
Abstract
Purpose of Review
Over the last few years, the scientific community has made significant progress in understanding the etiology of rheumatoid arthritis (RA). In this review, we summarize those key findings and trends.
Recent Findings
New data strongly implicates respiratory exposures, obesity, diet and microbiome, genetics, and their interactions in the etiology of RA. Furthermore, anti-posttranslationally modified protein antibodies (AMPAs) and abnormal glycosylation may be additional biomarkers for RA. Finally, functional genomics techniques implicate loss of certain macrophage populations and proliferation of synovial fibroblasts in RA.
Summary
These findings support the notion that RA originates at mucosal sites, augmented by genetic predisposition, and mediated by certain cell types including macrophages and fibroblasts. Weight loss, physical activity, and diet are additional modifiable factors beyond smoking cessation that can reduce risk of RA. Future epidemiologic and translational studies leveraging multi-omics approaches will help map the precise sequence of events in RA pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is one of the most common autoimmune diseases, and globally, its incidence is rising [1]. Although RA has been described for thousands of years, its etiology remained elusive until Caplan et al. described the first strong risk factor in the 1950s: coal exposure [2]. Three decades later in 1983, one of the first observations of a possible genetic association was published, showing 22% of RA patients had a first-degree relative with RA [3]. We now know that indeed having a first-degree relative with RA increases risk for RA nearly threefold [4, 5]. In 1987, another respiratory exposure, silica, was also found to be a risk factor for RA [6], with a recent studies confirming it increases odds of RA more than twofold [7, 8]. That same year in 1987, tobacco smoking was first found to increase risk of RA [9]. Multiple subsequent studies have shown tobacco smoking increases odds of RA over twofold, especially for individuals with increased smoking duration as compared to intensity [10, 11].
Compared to this relatively slow progress in understanding RA etiology from the twentieth century, recent years have seen a relative explosion in risk factors and mechanisms for RA etiology. In this manuscript, our goal is to synthesize the key findings and most significant trends from the past 5 years.
Respiratory Exposures
As described above, the first well-established risk factors for RA were respiratory exposures. One of the most significant trends from the past several years has been the discovery of additional respiratory triggers for RA. For example, an increasing number of studies have shown an association between asthma and development of incident RA [12], with a recent meta-analysis confirming this association [13]. The same group performed a meta-analysis of allergic rhinitis, finding a mild association with incident RA when high-quality studies alone were examined [14]. Viral infections were recently shown to increase risk of RA as well, which may be of particular relevance given the ongoing coronavirus pandemic [15]. Mycoplasma pneumonia was also recently linked to incident RA, especially in the elderly and the two years following infection [16]. Indeed, a large study within the Epidemiological Investigation of RA (EIRA) cohort in Sweden showed all types of respiratory diseases (acute and chronic, upper and lower) to be associated with incident RA with odds ratios of two- to threefold in nonsmokers [17].
Not only respiratory diseases but also other types of respiratory irritants have been increasingly shown to be associated with RA. A cohort study of 7600 Canadians showed an association between industrial fine particulate matter and ACPA positivity [18], whereas another cohort study in China showed a significant association between traffic-related air pollutants and RA readmissions [19]. In two large cohort studies, passive smoke [20] and fertilizer exposure [21] in childhood were also linked to RA. Indeed, among adults, inhaled respiratory exposures including not only fertilizers but also solvents and painting were associated with RA [22]. Furthermore, occupations with inhalational exposures such as bricklayers, concrete workers, and electrical workers are also associated with RA [23]. Together, these data provide further evidence for the importance of pulmonary irritation and/or inflammation in the pathogenesis of RA (Table 1).
Obesity
Another significant trend has been the increasing awareness that obesity contributes to increased risk of RA. A prior study had showed obesity might account for over half the increase in RA in the last several decades [24]. More recent studies have now shown elevated body mass index (BMI) not only increases risk of RA but also decreases time to RA [25]. In fact, among individuals with elevated BMI and at least two anti-citrullinated peptide antibodies (ACPA), odds for RA increased 23-fold compared to controls [25]. Another study also demonstrated that even being overweight (BMI>25) increased risk of RA, especially in adults less than 50 years of age [26]. Others have suggested the association between increased BMI and RA is greatest for women [27,28,29] or smokers [29].
Factors related to obesity may also increase risk of RA. A more recent meta-analysis showed not only BMI but also increased weight circumference increased risk of RA [27]. Along similar lines, a large cohort study within the Nurse’s Health Study showed that physical activity dramatically reduced the risk of later developing RA, with BMI mediating only 14% of this effect [30•]. Although no studies have demonstrated that weight loss reduces RA risk, a recent cohort study of 114,000 individuals showed that adherence to metformin was associated with decreased risk of RA [31]. Together, these findings point towards the exciting discovery that excess weight potentiates RA development. Thus, weight loss may be another modifiable risk factor for RA besides smoking.
Diet
Prompted by the observation that some patients with RA seem to flare with certain foods, studies in the last few decades began to investigate whether dietary factors could influence risk of RA. Indeed, some early studies showed that fish [32], omega-3 fatty acids [33], and modest alcohol consumption [34] all decreased risk of RA. However, recent studies have dampened early enthusiasm that specific foods may potentiate or protect against RA, showing no association with fish [35], alcohol [36], coffee [37], meat [38], or dairy products [38]. A study within the Studies of Etiology of RA (SERA) cohort found one potential reason for discordant results. They showed omega-3 fatty acids were inversely associated with rheumatoid factor (RF) and ACPA positivity, but only in patients with the human leukocyte antigen (HLA) shared epitope alleles [39]. Thus, diet and genetics may interact to incite RA in some fashion.
