered by positive emotions like laughing or joking [8] (Fig. 1: possible trigger factors for cataplexy).

Cataplexy triggers in patients with hypocretin-1 deficiency (n = 109). The triggers are sorted in descending order, based on the frequency of answers scored as “often” or “always.” Reprinted from Sleep Medicine, 12, Overeem et al. The clinical features of cataplexy: a questionnaire study in narcolepsy patients with and without hypocretin-1 deficiency, 12–18, 2011, with permission from Elsevier.
The evolution of cataplexy usually lasts a few seconds, rarely minutes, with a cranio-caudal spread. The loss of muscle tone mostly starts with the face and neck and then spreads to the trunk and limbs bilaterally.
Muscles associated with breathing are spared, although some patients report limited capacity to breath during a cataplectic episode. Hyperkinesias, in the form of phasic (twitching of facial muscles), tonic (tongue protrusion, grimacing, or neck extension), and repetitive motor activities, can be superimposed on muscle atonia [9]. These positive motor phenomena might be more common in children and at disease onset [10]. Injuries associated with cataplexy are not common but may occur when the attack is involving the whole body (complete cataplexy), although falls are reported by one-third of patients [6••]. Partial cataplexies usually include only a few muscles, mostly facial or neck muscles. Often only the affected person notices these partial cataplexies. In children with cataplexy, episodes can be long-lasting with a low muscle tone accompanied with wobbly gait and perioral movements like grimacing and tongue protrusion [6••].
The frequency ranges from several attacks per day to few episodes per year [11•]. The number of cataplexies seems to decrease with disease duration, whether this is due to compensatory strategies or the natural course of the disease is still an unsolved issue. Status cataplecticus is a rare condition in narcolepsy with cataplexies. It can occur after sudden cessation of antidepressive drugs or by strong emotions. Cataplexies continue for hours or days without a refractory period.
During a cataplexy, deep tendon reflexes are extinct [12], whereas the Babinski sign can transiently appear [11•]. Cataplectic episodes can furthermore be associated with excessive daytime sleepiness (EDS), hallucinations, sleep paralysis, dreaming, and REM sleep behavior disorder, particularly when the duration of the attack is long [13].
The occurrence of cataplexies typically follows other narcolepsy symptoms such as EDS within a time delay ranging between one and 40 years [14]. In rare cases, cataplexies precede the onset of other symptoms of NT 1. In the European Narcolepsy Network database, only 24/542 (4.43%) cases with NT 1 reported cataplexy as the first symptom. A late onset of cataplexy probably underlies the diagnostic delay of narcolepsy (10.4–0.8 years in men and 9.8–0.8 years in women). The diagnosis of NT1 presumes the knowledge of cataplexy and other facultative symptoms of narcolepsy like hypnagogic/hypnopompic hallucination and sleep paralysis [15].
Cataplexy as a dynamic process
Huang et al. described cataplexy as a dynamic process divided into four phases: triggering, resisting, atonic, and recovering. Phase 1 describes the loss of muscle tone, which typically manifests as face drooping, eyelid closure, phase 2 includes sagging of the jaw, dysarthria, passive tongue protrusion, which might be a the attempt the resist, during phase 3 the bilateral loss of motor control of the extremities emerges [16]. This phase includes a complete inability to move (termed cataplectic immobility [11•]). The last phase, phase 4, refers to the recovery, the recurrence of the complete muscle activity. Not in all occasions the whole process is run through, explaining the differentiation between partial and complete cataplexy [16].
This division of cataplexy into four stages reflects the dynamic character of this process. The authors assert common grounds with the classification of complete and partial types while a partial cataplexy just does not pass through all stages or rather passes through very fast. In their opinion the resisting stage (phase 2) shows directly the ability to compensate and avoid the evolution of the following stages. This hypothesis was reinforced by electrophysiological findings, investigating 34 patients with NT1 with transcranial magnetic stimulation motor evoked potential (TMS MEP) during a) quiet wakefulness, b) laughter without cataplexy, and c) laughter with cataplexy, trying to examine the four stages separately. The results revealed changes of the MEP amplitude and a prolonged MEP latency in the primary motor cortex (PMC). More specifically, patients showed significantly decreased intracortical facilitation during quiet wakefulness, whereas the MEP amplitude during laughter without cataplexy did not differ from healthy controls. During cataplexy, MEP amplitude significantly increased during the first two stages (triggering, resistance) and decreased in stage 3. During this stage, MEP latency was prolonged compared with “normal” laughter and quiet wakefulness. The authors concluded that the PMC and motor control pathways play a crucial role in the evolution of cataplexy. They further explain that increased MEP amplitude during phase 1 (triggering) and phase 2 (resisting) reflects the role of the PMC as the resisting regulator to struggle against the loss of muscle tone or postural collapse with full consciousness. Therefore the activity of the PMC may prevent the evolution of a complete cataplexy and thus might represent a compensatory mechanism in the brain during cataplexy [17]. So far, the four-process model has not been implemented in clinical practice.
