
Abstract
Purpose of the Review
Even as immune checkpoint inhibitors (ICIs) have transformed the lifespan of many patients, they may also trigger acceleration of long-term cardiovascular disease. Our review aims to examine the current landscape of research on ICI-mediated atherosclerosis and address key questions regarding its pathogenesis and impact on patient management.
Recent Findings
Preclinical mouse models suggest that T cell dysregulation and proatherogenic cytokine production are key contributors to plaque development after checkpoint inhibition. Clinical data also highlight the significant burden of atherosclerotic cardiovascular disease (ASCVD) in patients on immunotherapy, although the value of proactively preventing and treating ASCVD in this population remains an open area of inquiry. Current treatment options include dietary/lifestyle modification and traditional medications to manage hypertension, hyperlipidemia, and diabetes risk factors; no current targeted therapies exist.
Summary
Early identification of high-risk patients is crucial for effective preventive strategies and timely intervention. Future research should focus on refining screening tools, elucidating targetable mechanisms driving ICI atherosclerosis, and evaluating long-term cardiovascular outcomes in cancer survivors who received immunotherapy. Moreover, close collaboration between oncologists and cardiologists is essential to optimize patient outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Opinion statement
The current treatment options for ICI-mediated atherosclerosis require extensive further investigation given the comparatively recent awareness of this phenomena. Targeted therapies are limited and management is still heavily informed by evidence for traditional age and lifestyle-related ASCVD, although preclinical evidence suggests that immune dysregulation plays a much more prominent role in ICI-mediated atherosclerosis. One key component is monitoring and optimizing baseline cardiovascular risk factors such as hypertension, hyperlipidemia, and diabetes, while engaging in lifestyle modifications such as smoking cessation, a low-fat diet, and regular exercise as tolerated. Radiographic imaging remains an underutilized tool for assessing baseline plaque burden, identifying future lesional areas, and predicting the risk of severe cardiovascular events in patients on ICIs. However, further research is needed on how to best utilize and hybridize standard-of-care CT/FDG-PET scans for oncology patients with cardiac imaging modalities that track progression of coronary artery calcium and vascular wall inflammation. Therapeutically, standard medical therapy including statins, ezetimibe, PCSK-9 inhibitors, and antiplatelet agents can be employed as tolerated. There is some evidence that statins in particular enhance the effectiveness of ICI while attenuating plaque progression; however, clinicians should be aware of the potential increased risk for myopathy. Nonspecific immunomodulatory agents such as steroids are not recommended for prevention or suppression of ICI-induced plaque development, as they may worsen atherosclerosis. While targeted therapies that address the underlying immune-mediated mechanisms are still being developed, optimal management of ICI atherosclerosis should emphasize recognition of immunotherapy as a major ASCVD risk factor, early risk stratification/optimization, and a multidisciplinary approach involving both oncologists and cardiologists to determine appropriate screening and medical management.
Introduction
Immune checkpoint inhibitors (ICIs) have dramatically changed cancer therapy over the last decade, allowing patients with many malignancies to live longer with better quality of life, particularly those with melanoma and non-small cell lung cancer (NSCLC). Over thirty percent of all cancer patients are now eligible for ICI therapy, while up to 70% of ICI-treated patients may go on to develop an immune-related adverse event (IRAE) [1]. By reducing immune tolerance of malignant cells, ICIs inherently carry the risk of off-target T-cell autoreactivity in other organs. Much research on ICI cardiotoxicity has focused on lymphocytic myocarditis, a rare IRAE with a high mortality rate [2]. A growing body of basic science, translational, and clinical evidence now suggests that myocarditis may be the tip of the iceberg in terms of cardiac IRAEs, with ICI-induced acceleration of atherosclerotic cardiovascular disease (ASCVD) contributing significantly to vascular toxicity in the long term.
Disentangling the relationship between cardiovascular disease, cancer, and cancer treatment is complicated by shared risk factors such as older age, comorbid conditions (e.g., hypertension, diabetes, and dyslipidemia) and a chronic inflammatory state. Initially viewed as a bland lipid storage disorder, atherosclerosis is now thought to be exacerbated by an inflammatory milieu. Observational and prospective epidemiological studies have demonstrated that higher levels of IL-6, TNF-α, fibrinogen, and CRP are associated with increased cardiovascular risk [3,4,5,6,7]. Atherosclerosis-specific self-antigens include LDL, oxidized LDL (oxLDL), heat shock protein, and ApoB [8,9,10,11]. In both human and mouse models, endothelial expression of vascular cell adhesion molecules and T-cell chemoattractants is upregulated within early atheromas, which in turn facilitate binding and migration of macrophages and T-lymphocytes at these sites [12,13,14,15]. Once inside the arterial wall, immune cells promote evolution of plaques and contribute towards their acute thrombotic complications: macrophages ingest lipids to become foam cells, while T cells secrete inflammatory cytokines that further stimulate macrophages and smooth muscle cells (SMCs) that make up plaque. These cytokines—particularly IFN-γ—ultimately contribute to risk of acute thromboses through growth and destabilization of plaques and their fibrous cap [16,17,18,19,20,21,22]. Systemically circulating acute phase reactants such as fibrinogen and plasminogen may also alter thrombotic risk [19].
Targeting inflammation to reduce ASCVD risk in humans has been previously tested with mixed results. CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study), LoDoCo (Low-Dose Colchicine), and COLCOT (Colchicine Cardiovascular Outcomes Trial) support the theory that inhibition of innate immunity slows the progression of ASCVD [23]. However, the CIRT trial (Cardiovascular Inflammation Reduction Trial) did not find that low-dose methotrexate reduced cardiovascular events or levels of IL-1β, IL-6, or CRP. CANTOS also showed only modestly (− 15%) reduced cardiovascular events in post-myocardial infarction patients [24]. These trials highlight how immunomodulatory therapies for atherosclerosis must target specific points of immune dysregulation, which are still not fully understood.
The majority of cancers are associated with increased systemic inflammation, which is thought to contribute to the significantly increased risk of thrombosis in patients with active cancer. These risks are heightened by radiation therapy, which accelerates plaque development and calcification, as well as certain tyrosine kinase inhibitors which have direct vascular toxicities [25,26,27,28,29,30]. Certain malignancies, such as pancreatic or advanced genitourinary cancer, also carry an independently high thrombogenic risk [31]. More recently, accelerated ASCVD has emerged as an important frontier in understanding ICI cardiotoxicity beyond myocarditis [23].
Here, we review the latest clinical and preclinical literature on the epidemiology and mechanisms of ICI atherosclerosis. With heart disease a leading cause of death among cancer survivors, and ICIs increasingly used as adjuvant treatment, understanding the causative pathways behind ICI-mediated ASCVD can help clinicians thread the needle between maximizing cancer treatment response while mitigating serious potential long-term cardiac toxicities.
Clinical evidence supporting ICI-mediated atherosclerosis
Several recent studies suggest an association between ICI initiation and rates of subsequent atherosclerotic cardiovascular events [23]. In one large matched cohort study involving 2842 patients who started immunotherapy and 2842 controls matched by age, the 2-year risk of myocardial infarction (n = 37, 1.30%), coronary revascularization (n = 22, 0.77%), or ischemic stroke (n = 35, 1.23%) after starting an ICI was threefold higher, with > threefold higher rate of plaque progression on imaging [32•]. Interestingly, this association was attenuated by concomitant use of statins or corticosteroids, though interpretation of these results is limited by potential confounding by indication. Another study by Bar et al. reported that among 1215 cancer patients receiving ICI therapy, 31 (2.6%) developed acute vascular events and 8 (0.66%) developed myocardial infarction within 6 months of treatment initiation [33].
Smaller scale studies and prior meta- or pooled-analyses differ in their conclusions on the burden of ICI-mediated ASCVD. In patients with non-small cell lung cancer, prior studies have shown no definitive increase in cardiovascular or venous/arterial events relative to traditional cytotoxic therapy [33, 34]. One meta-analysis of treatment-related deaths in FDA-approved PD1/PDL1 clinical trials (n = 82) found a 9.8% incidence of deaths attributable to cardiovascular events, although incidence of myocardial infarction (n = 1, 1.2%) and acute coronary syndrome (n = 1, 1.2%) was low [35]. Conversely, an FDA pooled analysis of patients on ICI therapy (n = 21,644) suggested up to a 35% (95% CI: 0.76 to 2.4) increase in isolated coronary ischemia over 6 months for patients on an ICI versus traditional cytotoxic therapy [34].
Radiographically, patients with lung cancer and melanoma do appear to develop more unstable plaque phenotypes after immunotherapy. Non-calcified plaques are associated with greater cardiovascular and stroke risk than calcified plaques, while carotid plaque inflammation has been linked to less calcification on imaging [36]. One case–control study (40 cases with ICI and 20 controls without ICI) by Drobni et al. found that ICI use in lung cancer was associated with a significantly higher progression rate of noncalcified plaque volume (11.2% vs. 1.6% per year, p = 0.001). Patients who did not receive immunotherapy showed greater progression in calcified plaque volume (25% vs. 2% per year, p = 0.017) [37]. Similarly, in a cohort of 35 patients with melanoma and pre-existing aortic calcification, post-ICI plaque composition showed increased non-calcified plaque volume, which suggests a more vulnerable, rupture-prone state [36]. While existing studies often include a low proportion of patients on combination ICIs, making it difficult to assess if combination immunotherapy enhances ICI-mediated atherosclerosis, Drobni et al. reported that patients on combination ICIs exhibited greater plaque progression, suggesting that multiple checkpoint blockades have an additive effect on vascular inflammation [32•].
Atherosclerosis is a gradual process in which the disease burden progresses over many years or even decades. Improved oncologic outcomes have extended survival times, meriting closer attention to the potential health complications of longer term cancer survivorship. Follow-up evaluation of patients over a greater time window is needed to fully understand the chronic impact of ICI therapy on ASCVD and how it interacts with other cardiovascular risk factors such as age, hypertension, and diabetes.
Preclinical models for ICI atherosclerosis
The role of T cells in atherosclerosis has come to attention in recent years, particularly as innovative single-cell analyses in human and mouse models shed new light on the immune diversity of plaques beyond macrophages [38,39,40,41,42,43]. T cells are the predominant lymphocyte type in plaque, but murine models have reported both pro- and anti-atherogenic functions of different T cell subsets [8]. In mice, CD4+ T helper 1 (Th1) cells appear to be pro-atherogenic in large part due to production of IFN-γ and TNF-α [44,45,46,47,48,49]. The role of other Th subsets, CD8+ T cells, and Tregs remains equivocal, potentially due to differences in mice strain, T cell depletion strategy, gene knockout strategy, and study diet [8, 50]. There is also a lack of knowledge around antigen specificity for these T cell subsets, so it cannot be ruled out whether a particular Th subset is pro- or anti-atherogenic depending on the type of antigens or signaling present. For example, while generally thought to be atheroprotective in mice due to secretion of TGF-β and IL-10, Tregs’ initial protective response may shift towards pro-inflammatory as atherosclerosis progresses [51,52,53,54,55,56,57]. In humans, there is also not a consistent clinical correlation between blood/plaque Treg level and atherosclerosis [58,59,60,61]. Depuydt et al. did find that autoreactive CD4+ T cells may contribute to atherosclerosis in humans [43]. Fernandez et al. further suggest that T cell activation and exhaustion reprogramming play an important role, with exhausted human plaque T cells expressing more PD-1 than their blood counterparts [41•]. This has sparked interest in the potential impact of immunotherapy on plaque T-cell activity.
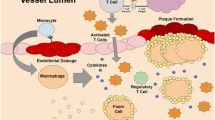
By releasing the innate brake on T cell activation, ICIs promote a systemic, T-cell mediated antitumor response. However, disrupting this key regulatory step can also result in many off-target immune reactions. Figure 1 and Table 1 summarize preclinical studies that have used genetic knockout models and co-stimulation/co-inhibition of ICI targets to shed light on the role of immune checkpoints in atherosclerosis.

This figure shows the different cardiac toxicities of immunotherapy and the different mouse models that have been used to investigate ICI-associated atherosclerosis. Abbreviations: PDL-1, programmed cell death ligand 1; Lag-3, lymphocyte activation gene 3; CTLA-4, cytotoxic T-lymphocyte antigen 4; Tim-3, T-cell immunoglobulin mucin; ApoE, apolipoprotein E; Ldlr, low-density lipoprotein receptor.
The PD-1/PDL-1 pathway was one of the original targets of ICI therapy, but also plays an important role in downregulating T-cell activity. Bu et al. showed that Pd1−/−Ldlr−/− mice had not only worsened atherosclerotic disease but also enhanced infiltration of cytotoxic T cells and macrophages in plaques and arterial walls, as well as elevated levels of serum IFN-γ and TNF-α, suggesting continued immune dysregulation [62]. Similarly, other studies in PD1-deficient mice or mice treated with anti-PD1 antibodies have reported that these mouse model experience worsened plaque burden with significant T cell and macrophage expansion both in atherosclerotic lesions and systemically [63,64,65]. Bu et al. also reported that Pd-l1/2-/- Ldlr -/- bone marrow chimeric mice had significantly more CD4+ T cells, CD8+ T cells, and macrophages in plaque lesions compared to controls; this difference was most notable for CD8+ T cells [62]. These results are consistent with Gotsman et al.’s prior work, which showed that Pd-l1/2-/- Ldlr-/- mice had more lesions and lesional inflammation [66]. Stimulation of the PD-1 pathway in a drug-treated mouse model appeared to have an opposite effect on atherosclerosis by decreasing levels of monocytes and T cells that secrete IFN-γ in the peripheral blood, while improving biomarkers such as B1 cells, Bregs, and oxLDL IgM levels considered to be atheroprotective [67]. Beyond the role of PD-1/PDL-1 in T cell exhaustion, a PD1-deficient mouse model with selective targeting of myeloid cells suggested that this pathway also acts as an important negative regulator of cholesterol synthesis and uptake [68]. Collectively, these preclinical studies provide robust evidence that inhibiting the PD1/PDL-1 pathway accelerates atherosclerotic plaque progression.