Differential diagnosis
Although cataplexy is the pathognomonic symptom of NT 1, it can also be found in few rare genetic diseases. These include Niemann-Pick type C disease, Angelmann Syndrome, Moebius Syndrome, Norrie Disease, Coffin-Lowry Syndrome, and Prader-Willi Syndrome. Pathophysiologically, the involvement of the hypothalamic/pontine structures mostly explains the incidence of cataplexy in these diseases [18]. In such genetic disorders, reduced CSF hypocretin − 1 levels are common (even though not always measured) but also patients with the absence of HLA-DQB1*0602 and normal hypocretin levels have been observed [18, 19]. For example, in Niemann Pick, the accumulation of lipid storage products in the nervous system could affect the hypocretin-producing cells in the hypothalamus and reduce their levels [20].
Cataplexy itself regardless of which origin should be differentiated from hypotension, transient ischemic attacks, drop attacks, akinetic seizures, neuromuscular disorders, vestibular disorders, sleep paralysis, and psychiatric disorders [21]. Even in patients with typical cataplexy in the context of NT1, pseudo cataplexies can emerge, typically characterized by the absence of emotional triggers. In some cases, negative emotional triggers such as crying, generalized weakness, abrupt onset, and preserved ability to communicate can be found. The most important distinction seems to be the involvement of the face and the preservation of deep tendon reflexes. In addition, pseudo cataplexies occur more frequently in public places [22].
Case reports suggest cataplexies as a possible side effect of medications including prazosin [23], lamotrigine [24], clozapine [25], and modafinil [26].
With the introduction of dual orexin receptor antagonist for the treatment of insomnia, cataplexy was present as an anticipated side effect, which should resolve with stopping the intake [27]. In a few cases, it can also appear with the abrupt withdrawal of antidepressants with partum [17].
Pathophysiology
The discovery of hypocretin/orexin loss as the central cause of narcolepsy opened the window to design animal models to study the complex function of this neuropeptide and its role in different brain circuits. About 90% of patients with cataplexy are hypocretin/orexin-deficient [7]. Hypocretinergic neurons are located in the tuberal hypothalamus. It does not only contribute to the regulation of arousal and sleep-wake cycles but also in the control of rewarded behavior [28]. It is also involved in brainstem circuits responsible for involuntary muscle atonia during cataplexy, which normally is restricted to REM sleep [29]. In wild type mice, it was shown that an activation of cells in the subdorsolateral nucleus (SLD), which underlies muscle atonia only during REM-sleep, induced cataplexy whereas an inactivation of these cells stopped cataplexy [30]. These findings corroborate that circuits which control REM sleep atonia also play a functional role in regulating muscle paralysis in cataplexy. This function of the SLD is, beside others, under hypocretinergic/orexinergic control. The release of a cataplectic attack can be explained by an activation of the medial prefrontal cortex (mPFC) via positive emotions, which activates GABAergic neurons in the central nucleus of the amygdala (CeA), which, in turn, inhibit the locus coeruleus (LC), laterodorsal tegmentum (LPT), and ventrolateral periaqueductal gray (vlPAG). This inhibition leads to a disinhibition of the SLD and generates muscle atonia [5••].
The dysregulation of LC neurons contributes to pathologic changes in arousal/sleep and is associated to other phenomena such as cataplexy. While ascending norepinephrine neurons of the LC (LC NE) project to the forebrain and mediate cortical arousal, descending projections to the spinal cord affect muscle tone. It has been suggested that the activity of LC NE neurons can be halted during limbic activation and can lead to cataplexy [31]. For this reason, it is assumed that the therapeutic regulation of LC activity plays an important role in the treatment of patients with cataplexy.
Human neuroimaging studies have revealed abnormal hypothalamic and amygdala activity in patients with narcolepsy during the processing of humorous or emotional stimuli [32,33,34] indicating the crucial role of the limbic system in the pathophysiology of narcolepsy with cataplexy. Additionally, changes in the subcortical mesolimbic reward network have been observed [35]. So far, few studies investigated neural correlates during cataplectic attacks in humans. Meletti et al. [36] found that laughter-induced cataplexy is associated with an increase in neural activity in the amygdala, the anterior insula, the nucleus accumbens, and the ventromedial prefrontal cortex. This study suggests that among others limbic system changes may underly the release of cataplectic attacks in narcolepsy.