The CTLA-4 pathway plays an important atheroprotective role by competing with CD28 in T cell activation and acting as a co-inhibitor of T cell migration into nonlymphoid tissues such as arterial walls or tumors [69, 70]. CTLA-4 Tg/Apoe-/- mice, which constitutively over-express CTLA-4, have been found to have less plaque formation as well as markedly reduced intralesional and vessel wall accumulation of macrophages and CD4+ T cells [71]. Treatment with abatacept, which inhibits CD28 mediated T-cell activation, also mitigated the accelerated atherosclerosis and elevated serum levels of IFN-γ seen in ApoE3-Leiden and Apoe -/-mice [72, 73]. Conversely, Poels et al. reported that antibody-mediated inhibition of the CTLA-4 pathway in Ldlr-/- mice induced an activated T cell profile and increased ICAM1 endothelial expression along with increases in aortic plaque area [74].
PD-1/PDL-1 and CTLA4 remain the most common and widely used targets for cancer immunotherapy. Short-term combination immunotherapy led to unfavorable plaque and artery wall profiles in Ldlr-/- mice in the setting of T-cell, not myeloid, driven inflammation: larger necrotic core size, increased intraplaque CD8 T cells, increased CD4 effector/CD8+ T cells in arterial walls, and more endothelial activation as measured by Icam1 expression [75].
More recently, combination therapy with PD-1 and LAG3 blockade was approved for patients with unresectable or metastatic melanoma [76]. LAG3 is expressed across multiple leukocyte subtypes including T cells. Within T cells, it negatively regulates T cell activation and proliferation and is expressed at higher levels on exhausted T cells [77]. Mulholland et al. used Ldlr-/- mice as a background to test the effect of a Lag3 genetic knockout, as well as treatment with an anti-Lag3 antibody. In both models, there was no change in plaque burden, but recruitment of T cells to plaque lesions and increased levels of T cell activation/cytokine production were observed. They also noted an additive effect of dual LAG3/Pd-1 blockade on T cell activation [77].
TIM-3, a co-inhibitory immune checkpoint expressed on CD8 T cells, may also be a negative regulator of atherosclerosis. Foks et al. report that in Ldlr- mice fed a lipid-rich diet, TIM-3 antibody blockade increased macrophage content and decreased Treg cells in atherosclerotic plaque [23]. This complements findings in human subjects treated with dual inhibition of PD-1 and TIM-3, who had increased pro-inflammatory cytokine levels (IFN-g and TNF-a) compared to singular blockade of either immune checkpoint [78].
Conversely, magrolimab, an anti-CD47 monoclonal antibody that is part of the macrophage checkpoint inhibitor class, appears to exert anti-atherogenic effects in recent human and animal studies. Macrophages play an important role in programmed cell removal of apoptotic or diseased cellular debris through a process called efferocytosis, leading to interest in magrolimab’s potential to induce tumor cell phagocytosis [79]. A retrospective study of 9 lymphoma patients treated with magrolimab and rituximab also showed suppression of vascular inflammation on FDG-PET, although it did not assess how plaque composition or efferocytosis rates were directly modified by magrolimab [80]. The pro-atherogenic effects of CD47 are thought to be from nonspecific downregulation of efferocytosis, as a key feature of plaque is the formation of a necrotic core of apoptotic lipid-laden cells [81]. Kojima et al. recently reported that treatment with CD47 antibodies attenuated atherosclerosis across multiple mouse models, consistent with their findings that CD47 is also upregulated in atherogenesis and localizes intensely to plaque necrotic core [82].
Preclinical models of atherosclerosis in other autoinflammatory conditions offer further insight into the cardiovascular sequelae of dysregulated adaptive immune activity that ICIs may mimic. Rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) have well-established associations with the development of cardiovascular disease. MRL-Faslpr mice, which develop an SLE-like phenotype, are at higher risk of atherosclerotic disease and vascular damage, potentially driven by leukocyte infiltration and over-expression of inflammatory cytokines such as IL-6 and TNF-α [83, 84]. While MRL-Pdcd1-/- mice have been found to be predisposed to lymphocytic myocarditis, there are no current studies which examine atherosclerosis in MRL-Pdcd1-/- mice [85]. The B6.SLE mouse model also leads to a lupus phenotype involving T cell activation. Transfusion of isolated CD4+ T cells from this model into Ldlr-/-, Rag-/- mice (lacking functional B and T cells) dramatically accelerated atherosclerosis without causing lupus in the recipient [86].
In summary, recent preclinical models of atherosclerosis suggest that immune checkpoint pathways play a vital role in down-regulating T cell activation and migration. Further research is required to understand whether T cell activation and plaque progression worsen with a “dual-hit” of checkpoint blockade compared to ICI monotherapy, as risk of checkpoint myocarditis has been shown to. Finally, most studies utilized complete genetic knockout/chimeric mice or administration of an antibody drug not used for treatment of human subjects. This may limit their generalizability due to potential treatment effects on unexamined cell subtypes. For example, the role of myeloid versus T cell PD-1 blockade is still not well-understood. Strauss et al. found that targeted PD1 deficiency in myeloid-specific (Pd1 f/fLysMcre) mouse cells appeared to decrease tumor growth more than T cell specific (Pd1 f/fCD4cre) targeting [68]. At the same time, the myeloid-targeted mice had significantly elevated cholesterol levels, which may be consistent with clinical research showing baseline hypercholesterolemia and obesity’s association with improved survival in human patients treated with ICIs [87,88,89].
Proposed mechanisms for ICI atherosclerosis
No direct mechanistic studies in humans have been performed to evaluate accelerated atherosclerosis in the setting of ICI therapy. However, extrapolating from mouse models for atherosclerosis and ICI myocarditis provides insight into several plausible pathways for immune checkpoint blockade to cause plaque progression. Single-cell sequencing and mass cytometry showed that T-cells are active contributors in human and mice atherosclerotic lesions [51, 90]. These plaques exhibit significant heterogeneity in CD4+ and CD8+ T-cell phenotypes, ranging from highly active T cells expressing inflammatory molecules like granzymes and cytokines, to exhausted T cells characterized by high PD1 expression. Interestingly, in late-stage atherosclerosis, three major T cell subsets appear to undergo clonal expansion, indicating that generation of autoreactive CD4, CD8, and Treg cells may play an important role in ASCVD progression [91]. Conversely, inhibition of costimulatory T cell activation in mice through targeting of CD40/CD40L and B7 led to reduced plaque size and formation [92,93,94]. However, while the importance of T cell immunity in atherosclerosis is well established, the exact site of T-cell activity-associated atherosclerosis remains unclear.
One theory is that checkpoint inhibition of PD1 could activate T cells and decrease peripheral tolerance within intimal plaques [38, 95], leading to increased risk of plaque rupture and MACE. This has not been experimentally verified and may not be generalizable to explain higher rates of ASCVD seen in patients undergoing non-PD-1/PDL-1 treatments. Furthermore, in patients with basal or squamous cell carcinoma treated by PD-1 blockade, pre-existing tumor-specific T cells appeared to have limited reactivation capacity, and response to therapy may have resulted instead from constant attraction of new T cells [96].
Another proposed mechanism for ICI atherosclerosis is the unleashing of antigen-experienced T cells to recognize atherosclerosis-specific autoantigens. Recent mouse model studies of myocarditis have confirmed the presence of antigen-specific effector-like cytotoxic CD8 T cells which recognize cardiac autoantigens such as alpha-myosin [97,98,99]. It is still unclear whether these findings can be extrapolated to ICI atherosclerosis, and if the latter is indeed an antigen-driven process, what the identities of these specific antigens might be [91]. Single-cell RNA analysis of human coronary atherosclerotic plaque has demonstrated clonal expansion of primarily antigen-experienced T-cells, highlighting one potential pathway for self-reactive epitopes to accelerate atherosclerosis by interacting with smooth muscle cells and macrophages in the plaque microenvironment [100].
Recent research in Apoe-/- mice with advanced atherosclerosis found patterns of deteriorating peripheral T-cell tolerance, most notably in plaques followed by artery tertiary lymph node organs [91]. The large number of immune checkpoint pathways beyond PD-1/PD-L1 and CTLA4 has not been as closely examined for their role in modulating peripheral T cell tolerance in the setting of cancer and atherosclerosis. Further research is required to determine how much T-cell recruitment versus T cell reactivation drives the accumulation and clonal expansion of various T cell subsets within ICI atherosclerosis.
Functional cross-talk between T cells and macrophages also deserves further investigation in both the plaque and solid tumor immune microenvironments. Lipid-laden macrophages, or foam cells, directly contribute to plaque volume and necrotic core formation. After infiltrating vessel walls, foam cells are thought to enhance activation of the inflammasome and CD4 T cells, particularly Th1 cells, which in turn secrete proatherogenic IFN-γ and TNF-α. [63, 101] Interferons are particularly important regulators of atherosclerosis due to their role in enhancing endothelial activation, regulation of apoptosis, and foam cell formation [102]. It will be important for clinicians to understand how ICI-mediated systemic T-cell activation impacts CD4-monocyte interactions within plaque, whether foam cells in particular retain some degree of monocyte antigen-presenting capabilities, and how T cells may play a reciprocal role in ASCVD-related monocyte function. For example, while CD8 T cells’ contribution to atherosclerosis is less clear, Cochain et al. demonstrated in a Ldlr-/- mouse model that CD8+ T cell activity promoted atherosclerosis through modulation of monopoiesis and serum monocyte levels [103, 104]. Similarly, other murine models in PD1-deficient mice show a shift from CD4+ to CD8+ T cells within vessels affected by plaque, while CD8 T-cell depletion in Apoe- and Ldlr-/- mice reduced atherosclerosis [62, 66]. This was hypothesized to be due to CD8 T cells’ cytotoxic function in macrophage cell death and necrotic core formation within atherosclerotic plaques. ScRNA-sequencing profiles also suggest that dysfunctional CD8 T cell tolerance in mouse plaques is shared by human coronary/carotid arterial plaques [91].
The role of Tregs in ICI atherosclerosis remains an open question. Tregs secrete TGF-b and IL-10 which encourage an anti-inflammatory macrophage phenotype, while constitutively expressing CTLA-4 and PD-1/PDL-1 [105,106,107]. Increased number of Treg cells is associated with smaller human plaque size and improved plaque stability [107]. In the context of atherosclerosis, however, Treg cells may switch to exTreg cells through Treg/Th17 conversion in plaques [91]. One PD1-/Ldlr- mouse model found that PD1 knockout stimulated the Foxp3 + Treg response systemically and in atherosclerotic vessels, but overall, a pro-atherogenic CD4+ T cell response dominated [63]. Although studies have shown differing conclusions on PD-1/PDL-1’s regulation of Treg function, Tregs appear to play an overall negative regulatory role in T cell activation that may counteract the pro-inflammatory cascade triggered by blockade of CTLA-4 or PD1/PDL1 [108, 109]. Current evidence on how Treg function is affected by other checkpoints targeted in ICI therapy, such as LAG-3 is sparse. Data from genetic knockout mice suggest that LAG-3 promotes autoimmunity by limiting Treg cell proliferation/function [110].
Beyond primary ICI atherosclerosis, immunotherapy may exacerbate atherosclerosis through alternative IRAEs such as vasculitis. Among the systemic vasculitides, accelerated atherosclerosis has been clearly noted in Takayasu arteritis and ANCA-associated vasculitis (AAV) [111]. Systematic reviews and pharmacovigilance studies have reported that anti-PD-1 and CTLA-4 directed therapy have been associated with large-vessel vasculitis and vasculitis of the nervous system [112,113,114]. Interestingly, a humanized mouse model of GCA found that treatment with anti-PD-1 antibodies led to arterial infiltration with T cells and macrophages even from healthy subjects and development of fulminant arteritis [65]. Native AAV has been linked to single nucleotide polymorphisms in the CTLA-4 gene, highlighting one possible mechanism through which CTLA-4 inhibition may also lead to vasculitis [115].
Collectively, these studies point to immune checkpoint pathways as important negative regulators of T-cell activation, function, and recruitment. Long-term, ICI-mediated ASCVD may result from disrupting these mechanisms’ role in artery walls and plaques.
Treatment options
Prevention and monitoring
Several recently completed trials suggest that the adjuvant/neoadjuvant use of immunotherapy leads to additional improvement in survival rates for patients with resectable disease, particularly those with NSCLC or melanoma [116,117,118,119,120,121], [122,123,124,125,126]. As immunotherapy shifts towards curative or long-term adjuvant use for some patients, it becomes increasingly important that clinicians recognize and implement ICI-mediated ASCVD screening as part of routine cancer treatment. This may require coordination between primary care providers, oncologists, and potentially cardiologists to screen for and manage modifiable risk factors such as cholesterol levels, diabetes mellitus, and hypertension.
Cross-specialty collaboration is also needed to determine the optimal use and timing of imaging techniques. Plaque progression is a strong predictor of future cardiovascular events. Drobni et al. found that among 40 patients with melanoma, there was a greater progression rate of thoracic plaque burden (2.1% per year to 6.7% per year, p = 0 0.02) on CT after initiation of ICI [32•]. In a smaller cohort of 11 patients with pre-existing ASCVD on nivolumab in Italy who received contrast-enhanced CT scans at baseline and a minimum of 8 weeks, 63.6% had no significant changes and 27.3% experienced significant improvement in plaque burden; only one patient showed modest worsening of atherosclerotic lesions, and one patient demonstrated repeated, dramatic resolution of plaques on treatment with PD1/PDL1 inhibitors [127, 128]. Interestingly, all cases with significant improvement in plaque burden also reported grade > 2 IRAEs [127]. The imaging results of FDG PET-CT studies assessing systemic inflammation and plaque burden after ICI in small-scale cohorts of melanoma patients and mouse models have also been inconclusive, although Calabretta et al. do suggest that immunotherapy may trigger low-grade vascular wall inflammation primarily affecting the earlier, more vulnerable non-calcified coronary plaques, which could increase risk of future rupture [129, 130].
Going forward, it is important for clinicians to recognize the benefits and limitations of standard oncologic imaging approaches as a means of screening for and monitoring ASCVD in patients on immunotherapy [131]. Incidental coronary artery calcium (CAC) seen on non-gated CT imaging has been found to be predictive of major adverse cardiac events, particularly when the CAC score was over 100. However, prospective randomized trials examining the impact of non-gated CAC on ASCVD outcomes are needed [132, 133]. Future studies incorporating the use of coronary CT angiography to evaluate for ICI-mediated atherosclerosis are also needed. Novel imaging approaches such as immune-PET tracers are currently uncommon in clinical practice, but have proven valuable in human and mouse models for detecting vulnerable atherosclerotic lesions generally [134,135,136,137].
Pharmacologic therapy
Treatment strategies for ICI-mediated ASCVD remain in the exploratory phase. Statins, ezetimibe, fibrates, and PCSK9 inhibitors are safe, effective, and well-established treatments for atherosclerosis. However, their impact in the setting of ICIs and cancer requires further exploration to fully characterize risks and benefits to this complex patient population.