A functional magnetic resonance imaging (fMRI) study in 21 children/adolescents with recent onset of NT1 revealed an involvement of different brain circuits including the left subthalamic nucleus/zona incerta (STN/ZI) during laughter expression without cataplexy [37]. Interestingly, neurons located in the ZI and lateral hypothalamus produce melanin-concentrating hormone, another neuromodulator involved in the wake-sleep cycle [38, 39]. Thus, ZI has become another potential source in the pathophysiology of cataplexy.
In an animal model, an orexin gene transfer into the ZI and lateral hypothalamus significantly decreased cataplexy [40]. Moreover, the ZI is known to have efferents to different strategic brain areas, including the hypothalamus, the brainstem (including LC, vlPAG), and spinal cord and receives afferents from the amygdala, anterior cingulate, and sensory-motor cortex [41]. In the above mentioned fMRI study, an overactivation of the ZI in patients with NT1 was confirmed. In addition, a region of interest (ROI) analysis showed that ZI recruitment is increased when laughter is not followed by cataplexy suggesting that there might be a mechanism to block cataplexy in NT1 which relies on the activation of the ZI neurons, and in turn stabilizes the nuclei of the dorsolateral pons.
The absence of ZI activity during laughter in healthy controls in the present study and in previous fMRI studies on laughter suggests that its involvement is specific to patients with NT1 [37].
Measures for cataplexy
Objective measures for cataplexy are missing. In clinical practice, cataplexy is hard to be provoked by a clinician. Therefore, the presence of cataplexy is mostly identified by clinical history alone. Patients report that cataplexies are usually provoked by unsuspected, funny situations, mostly with familiar people, and less commonly aggressive emotions, or sexual intercourse [42]. The frequency, severity, and intensity subjectively depend on or are influenced by the actual way of living, coping strategies, specific medication, and comedication. Diaries can help to capture this data. To provoke cataplectic episodes during polysomnographic recordings, “funny” videos or video games are frequently used to confirm the presence of cataplexy. A failure in verification of cataplexy in this setting does of course not exclude their presence.
To evaluate the frequency and severity of cataplexies diaries and questionnaires are used. In general, there are only a few questionnaires assessing the prevalence of cataplexy mostly in the context of narcolepsy. One frequently used questionnaire is the Ullanlinna Narcolepsy Scale which includes an 11-item scale designed to evaluate a variety of symptoms relating to narcolepsy, including frequency of daytime narcoleptic episodes, muscle weakness associated with powerful emotions, and nighttime sleep latency [43]. Another one is the Swiss Narcolepsy Scale, which also focuses on the ability to sleep and stay awake and muscle weakness [44]. These questionnaires were established to distinguish narcolepsy from other central disorders of hypersomnolence. Its use to evaluate treatment efficacy especially in cataplexy is not validated so far. The only validated questionnaire introduced to evaluate effects of treatment is the Narcolepsy Severity Scale (NSS). It includes among others two questions addressing the frequency of generalized and partial cataplexy [45].
Pharmacologic treatment of cataplexy
Widely used anti-cataplectic treatments are antidepressants (selective serotonin reuptake inhibitors, serotonin and norephinephrine reuptake inhibitors, tricyclics), sodium oxybate (agonist to the gamma-aminobutyric acid receptor B; GABA-B), and pitolisant (histamine H3-autoreceptor agonist). Treatment guidelines of both the American Academy of Sleep Society (AASM) and the European Federation of Neurological Societies (EFNS) recommend sodium oxybate as first-line therapy for cataplexy and antidepressants as second-line treatment. [46,47,48]
These recommendations rely on few well-designed level 1 studies for some agents. But there is a lack of comparative and long-term studies for the treatment of cataplexy.
Antidepressants
Selective Serotonin Reuptake Inhibitors (SSRIs) (e.g., Citalopram, Escitalopram, Fluoxetine) selectively inhibit 5-HT reuptake transporters. Effective doses for fluoxetine range between 20 and 60 mg with mild effect on cataplexy. Citalopram (20–40 mg) and escitalopram (10–20 mg) reduced cataplexy significantly in small case series [49, 50]. Common adverse effects of SSRIs are irritation, gastrointestinal and sexual dysfunction, and movement disorders like restless legs syndrome. SNRIs (e.g., Venlafaxin) enhance the availability of NE and 5-HT activity. Venlafaxin is used in doses between 150 and 375 mg/day. It was shown to improve cataplexy in four patients. The effect was recently confirmed in a small number of children [51]. In two other SNRIs, atomoxetine and duloxetine, a change of frequency of cataplexy was reported [52]. Typical side effects of SNRIs include sweating, tachycardia, and hypertension. In both, SSRI and SNRI, status cataplecticus can emerge with a fast withdrawal [53]. Interestingly, the efficacy of SSRIs and SNRIs for cataplexy treatment has not been investigated in randomized placebo-controlled trials. The only approved antidepressant in the treatment of cataplexy is clomipramine, a tricyclic antidepressant (TCA). Clomipramine inhibits the reuptake of 5-HT transporter. Its fast metabolization into desmethylclomipramine adds an inhibition of the adrenergic reuptake. Clomipramine was shown to reduce frequency and severity at a dose of 25–75 mg [54]. The anticholinergic effects of clomipramine are responsible for its adverse effects, including dry mouth, sweating, constipation, high hart rate, gain of wait, increase of blood pressure, urinary problems, and sexual dysfunction. The abrupt stop of medication can lead to status cataplecticus. Long-term treatment with clomipramine can induce tolerance.
Sodium oxybate
Sodium oxybate is the sodium salt of gamma hydroxybutyrate (GHB). It acts as an agonist to the gamma-aminobutyric acid receptor B [55]. Because of a short half-time (< 60 min) sodium oxybate needs to be taken twice a night. Its mechanism of acting is not fully understood but it appears to suppress LC NE neurons during sleep, thereby promoting slow wave sleep. The cessation of LC activity is suspected to result in rebound activation with increased responsiveness during daytime with a positive effect on EDS and cataplexy [56]. A dose dependent effect on cataplexy was proven in several randomized trials [55] with a moderate decrease of anticataplectic effect by time. Side effects are nausea, dizziness, vomiting, somnolence, enuresis, and tremor. A cessation of the therapy is not associated with a rebound of cataplexy. The delay of maximal response to treatment can vary between 94 and 279 days [55]. In a meta-analysis evaluating the efficacy of GHB in all clinical symptoms of narcolepsy, the reduction of cataplexies was confirmed. With a daily intake of 9 g GHB/night, a reduction of cataplexies up to 70% was observed [57]. This dose-dependent effect was verified in a randomized controlled trial with a reduction of cataplexies on a weekly basis [58]. Interestingly, the abrupt cessation of GHB does not lead to a withdrawal syndrome or status cataplecticus, a typical adverse effect after stopping the intake of antidepressants [59].
Pitolisant
The inverse histamine H3-autoreceptor agonist pitolisant primarily increases histaminergic action. It additionally activates LC NE neurons, increases the acetylcholine and dopamine release in the prefrontal cortex and acetylcholine release in the hippocampus [60]. Pitolisant shows positive effects on wakefulness and narcolepsy-like symptoms in narcoleptic rats [61]. In humans, a decrease of cataplexy was proven, even if its anticataplectic mechanism is still not fully understood.
One possible explanation might be the presence of a high density of H3 receptors in the amygdala, receiving histaminergic inputs from the tuberomammillary nucleus, for the modulation of cataplexy [62, 63]. Cataplexy severity is probably enhanced in patients with narcolepsy in whom the number of tuberomammillary neurons is twice as high as in controls, which might reflect a compensatory effect to the loss of hypocretinergic/orexinergic input. The half-life of pitolisant ranges between 10 and 12 h and requires only a single dosage per day [64]. Dosages between 4.5 and 36 mg are acquired. Steady plasma levels are reached within 1 week. It is well tolerated and improves not only cataplexy but also EDS and hallucinations in narcolepsy. These narcolepsy indicate that pitolisant might constitute a useful first-line therapy for narcolepsy. Adverse events include insomnia, anxiety, nausea, headache, and irritability [64]. Its efficacy to decrease cataplexies was proven in two class-I evidence studies so far. The first was a post-hoc analysis of the safety and efficacy trial of pitolisant focusing on the effect on cataplexy frequency on a weekly basis. In this post-hoc analysis, pitolisant was superior to placebo but not non-inferior to modafinil in terms of improvement in cataplexy rate from baseline [65]. The second placebo controlled trial including 106 patients observed a reduction of weekly cataplexy rate up to 75% [64]. Both were short-term trials and there are no head to head studies comparing pitolisant with GHB for example so far.
Reported treatment options
Opioids are reported to reduce cataplexy through the activation of opioid receptors and an enhancement of monoamine neurotransmission. They additionally suppress REM sleep, which might explain its effects in the context of REM sleep associated muscle atonia [66,67,68].
Interestingly, brains from heroine-addicted patients have on average 54% more immunohistochemically detected neurons producing hypocretin than controls. Whereas the melanin-concentrating hormone cells, another cell population involved in the NT1 pathophysiology of the hypothalamus, did not change in response to morphine administration. Even more morphine increased the number of hypocretin cells to normal numbers in partially depleted transgenic mice. Morphine was shown to decrease cataplexy in narcoleptic mice. This underlines the potential role of opiate agonists in the treatment of narcolepsy and points to the role of hypocretin in addiction [69].