Statins
Despite hyperlipidemia control’s well-known role in ASCVD risk reduction, prospective randomized control trials have not yet evaluated the efficacy of statins from both an onco- and atheroprotective perspective during ICI therapy. In a recent retrospective study, Drobni et al. showed that statin or aspirin use by patients receiving ICI therapy was not associated with differences in a composite outcome of cardiovascular events, but statins were associated with significantly slower annual rate of progression in both total and noncalcified plaque volume in an imaging substudy of 40 patients [32•]. Because patients on statins were more likely to have ASCVD risk factors, it is difficult to interpret Drobni et al.’s findings in isolation. Interestingly, several recent studies report that concomitant use of common cardiovascular medications such as statins or aspirin also improves ICI activity, with enhanced activity of cytotoxic CD8 T cells and reduction of pro-inflammatory cytokines as possible mechanisms [138,139,140]. Adjuvant statin use during ICI therapy may carry important risks however. Drobni et al. subsequently conducted another retrospective analysis that suggested a > twofold risk of inflammatory or non-inflammatory skeletal myopathy in such cases, with a 2.5-fold higher risk of inflammatory myopathy, although there was not an increased risk of transaminitis [141].
PCSK-9 inhibitors
PCSK9 inhibitors are a newer class of monoclonal antibodies (and RNAi) used to treat patients at high risk for ASCVD refractory to statins. Similar to statins, recent studies have found that adjuvant PCSK9 use with ICIs, leading to enhanced intratumoral cytotoxic T cell infiltration, antigen presentation, and expression of co-inhibitory checkpoint molecules [142,143,144].
Steroids
In retrospective studies, steroid use during ICI therapy has been associated with a lower annual rate of plaque progression (3.5% vs. 6.9%, p = 0.04); however, there is a lack of prospective data supporting this association [32•]. Several confounders may still be present including study matching by cancer type, degree of pre-existing ASCVD, and length of steroid or ICI therapy. More importantly, chronic glucocorticoid excess has been associated with worse cardiovascular outcomes, most strikingly in patients with Cushing’s and metabolic syndrome, but also in those with autoimmune conditions requiring exogenous steroids such as lupus or rheumatoid arthritis [145,146,147,148,149]. Inflammation from autoimmune disease itself likely contributes to ASCVD. However, glucocorticoids have non-specific immunosuppressive effects, while also carrying their own increased risk of long-term atherosclerotic complications. Several studies have reported that even low levels of steroids confer an increased risk of ASCVD in these patients when used chronically [148,149,150]. From an oncologic standpoint, some studies raise the concern that steroids may blunt ICI treatment response, though this is not entirely clear [151,152,153,154,155,156]. Overall, steroids are not recommended for prevention or treatment of ICI-mediated ASCVD, and more research on targeted steroid-sparing therapies is needed.
Emerging therapies
Just as ICIs have transformed oncology, they have also changed clinical considerations for patients who experience cardiotoxic IRAEs. While existing literature suggests that ICIs cause a net acceleration in atherosclerosis, we lack a complete mechanistic understanding of their effects on plaque formation and progression. Induction of a pro-inflammatory, T cell-rich environment likely plays an important role. However, ICIs have heterogeneous effects on immune activation, some of which may even be atheroprotective, and newer checkpoint targets such as TIM3 inhibitors remain less studied. Although active research is ongoing into focused immune targets behind plaque formation in ICI-associated atherosclerosis, specific molecular and cell-based therapies remain needed and lacking.
Sex-related differences in ICI cardiotoxicity remain one knowledge gap with implications for ICI atherosclerosis risk stratification. Both clinical and mouse studies suggest that females may be at higher risk of ICI myocarditis, but the literature on sex disparities in ICI atherosclerosis is more equivocal, limited by small sample sizes or borderline significance [32, 33, 157, 158]. In the general population, sex differences in cardiovascular biomarkers (e.g. adipokines, inflammatory markers, fibrosis, and metalloproteinase inhibitors) have been found and were most pronounced between pre-menopausal women versus men [159]. There is also a well-established sex-based dimorphism in risk of autoimmune disease and IRAEs that is increased for women [160,161,162]. This highlights the complex interplay between several cardiovascular risk factors that may help determine individuals’ risk for ICI atherosclerosis and response to standard therapy.
Moving forward, eliciting the interaction of genetic, hormonal, and immune-related factors in ICI atherosclerosis is critical to preserving cardiovascular health among cancer patients who receive immunotherapy. Expanding our knowledge of these pathways will allow for the basic and translational insights needed to develop novel preventive and therapeutic strategies. For this complex patient population, developing more precise ASCVD risk stratification and targeted therapies while defining current imaging and pharmaceutical best practices will require close collaboration between cardio-oncologists, oncologists, and basic/clinical/translational researchers.
Conclusion
In addition to their remarkable success in treating many malignancies, ICIs have been linked to increased risk of ASCVD, one of the most common chronic conditions with an underlying inflammatory component. As immunotherapy moves into the curative/adjuvant setting, advanced cancer survivorship is also expected to grow. This population of patients who require ongoing, potentially lifetime, use of ICI therapy to control their advanced or metastatic disease carry unknown cardiovascular risks that can significantly impact mortality beyond their cancer diagnosis. It is essential for all clinicians who treat cancer patients longitudinally to recognize ASCVD as a potential immune-related complication that would benefit from close monitoring and control of risk factors such as hypertension, hyperlipidemia, and diabetes mellitus, especially in those with a prior ASCVD history or risk factors.
The precise mechanisms underlying the pathogenesis of ICI atherosclerosis remain in need of further preclinical and clinical study. Several mouse models of ICI atherosclerosis suggest that checkpoint inhibition disrupts negative regulation of T-cell activity and serum levels of proatherogenic cytokines in particular. Still, it remains necessary to distinguish the extent of T-cell reactivation versus T-cell recruitment during this process, as well as whether Treg function leads to a net atherogenic or atheroprotective effect in the setting of ICI use. Finally, cross-talk between CD4/CD8 T cells and myeloid populations such as macrophages/monocytes also appear to play an important role in plaque development, although the precise impact of immunotherapy on their activity remains under-explored.
Further characterization of the mechanisms underlying plaque progression is needed to determine appropriate timing of atherosclerotic imaging as well as medical interventions. Existing observational studies have limited follow-up timelines and likely do not capture the full extent of the association between ICI use and lifetime ASCVD risk or other adverse cardiovascular events. Longer-term trials using larger cohort sizes are also needed to understand how standard medical management of ASCVD risk factors affects cancer patients in heterogeneous ways. Our review underscores the importance of further preclinical and clinical investigation into this growing patient population to explore targeted therapeutic targets.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance
Suero-Abreu GA, Zanni MV, Neilan TG. Atherosclerosis with immune checkpoint inhibitor therapy: Evidence, diagnosis, and management: JACC: CardioOncology State-of-the-Art Review. JACC Cardio Oncol. 2022;4:598–615.
Palaskas N, Lopez-Mattei J, Durand JB, Iliescu C, Deswal A. Immune checkpoint inhibitor myocarditis: Pathophysiological characteristics, diagnosis, and treatment. J Am Heart Assoc. 2020;9:e013757.
Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43.
Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–43.
Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–72.
Harris TB, et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am J Med. 1999;106:506–12.
Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–34.
Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17:387–401.
Moon JJ, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–13.
Kimura T, et al. Regulatory CD4+ T cells recognize major histocompatibility complex class II molecule-restricted peptide epitopes of apolipoprotein B. Circulation. 2018;138:1130–43.
Stemme S, et al. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1995;92:3893–7.
Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–7.
Gu L, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–81.
Cybulsky MI, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–62.
Li H, Cybulsky MI, Gimbrone MA Jr, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb. 1993;13:197–204.
Smith JD, et al. Decreased atherosclerosis in mice deficient in both macrophage colony-stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci USA. 1995;92:8264–8.
Qiao JH, et al. Role of macrophage colony-stimulating factor in atherosclerosis: Studies of osteopetrotic mice. Am J Pathol. 1997;150:1687–99.
Libby P, et al. Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol. 1996;7:330–5.
Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365–72.
Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb. 1991;11:1223–30.
Nikkari ST, et al. Interstitial collagenase (MMP-1) expression in human carotid atherosclerosis. Circulation. 1995;92:1393–8.
Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–97.
Vuong JT, et al. Immune checkpoint therapies and atherosclerosis: Mechanisms and clinical implications: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022;79:577–93.
Ridker PM, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. 2019;380:752–62.
Maraldo MV, et al. Cardiovascular disease after treatment for Hodgkin’s lymphoma: An analysis of nine collaborative EORTC-LYSA trials. Lancet Haematol. 2015;2:e492–502.
Zhang X, et al. Clinical outcomes of radiation-induced carotid stenosis: A systematic review and meta-analysis. J Stroke Cerebrovasc Dis. 2020;29:104929.
Huang T-L, et al. Long-term effects on carotid intima-media thickness after radiotherapy in patients with nasopharyngeal carcinoma. Radiat Oncol. 2013;8:261.
Brown KN, Hussain K, Richards JR. Radiation-Induced Coronary Artery Disease. 2023 Jun 12. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 30725633.
Orphanos GS, Ioannidis GN, Ardavanis AG. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol. 2009;48:964–70.
Wu MD, Moslehi JJ, Lindner JR. Arterial thrombotic complications of tyrosine kinase inhibitors. Arterioscler Thromb Vasc Biol. 2021;41:3–10.
Solinas C, Saba L, Sganzerla P, Petrelli F. Venous and arterial thromboembolic events with immune checkpoint inhibitors: A systematic review. Thromb Res. 2020;196:444–53.
• Drobni ZD, et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation. 2020;142:2299–311. Findings from this study suggest that the risk of atherosclerotic cardiovascular complications is significantly higher after starting immunotherapy.
Bar J, et al. Acute vascular events as a possibly related adverse event of immunotherapy: A single-institute retrospective study. Eur J Cancer. 2019;120:122–31.
Chitturi KR, et al. Immune checkpoint inhibitor-related adverse cardiovascular events in patients with lung cancer. JACC Cardio Oncol. 2019;1:182–192.
Wang Y, et al. Treatment-related adverse events of PD-1 and PD-L1 inhibitors in clinical trials: A systematic review and meta-analysis. JAMA Oncol. 2019;5:1008–19.
Turker I, et al. Immune checkpoint inhibitors’ effects on calcified aortic plaques in melanoma survivors: A retrospective cohort study. JACC Cardio Oncol. 2023;5:536–8.
Drobni ZD, et al. Impact of immune checkpoint inhibitors on atherosclerosis progression in patients with lung cancer. J Immunother Cancer. 2023;11: e007307. https://doi.org/10.1136/jitc-2023-007307.
Winkels H, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res. 2018;122:1675–88.
Lin J-D, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. 2019;4(4):e124574.
Cole JE, et al. Immune cell census in murine atherosclerosis: cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc Res. 2018;114:1360–71.
• Fernandez DM, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–88. Results from this study suggest that exhausted T-cells in plaque express more PD1, highlighting one potential mechanism by which PD1 inhibition may affect plaque development.
Depuydt MAC, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. 2020;127:1437–55.
Depuydt MAC, et al. Single-cell T cell receptor sequencing of paired human atherosclerotic plaques and blood reveals autoimmune-like features of expanded effector T cells. Nat Cardiovasc Res. 2023;2:112–25.
Emeson EE, Shen ML, Bell CG, Qureshi A. Inhibition of atherosclerosis in CD4 T-cell-ablated and nude (nu/nu) C57BL/6 hyperlipidemic mice. Am J Pathol. 1996;149:675–85.
Zhou X, Robertson A-KL, Rudling M, Parini P, Hansson GK. Lesion development and response to immunization reveal a complex role for CD4 in atherosclerosis. Circ Res. 2005;96:427–34.
Li J, et al. CCR5+T-bet+FoxP3+ effector CD4 T cells drive atherosclerosis. Circ Res. 2016;118:1540–52.
Frostegård J, et al. Cytokine expression in advanced human atherosclerotic plaques: Dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 1999;145:33–43.
Gupta S, et al. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–61.
Laurat E, et al. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 2001;104:197–202.
Cochain C, Zernecke A. Protective and pathogenic roles of CD8+ T cells in atherosclerosis. Basic Res Cardiol. 2016;111:71.
Winkels H, Wolf D. Heterogeneity of T cells in atherosclerosis defined by single-cell RNA-sequencing and cytometry by time of flight. Arterioscler Thromb Vasc Biol. 2021;41:549–63.
Gaddis DE, et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun. 2018;9:1095.
Klingenberg R, et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J Clin Invest. 2013;123:1323–34.
Lin J, et al. The role of CD4+CD25+ regulatory T cells in macrophage-derived foam-cell formation. J Lipid Res. 2010;51:1208–17.
Ait-Oufella H, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–80.
Mallat Z, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85:e17–24.
Robertson A-KL, et al. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112:1342–50.
George J, et al. Regulatory T cells and IL-10 levels are reduced in patients with vulnerable coronary plaques. Atherosclerosis. 2012;222:519–23.
Wigren M, et al. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler Thromb Vasc Biol. 2012;32:2000–4.
Barth SD, et al. The ratio of regulatory (FOXP3+) to total (CD3+) T cells determined by epigenetic cell counting and cardiovascular disease risk: A prospective case-cohort study in non-diabetics. EBioMedicine. 2016;11:151–6.
Ammirati E, et al. Circulating CD4+CD25hiCD127lo regulatory T-cell levels do not reflect the extent or severity of carotid and coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1832–41.
Bu D-X, et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1100–7.
Cochain C, et al. Programmed cell death-1 deficiency exacerbates T cell activation and atherogenesis despite expansion of regulatory T cells in atherosclerosis-prone mice. PLoS One. 2014;9:e93280.
Xia W, Zou C, Chen H, Xie C, Hou M. Immune checkpoint inhibitor induces cardiac injury through polarizing macrophages via modulating microRNA-34a/Kruppel-like factor 4 signaling. Cell Death Dis. 2020;11:575.
Zhang H, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci USA. 2017;114:E970–9.
Gotsman I, et al. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. 2007;117:2974–82.
Grievink HW, et al. Stimulation of the PD-1 pathway decreases atherosclerotic lesion development in Ldlr deficient mice. Front Cardiovasc Med. 2021;8:740531.
Strauss L, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol. 2020;5(43):eaay1863.