In a case report of two patients with NT1, a motoric trick was described to be effective to terminate cataplexy. This trick was a 1) painful stimulus and 2) a specific movement of the head and arm performed by a patients' relative [70]. The authors argued that the modulation of pain reception in the brainstem may interfere with the neuronal circuits involved in cataplexy.x
Conclusion
Due to their subtle manifestation in some patients with narcolepsy, cataplexies are under-recognized. Moreover, standardized diagnostic measures for cataplexy are still missing. Understanding cataplexy as a dynamic process including attempts to compensate for loss of muscle tone helps to explain differences in the clinical semiology within and between patients. The role of hypocretin/orexin in the pathophysiology of cataplexy has been well established and latest research strongly supports the hypothesis of an immune-based degeneration of orexin cells. Identifying the disease early in its course remains a major challenge before immune-based therapies may be successfully transferred into clinical practice. Alternatively, orexin substitution therapies are a promising therapeutic option for the near future. For the assessment of treatment efficacy, objective measures for severity, frequency, and intensity are highly warranted.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Löwenfeld L. Über Narkolepsie. Münchener medizinische Wochenzeitschrift. 1902;49:1041–5.
Overeem S, Lammers GJ, van Dijk JG. Weak with laughter. Lancet. 1999;354(9181):838. https://doi.org/10.1016/s0140-6736(99)80023-3.
Yoss RE, Daly DD. Narcolepsy. Med Clin North Am. 1960;44:953–68. https://doi.org/10.1016/s0025-7125(16)33982-7.
Akimoto H, Honda Y, Takahashi Y. Pharmacotherapy in narcolepsy. Dis Nerv Syst. 1960;21:704–6.
•• Szabo ST, Thorpy MJ, Mayer G, Peever JH, Kilduff TS. Neurobiological and immunogenetic aspects of narcolepsy: implications for pharmacotherapy. Sleep Med Rev. 2019;43:23–36. https://doi.org/10.1016/j.smrv.2018.09.006. This review gives an detailed discription of the mechanism of action in the most important agents used in narcolepsy.
•• Overeem S, van Nues SJ, van der Zande WL, Donjacour CE, van Mierlo P, Lammers GJ. The clinical features of cataplexy: a questionnaire study in narcolepsy patients with and without hypocretin-1 deficiency. Sleep Med. 2011;12(1):12–8. https://doi.org/10.1016/j.sleep.2010.05.010. This study represents the diversity of catapley and its different trigger factor.
Dauvilliers Y, Arnulf I, Mignot E. Narcolepsy with cataplexy. Lancet. 2007;369(9560):499–511. https://doi.org/10.1016/S0140-6736(07)60237-2.
Dauvilliers Y, Billiard M, Montplaisir J. Clinical aspects and pathophysiology of narcolepsy. Clin Neurophysiol. 2003;114(11):2000–17. https://doi.org/10.1016/s1388-2457(03)00203-7.
Pizza F, Antelmi E, Vandi S, Meletti S, Erro R, Baumann CR, et al. The distinguishing motor features of cataplexy: a study from video-recorded attacks. Sleep. 2018;41(5). https://doi.org/10.1093/sleep/zsy026.
Plazzi G, Pizza F, Palaia V, Franceschini C, Poli F, Moghadam KK, et al. Complex movement disorders at disease onset in childhood narcolepsy with cataplexy. Brain. 2011;134(Pt 12):3477–89. https://doi.org/10.1093/brain/awr244.
• Bassetti CLA, Adamantidis A, Burdakov D, Han F, Gay S, Kallweit U, et al. Narcolepsy - clinical spectrum, aetiopathophysiology, diagnosis and treatment. Nat Rev Neurol. 2019;15(9):519–39. https://doi.org/10.1038/s41582-019-0226-9. This manuscript summerizes the current state of knowlege in narcolepsy and gives a comprehensive overview of the clinical work up and therapy.
Barateau L, Pizza F, Lopez R, Antelmi E, Plazzi G, Dauvilliers Y. Persistence of deep-tendon reflexes during partial cataplexy. Sleep Med. 2018;45:80–2. https://doi.org/10.1016/j.sleep.2017.12.011.
Attarian HP, Schenck CH, Mahowald MW. Presumed REM sleep behavior disorder arising from cataplexy and wakeful dreaming. Sleep Med. 2000;1(2):131–3. https://doi.org/10.1016/s1389-9457(99)00013-1.
van Dijk JG, Lammers GJ, Blansjaar BA. Isolated cataplexy of more than 40 years' duration. Br J Psychiatry. 1991;159:719–21. https://doi.org/10.1192/bjp.159.5.719.
Khatami R, Luca G, Baumann CR, Bassetti CL, Bruni O, Canellas F, et al. The European narcolepsy network (EU-NN) database. J Sleep Res. 2016;25(3):356–64. https://doi.org/10.1111/jsr.12374.
Huang B, Xu T, Wang Z, Chen K, Zhang J, Zhao Z, et al. The clinical characteristics of cataplectic attack in narcolepsy type 1. Sleep Med. 2019. https://doi.org/10.1016/j.sleep.2019.03.015.