Tivol EA, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7.
Taggart D, et al. Anti-PD-1/anti-CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8+ T cell trafficking. Proc Natl Acad Sci USA. 2018;115:E1540–9.
Matsumoto T, et al. Overexpression of cytotoxic T-lymphocyte-associated antigen-4 prevents atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2016;36:1141–51.
Ewing MM, et al. T-cell co-stimulation by CD28-CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int J Cardiol. 2013;168:1965–74.
Ma K, et al. CTLA4-IgG ameliorates homocysteine-accelerated atherosclerosis by inhibiting T-cell overactivation in apoE(-/-) mice. Cardiovasc Res. 2013;97:349–59.
Poels K, et al. Antibody-mediated inhibition of CTLA4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cells. 2020;9(9):1987.
Poels K, et al. Immune checkpoint inhibitor therapy aggravates T cell-driven plaque inflammation in atherosclerosis. JACC CardioOncol. 2020;2:599–610.
Tawbi HA, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386:24–34.
Mulholland M, et al. LAG3 regulates T cell activation and plaque infiltration in atherosclerotic mice. JACC: Cardio Oncol. 2022;4:635–645.
Qiu M-K, et al. PD-1 and Tim-3 pathways regulate CD8+ T cells function in atherosclerosis. PLoS One. 2015;10:e0128523.
Advani R, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med. 2018;379:1711–21.
Jarr K-U, et al. Effect of CD47 blockade on vascular inflammation. N Engl J Med. 2021;384:382–3.
Wang Y, et al. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Natl Acad Sci USA. 2020;117:15818–26.
Kojima Y, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90.
Marczynski P, et al. Vascular inflammation and dysfunction in lupus-prone mice-IL-6 as mediator of disease initiation. Int J Mol Sci. 2021;22(5):2291.
Qiao JH, Castellani LW, Fishbein MC, Lusis AJ. Immune-complex-mediated vasculitis increases coronary artery lipid accumulation in autoimmune-prone MRL mice. Arterioscler Thromb. 1993;13:932–43.
Waliany S, et al. Immune checkpoint inhibitor cardiotoxicity: Understanding basic mechanisms and clinical characteristics and finding a cure. Annu Rev Pharmacol Toxicol. 2021;61:113–34.
Wilhelm AJ, Rhoads JP, Wade NS, Major AS. Dysregulated CD4+ T cells from SLE-susceptible mice are sufficient to accelerate atherosclerosis in LDLr-/- mice. Ann Rheum Dis. 2015;74:778–85.
Perrone F, et al. The prognostic role of high blood cholesterol in advanced cancer patients treated with immune checkpoint inhibitors. J Immunother. 2020;43:196–203.
Young A, Evans L, Ng M. Middle ear salivary choristoma: A rare case report and update on congenital associations, facial nerve involvement, and treatment strategies. Case Rep Otolaryngol. 2020;2020:8435140.
Cortellini A, et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: When overweight becomes favorable. J Immunother Cancer. 2019;7:57.
Johnson DB, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–55.
Wang Z, et al. Pairing of single-cell RNA analysis and T cell antigen receptor profiling indicates breakdown of T cell tolerance checkpoints in atherosclerosis. Nat Cardiovasc Res. 2023;1–17.
Lacy M, et al. Cell-specific and divergent roles of the CD40L-CD40 axis in atherosclerotic vascular disease. Nat Commun. 2021;12:3754.
Seijkens TTP, et al. Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J Am Coll Cardiol. 2018;71:527–42.
Buono C, et al. B7–1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109:2009–15.
Baitsch L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60.
Yost KE, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25:1251–9.
Zhu H, et al. Identification of pathogenic immune cell subsets associated with checkpoint inhibitor-induced myocarditis. Circulation. 2022;146:316–35.
Axelrod ML, et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Nature. 2022;611:818–26.
Won T, et al. Cardiac myosin-specific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep. 2022;41:111611.
Chowdhury RR, et al. Human coronary plaque T cells are clonal and cross-react to virus and self. Circ Res. 2022;130:1510–30.
Westerterp M, et al. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. 2017;25:1294–1304.e6.
Boshuizen MCS, de Winther MPJ. Interferons as essential modulators of atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:1579–88.
Cochain C, et al. CD8+ T cells regulate monopoiesis and circulating Ly6C-high monocyte levels in atherosclerosis in mice. Circ Res. 2015;117:244–53.
Padgett LE, Araujo DJ, Hedrick CC, Olingy CE. Functional crosstalk between T cells and monocytes in cancer and atherosclerosis. J Leukoc Biol. 2020;108:297–308.
Kuipers H, et al. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. Eur J Immunol. 2006;36:2472–82.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Dietel B, et al. Decreased numbers of regulatory T cells are associated with human atherosclerotic lesion vulnerability and inversely correlate with infiltrated mature dendritic cells. Atherosclerosis. 2013;230:92–9.
Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol. 2013;14:152–61.
Francisco LM, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29.
Zhang Q, et al. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci Immunol. 2017;2(9):eaah4569.
Cohen Tervaert JW. Cardiovascular disease due to accelerated atherosclerosis in systemic vasculitides. Best Pract Res Clin Rheumatol. 2013;27:33–44.
Salem J-E, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018;19:1579–89.
Daxini A, Cronin K, Sreih AG. Vasculitis associated with immune checkpoint inhibitors-A systematic review. Clin Rheumatol. 2018;37:2579–84.
Boland P, Heath J, Sandigursky S. Immune checkpoint inhibitors and vasculitis. Curr Opin Rheumatol. 2020;32:53–6.
Lee KS, et al. Genetic variants in antineutrophil cytoplasmic antibody-associated vasculitis: A Bayesian approach and systematic review. J Clin Med Res. 2019;8(2):266.
Chmielewska I, Stencel K, Kalinka E, Ramlau R, Krawczyk P. Neoadjuvant and adjuvant immunotherapy in non-small cell lung cancer-clinical trials experience. Cancers. 2021;13(20):5048.
Forde PM, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–86.
Sidaway P. Neoadjuvant pembrolizumab improves outcomes. Nat Rev Clin Oncol. 2023;20:284.
Patel SP, et al. Neoadjuvant-adjuvant or adjuvant-only pembrolizumab in advanced melanoma. N Engl J Med. 2023;388:813–23.
Weber J, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377:1824–35.
Luke JJ, et al. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): A randomised, double-blind, phase 3 trial. Lancet. 2022;399:1718–29.
Long GV, et al. Adjuvant therapy with nivolumab versus placebo in patients with resected stage IIB/C melanoma (CheckMate 76K). J of Skin. 2023;7:s163–s163.
Cercek A, et al. PD-1 blockade in mismatch repair-deficient, locally advanced rectal cancer. N Engl J Med. 2022;386:2363–76.
Chalabi M, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med. 2020;26:566–76.
Bajorin DF, et al. Adjuvant nivolumab versus placebo in muscle-invasive urothelial carcinoma. N Engl J Med. 2021;384:2102–14.
Sharma P, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18:312–22.
Lamberti G, et al. New disappearance of complicated atheromatous plaques on rechallenge with PD-1/PD-L1 axis blockade in non-small cell lung cancer patient: Follow up of an unexpected event. Ther Adv Med Oncol. 2020;12:1758835920913801.