Huang B, Qian Z, Wang Z, Zhang J, Chen K, Xu T, et al. Fluctuation of primary motor cortex excitability during cataplexy in narcolepsy. Ann Clin Transl Neurol. 2019;6(2):210–21. https://doi.org/10.1002/acn3.670.
Lima FCB, do Nascimento Junior EB, Teixeira SS, Coelho FM, Oliveira GDP. Thinking outside the box: cataplexy without narcolepsy. Sleep Med. 2019;61:118–21. https://doi.org/10.1016/j.sleep.2019.03.006.
Nishino S, Kanbayashi T. Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications in the hypothalamic hypocretin/orexin system. Sleep Med Rev. 2005;9(4):269–310. https://doi.org/10.1016/j.smrv.2005.03.004.
Vankova J, Stepanova I, Jech R, Elleder M, Ling L, Mignot E, et al. Sleep disturbances and hypocretin deficiency in Niemann-Pick disease type C. Sleep. 2003;26(4):427–30. https://doi.org/10.1093/sleep/26.4.427.
American Academy of Sleep Medicine. The international classification of sleep disorders, Third Edition (ICSD-3). Darien, IL. 2014.
Meinen CN, Smith KM, Tillema JM, Kotagal S. Pseudo status cataplecticus in narcolepsy type 1. J Clin Sleep Med. 2018;14(9):1625–7. https://doi.org/10.5664/jcsm.7350.
Aldrich MS, Rogers AE. Exacerbation of human cataplexy by prazosin. Sleep. 1989;12(3):254–6. https://doi.org/10.1093/sleep/12.3.254.
Alonso-Navarro H, Montes JM, Plaza-Nieto JF, Jimenez-Jimenez FJ. Cataplexy possibly associated with lamotrigine. J Clin Psychopharmacol. 2016;36(4):400–2. https://doi.org/10.1097/JCP.0000000000000532.
Desarkar P, Goyal N, Khess CR. Clozapine-induced cataplexy. J Neuropsychiatr Clin Neurosci. 2007;19(1):87–8. https://doi.org/10.1176/jnp.2007.19.1.87.
Lopes E, Pereira D, da Silva Behrens NS, de Almeida FH, Calvancanti PO, de Araujo Lima TF, et al. Cataplexy as a side effect of modafinil in a patient without narcolepsy. Sleep Sci. 2014;7(1):47–9. https://doi.org/10.1016/j.slsci.2014.07.015.
Sutton EL. Profile of suvorexant in the management of insomnia. Drug Des Devel Ther. 2015;9:6035–42. https://doi.org/10.2147/DDDT.S73224.
Sakurai T. The role of orexin in motivated behaviours. Nat Rev Neurosci. 2014;15(11):719–31. https://doi.org/10.1038/nrn3837.
Scammell TE. Narcolepsy. N Engl J Med. 2015;373(27):2654–62. https://doi.org/10.1056/NEJMra1500587.
Kohlmeier KA, Tyler CJ, Kalogiannis M, Ishibashi M, Kristensen MP, Gumenchuk I, et al. Differential actions of orexin receptors in brainstem cholinergic and monoaminergic neurons revealed by receptor knockouts: implications for orexinergic signaling in arousal and narcolepsy. Front Neurosci. 2013;7:246. https://doi.org/10.3389/fnins.2013.00246.
Wu MF, Gulyani SA, Yau E, Mignot E, Phan B, Siegel JM. Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience. 1999;91(4):1389–99. https://doi.org/10.1016/s0306-4522(98)00600-9.
Reiss AL, Hoeft F, Tenforde AS, Chen W, Mobbs D, Mignot EJ. Anomalous hypothalamic responses to humor in cataplexy. PLoS One. 2008;3(5):e2225. https://doi.org/10.1371/journal.pone.0002225.
Schwartz S, Ponz A, Poryazova R, Werth E, Boesiger P, Khatami R, et al. Abnormal activity in hypothalamus and amygdala during humour processing in human narcolepsy with cataplexy. Brain. 2008;131(Pt 2):514–22. https://doi.org/10.1093/brain/awm292.
Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Schwartz S, et al. Reduced amygdala activity during aversive conditioning in human narcolepsy. Ann Neurol. 2010;67(3):394–8. https://doi.org/10.1002/ana.21881.
Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Bassetti CL, et al. Abnormal activity in reward brain circuits in human narcolepsy with cataplexy. Ann Neurol. 2010;67(2):190–200. https://doi.org/10.1002/ana.21825.
Meletti S, Vaudano AE, Pizza F, Ruggieri A, Vandi S, Teggi A, et al. The brain correlates of laugh and cataplexy in childhood narcolepsy. J Neurosci. 2015;35(33):11583–94. https://doi.org/10.1523/JNEUROSCI.0840-15.2015.
Vaudano AE, Pizza F, Talami F, Plazzi G, Meletti S. The neuronal network of laughing in young patients with untreated narcolepsy. Neurology. 2019. https://doi.org/10.1212/WNL.0000000000006853.
Ferreira JGP, Bittencourt JC, Adamantidis A. Melanin-concentrating hormone and sleep. Curr Opin Neurobiol. 2017;44:152–8. https://doi.org/10.1016/j.conb.2017.04.008.
Verret L, Goutagny R, Fort P, Cagnon L, Salvert D, Leger L, et al. A role of melanin-concentrating hormone producing neurons in the central regulation of paradoxical sleep. BMC Neurosci. 2003;4:19. https://doi.org/10.1186/1471-2202-4-19.
Liu M, Blanco-Centurion C, Konadhode R, Begum S, Pelluru D, Gerashchenko D, et al. Orexin gene transfer into zona incerta neurons suppresses muscle paralysis in narcoleptic mice. J Neurosci. 2011;31(16):6028–40. https://doi.org/10.1523/JNEUROSCI.6069-10.2011.
Lambert C, Simon H, Colman J, Barrick TR. Defining thalamic nuclei and topographic connectivity gradients in vivo. NeuroImage. 2017;158:466–79. https://doi.org/10.1016/j.neuroimage.2016.08.028.
Poryazova R, Khatami R, Werth E, Bassetti CL. Weak with sex: sexual intercourse as a trigger for cataplexy. J Sex Med. 2009;6(8):2271–7. https://doi.org/10.1111/j.1743-6109.2009.01328.x.
Hublin C, Kaprio J, Partinen M, Koskenvuo M, Heikkila K. The Ullanlinna Narcolepsy Scale: validation of a measure of symptoms in the narcoleptic syndrome. J Sleep Res. 1994;3(1):52–9. https://doi.org/10.1111/j.1365-2869.1994.tb00104.x.
Sturzenegger C, Baumann CR, Lammers GJ, Kallweit U, van der Zande WL, Bassetti CL. Swiss Narcolepsy Scale: a simple screening tool for hypocretin-deficient narcolepsy with cataplexy. Clin Transl Neurosci. 2018;2(2):2514183X18794175. https://doi.org/10.1177/2514183x18794175.
Dauvilliers Y, Beziat S, Pesenti C, Lopez R, Barateau L, Carlander B, et al. Measurement of narcolepsy symptoms: the narcolepsy severity scale. Neurology. 2017;88(14):1358–65. https://doi.org/10.1212/WNL.0000000000003787.
Wise MS, Arand DL, Auger RR, Brooks SN, Watson NF, American Academy of Sleep M. Treatment of narcolepsy and other hypersomnias of central origin. Sleep. 2007;30(12):1712–27. https://doi.org/10.1093/sleep/30.12.1712.
Billiard M, Bassetti C, Dauvilliers Y, Dolenc-Groselj L, Lammers GJ, Mayer G, et al. EFNS guidelines on management of narcolepsy. Eur J Neurol. 2006;13(10):1035–48. https://doi.org/10.1111/j.1468-1331.2006.01473.x.
Billiard M, Dauvilliers Y, Dolenc-Groselj L, Lammers GJ, Mayer G, Sonka K. Management of narcolepsy in adults. In: Gilhus NE, Barnes MP, Brainin M, editors. European handbook of neurological management. 2nd ed. Oxford: Wiley-Blackwell; 2010. p. 513–28.
Thirumalai SS, Shubin RA. The use of citalopram in resistant cataplexy. Sleep Med. 2000;1(4):313–6. https://doi.org/10.1016/s1389-9457(00)00026-5.
Sonka K, Kemlink D, Pretl M. Cataplexy treated with escitalopram--clinical experience. Neuro Endocrinol Lett. 2006;27(1–2):174–6.
Moller LR, Ostergaard JR. Treatment with venlafaxine in six cases of children with narcolepsy and with cataplexy and hypnagogic hallucinations. J Child Adolesc Psychopharmacol. 2009;19(2):197–201. https://doi.org/10.1089/cap.2008.036.
Izzi F, Placidi F, Marciani MG, Zannino S, Torelli F, Corte F, et al. Effective treatment of narcolepsy-cataplexy with duloxetine: a report of three cases. Sleep Med. 2009;10(1):153–4. https://doi.org/10.1016/j.sleep.2007.11.014.
Wang J, Greenberg H. Status cataplecticus precipitated by abrupt withdrawal of venlafaxine. J Clin Sleep Med. 2013;9(7):715–6. https://doi.org/10.5664/jcsm.2848.