Gelsomino F, et al. Programmed death-1 inhibition and atherosclerosis: Can nivolumab vanish complicated atheromatous plaques? Ann Oncol. 2018;29:284–6.
Poels K, et al. Immune checkpoint inhibitor treatment and atherosclerotic cardiovascular disease: An emerging clinical problem. J Immunother Cancer. 2021;9(6):e002916.
Calabretta R, et al. Immune checkpoint inhibitor therapy induces inflammatory activity in large arteries. Circulation. 2020;142:2396–8.
Lopez-Mattei JC, et al. Cardiac computed tomography in cardio-oncology: JACC: CardioOncology Primer. JACC Cardio Oncol. 2021;3:635–49.
Yu C, et al. Incidentally identified coronary artery calcium on non-contrast CT scan of the chest predicts major adverse cardiac events among hospital inpatients. Open Heart. 2021;8(2):e001695.
Peng A, Dudum R, Maron DJ, Sandhu A, Rodriguez F. Opportunistic screening of incidental coronary artery calcium with deep-learning algorithm on non-ECG gated chest CT imaging and association with cardiovascular events and mortality. J Am Coll Cardiol. 2023;81:2123.
Meletta R, et al. CD80 is upregulated in a mouse model with shear stress-induced atherosclerosis and allows for evaluating CD80-targeting PET tracers. Mol Imaging Biol. 2017;19:90–9.
Mach F, Schönbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998;394:200–3.
Mani V, et al. Predictors of change in carotid atherosclerotic plaque inflammation and burden as measured by 18-FDG-PET and MRI, respectively, in the dal-PLAQUE study. Int J Cardiovasc Imaging. 2014;30:571–82.
Pérez-Medina C, Fayad ZA, Mulder WJM. Atherosclerosis immunoimaging by positron emission tomography. Arterioscler Thromb Vasc Biol. 2020;40:865–73.
Cantini L, et al. High-intensity statins are associated with improved clinical activity of PD-1 inhibitors in malignant pleural mesothelioma and advanced non-small cell lung cancer patients. Eur J Cancer. 2021;144:41–8.
Bird L. Statins as adjuvants. Nat Rev Immunol. 2018;18:669.
Xia Y, et al. The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell. 2018;175:1059–1073.e21.
Drobni ZD, et al. Association between incidental statin use and skeletal myopathies in patients treated with immune checkpoint inhibitors. Immunother Adv. 2021;1:ltab014.
Liu X, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588:693–8.
Wang R, et al. Inhibition of PCSK9 enhances the antitumor effect of PD-1 inhibitor in colorectal cancer by promoting the infiltration of CD8+ T cells and the exclusion of Treg cells. Front Immunol. 2022;13:947756.
Almeida CR, Ferreira BH, Duarte IF. Targeting PCSK9: A promising adjuvant strategy in cancer immunotherapy. Signal Transduct Target Ther. 2021;6:111.
De Leo M, et al. Cardiovascular disease in Cushing’s syndrome: Heart versus vasculature. Neuroendocrinology. 2010;92(Suppl 1):50–4.
Tune JD, Goodwill AG, Sassoon DJ, Mather KJ. Cardiovascular consequences of metabolic syndrome. Transl Res. 2017;183:57–70.
Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007;157:545–59.
Pujades-Rodriguez M, Morgan AW, Cubbon RM, Wu J. Dose-dependent oral glucocorticoid cardiovascular risks in people with immune-mediated inflammatory diseases: A population-based cohort study. PLoS Med. 2020;17:e1003432.
Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Intern Med. 2004;141:764–70.
Souverein PC, et al. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart. 2004;90:859–65.
Romão R, et al. Impact of immune-related adverse events on immune checkpoint inhibitors treated cancer patients’ survival: Single center experience and literature review. Cancers. 2023;15(3):888.
Kalfeist L, et al. Impact of glucocorticoid use in oncology in the immunotherapy era. Cells. 2022;11(5):770.
Scott SC, Pennell NA. Early use of systemic corticosteroids in patients with advanced NSCLC treated with nivolumab. J Thorac Oncol. 2018;13:1771–5.
Chasset F, et al. Single-center study under a French Temporary Authorization for Use (TAU) protocol for ipilimumab in metastatic melanoma: Negative impact of baseline corticosteroids. Eur J Dermatol. 2015;25:36–44.
Faje AT, et al. High-dose glucocorticoids for the treatment of ipilimumab-induced hypophysitis is associated with reduced survival in patients with melanoma. Cancer. 2018;124:3706–14.
Arbour KC, et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non-small-cell lung cancer. J Clin Oncol. 2018;36:2872–8.
Wei SC, et al. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discov. 2021;11:614–25.
Zamami Y, et al. Factors associated with immune checkpoint inhibitor-related myocarditis. JAMA Oncol. 2019;5:1635–7.
Lau ES, et al. Sex differences in circulating biomarkers of cardiovascular disease. J Am Coll Cardiol. 2019;74:1543–53.
Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int Immunol. 2007;19:337–43.
Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16:626–38.
Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J Neurosci Res. 2006;84:370–8.
Foks AC, Ran IA, Wasserman L, Frodermann V, Ter Borg MN, de Jager SC, van Santbrink PJ, Yagita H, Akiba H, Bot I, Kuiper J, van Puijvelde GH. T-cell immunoglobulin and mucin domain 3 acts as a negative regulator of atherosclerosis. Arterioscler Thromb Vasc Biol. 2013 Nov;33(11):2558-65. https://doi.org/10.1161/ATVBAHA.113.301879. Epub 2013 Aug 29. PMID: 23990206
Funding
Dr. H.Z. is supported by the National Institutes of Health grant 1K08HL16140501. P.C. is supported by AHA 20CDA35310303, NIH/NHLBI K08-HL153798. S.M.W. is supported by SMW the Joan and Sanford I Weill Scholar Award. P.K.N. reports grants from the American Heart Association (20TPA35500081) and NHLBI (5 R01 HL 134830-01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ronald Witteles serves as an advisory board participant for Pfizer, Alnylam, Ionis, AstraZeneca, Janssen, Intellia, NovoNordisk, and Alexion. Joel Neal reports personal fees from AstraZeneca, Exelixis, Eli Lilly and Company, Amgen, Iovance Biotherapeutics, Blueprint Pharmaceuticals, Regeneron Pharmaceuticals, Natera, Sanofi/Regeneron, D2G Oncology, Surface Oncology, Turning Point Therapeutics, Mirati Therapeutics, Gilead Sciences, Summit Therapeutics; personal fees and other from Genentech/Roche, Takeda Pharmaceuticals, Abbvie, Novartis, Novocure; other from Merck, Boehringer Ingelheim, Exelixis, Nektar Therapeutics, Adaptimmune, GSK, Janssen, CME Matters, Clinical Care Options CME, Research to Practice CME, Medscape CME, Biomedical Learning Institute CME, MLI Peerview CME, Prime Oncology CME, Projects in Knowledge CME, Rockpointe CME, MJH Life Sciences CME, Medical Educator Consortium, and HMP Education. Sarah Waliany serves as a consultant for AstraZeneca. The remaining authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chan, A., Torelli, S., Cheng, E. et al. Immunotherapy-Associated Atherosclerosis: A Comprehensive Review of Recent Findings and Implications for Future Research. Curr Treat Options Cardio Med 25, 715–735 (2023). https://doi.org/10.1007/s11936-023-01024-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11936-023-01024-0