Guilleminault C, Raynal D, Takahashi S, Carskadon M, Dement W. Evaluation of short-term and long-term treatment of the narcolepsy syndrome with clomipramine hydrochloride. Acta Neurol Scand. 1976;54(1):71–87. https://doi.org/10.1111/j.1600-0404.1976.tb07621.x.
Roth T, Dauvilliers Y, Guinta D, Alvarez-Horine S, Dynin E, Black J. Effect of sodium oxybate on disrupted nighttime sleep in patients with narcolepsy. J Sleep Res. 2017;26(4):407–14. https://doi.org/10.1111/jsr.12468.
Szabo ST, Gold MS, Goldberger BA, Blier P. Effects of sustained gamma-hydroxybutyrate treatments on spontaneous and evoked firing activity of locus coeruleus norepinephrine neurons. Biol Psychiatry. 2004;55(9):934–9. https://doi.org/10.1016/j.biopsych.2003.12.013.
A randomized, double blind, placebo-controlled multicenter trial comparing the effects of three doses of orally administered sodium oxybate with placebo for the treatment of narcolepsy. Sleep. 2002;25(1):42–9.
Xyrem International Study G. Further evidence supporting the use of sodium oxybate for the treatment of cataplexy: a double-blind, placebo-controlled study in 228 patients. Sleep Med. 2005;6(5):415–21. https://doi.org/10.1016/j.sleep.2005.03.010.
The abrupt cessation of therapeutically administered sodium oxybate (GHB) does not cause withdrawal symptoms. J Toxicol Clin Toxicol. 2003;41(2):131–5. https://doi.org/10.1081/clt-120019128.
Ligneau X, Perrin D, Landais L, Camelin JC, Calmels TP, Berrebi-Bertrand I, et al. BF2.649 [1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine, hydrochloride], a nonimidazole inverse agonist/antagonist at the human histamine H3 receptor: preclinical pharmacology. J Pharmacol Exp Ther. 2007;320(1):365–75. https://doi.org/10.1124/jpet.106.111039.
Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, et al. An inverse agonist of the histamine H(3) receptor improves wakefulness in narcolepsy: studies in orexin−/− mice and patients. Neurobiol Dis. 2008;30(1):74–83. https://doi.org/10.1016/j.nbd.2007.12.003.
Burgess CR, Oishi Y, Mochizuki T, Peever JH, Scammell TE. Amygdala lesions reduce cataplexy in orexin knock-out mice. J Neurosci. 2013;33(23):9734–42. https://doi.org/10.1523/JNEUROSCI.5632-12.2013.
Pollard H, Moreau J, Arrang JM, Schwartz JC. A detailed autoradiographic mapping of histamine H3 receptors in rat brain areas. Neuroscience. 1993;52(1):169–89. https://doi.org/10.1016/0306-4522(93)90191-h.
Szakacs Z, Dauvilliers Y, Mikhaylov V, Poverennova I, Krylov S, Jankovic S, et al. Safety and efficacy of pitolisant on cataplexy in patients with narcolepsy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(3):200–7. https://doi.org/10.1016/S1474-4422(16)30333-7.
Dauvilliers Y, Bassetti C, Lammers GJ, Arnulf I, Mayer G, Rodenbeck A, et al. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol. 2013;12(11):1068–75. https://doi.org/10.1016/S1474-4422(13)70225-4.
Wichniak A, Brunner H, Friess E, Pollmacher T. Successful suppression of cataplectic attacks with tramadol. Neurology. 2003;61(6):864–5. https://doi.org/10.1212/01.wnl.0000080367.25091.77.
Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL. Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. J Pharmacol Exp Ther. 1992;260(1):275–85.
Fry JM, Pressman MR, DiPhillipo MA, Forst-Paulus M. Treatment of narcolepsy with codeine. Sleep. 1986;9(1 Pt 2):269–74. https://doi.org/10.1093/sleep/9.1.269.
Thannickal TC, John J, Shan L, Swaab DF, Wu MF, Ramanathan L, et al. Opiates increase the number of hypocretin-producing cells in human and mouse brain and reverse cataplexy in a mouse model of narcolepsy. Sci Transl Med. 2018;10(447). https://doi.org/10.1126/scitranslmed.aao4953.
Matos N, Gaig C, Santamaria J, Iranzo A. Tricks to rapidly terminate episodes of cataplexy in narcolepsy. Sleep Med. 2016;20:129–30. https://doi.org/10.1016/j.sleep.2015.09.029.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Heidbreder declares that she has no conflict of interest.
Dr. Dirks declares that she has no conflict of interest.
Mr. Ramm declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Sleep Disorders
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heidbreder, A., Dirks, C. & Ramm, M. Therapy for Cataplexy. Curr Treat Options Neurol 22, 13 (2020). https://doi.org/10.1007/s11940-020-0619-5
Published:
DOI: https://doi.org/10.1007/s11940-020-0619-5