While specific foods may not yield strong associations with RA, certain dietary patterns have recently been shown to have a modest association with RA. For example, a large cohort study within the Nurse’s Health Study showed long-term dietary quality, as assessed by a scoring system for each food, was associated with reduced risk of RA in women. This association was especially true for women less than 55 years old and for seropositive RA [40]. A subsequent study showed that an inflammatory dietary pattern was again associated with increased risk of seropositive RA in women less than 55 years of age. However, this association was partially mediated by BMI [41]. Studies in other populations have also confirmed Mediterranean diet reduced risk of RA in smokers [42] as well as RA disease activity [43], whereas a Western dietary pattern increased risk of RA [44]. More research is needed to determine which components of these dietary patterns mediate the association with RA, and to what degree diet may mediate the association between obesity and RA.
Microbiome
One factor that may mediate the association between dietary pattern and RA risk is oral and/or intestinal dysbiosis, which has been increasingly implicated in RA pathogenesis. An increased prevalence of periodontal disease and altered oral microbiota profile in early-onset RA has been known for several years [45]. A recent work, however, showed that this alteration occurs before RA onset, with ACPA-positive at-risk individuals having a higher prevalence of periodontitis and P. gingivalis compared to both healthy controls and patients with early RA. Alterations of the gut microbiome in patients with RA have also been demonstrated for several years [46], especially with expansion of Prevotella species [47, 48]. In a recent landmark study, Alpizar-Rodriguez and colleagues expanded these findings to a pre-clinical RA group, demonstrating alterations of the gut microbiome, particularly enrichment of Prevotella species, compared to controls [49]. Observing these microbial changes before RA onset implicates oral/intestinal dysbiosis in the etiology of RA.
An important follow-up question is when and why the microbiome changes. While diet could be one reason as discussed above, antibiotics could be another. Two recent case-control studies showed that previous antibiotic exposure increased risk for RA in a dose-response fashion, with odds ratios of two- to threefold for RA for individuals with ten or more antibiotic prescriptions before RA onset [50, 51]. Importantly, this association was not mediated by the infections themselves, as respiratory infections without antibiotics were shown not to have as strong an association [50]. Chronic diarrhea was recently shown to increase risk of RA in a large cohort study within the E3N-EPIC study, especially in smokers [52].
Together, these findings support the notion that alterations in the microbiome may play a role in RA pathogenesis. More broadly, altered immunity at a mucosal site (e.g., intestines and/or lungs), in the context of a permissive genetic background, may be important for development of RA.
Genetics
A growing theme that has begun to permeate all the above trends is the pivotal role of genetics. Historically, twin studies suggested that the liability to RA was approximately 15% genetic [53]. However, increasing discovery of single nucleotide polymorphisms (SNPs) with genome-wide association studies (GWAS) show that genetics likely explain more, perhaps 30–40% of RA risk [54]. New risk loci for RA continue to be discovered [55, 56], including polymorphisms for interleukin-10 [57], IL1B [58], and T cell immunoglobulin and mucin domain 3 (TIM-3) [59]. Currently, the number of SNPs associated with RA totals over 269 [60]. Another study identified 243 phosphorylation-related SNPs, or missense SNPs that affect protein phosphorylation status [61].
As accessibility of genetic data expands, its usefulness has grown from serving not only as a risk factor for disease but also as a clinical and research tool. Importantly, a recently published genetic probability (“G-PROB”) tool calculates the probability of various types of inflammatory arthritis-causing diseases, improving correct diagnosis at presentation from 39 to 51% [62•]. This tool may be particularly useful for diagnosing individuals with inflammatory arthritis of unclear etiology, such as patients with seronegative RA. Genetic data has become an accessible research tool as well, for example, through Mendelian randomization studies [63]. These are observational studies that leverage the fact that SNPs are randomly assigned and always precede disease onset, thus acting similarly to a randomized controlled trial. For example, a recent Mendelian randomization study of over 850,000 Europeans confirmed that prediction of BMI based on a 806-gene profile did increase the risk of RA [64]. Finally, genetic data availability also enables creation of genetic risk scores [60]. Genetic risk scores are useful tools for performing gene-environment interaction studies, as outlined in the next section.
Gene-Environment Interactions
A significant trend in the last several years has been to study how the various genetic and environmental risk factors interact with each other, or so-called gene-environment interactions. The first to do this in the field of RA was Padyukov et al. in 2004, who identified an interaction between smoking and HLA-DRB1 for seropositive (RF-positive) RA [65]. Klareskog et al. expanded this discovery to the interaction between smoking and HLA-DRB1 shared epitope for ACPA-positive RA, which raised the risk for RA by an impressive 21-fold compared to nonsmokers without the shared epitope [66]. Many subsequent studies have replicated this interaction between the shared epitope and smoking, with recent studies suggesting that aryl hydrocarbon receptor crosstalk [67] and/or DNA methylation of cg21325723 [68] may underlie the mechanism of this interaction. The gene-smoking interaction in RA also varies by serological subset. That is, a recent study showed the effect of smoking on risk of RA varies by rheumatoid factor (RF) and anti-citric citrullinated peptide (CCP) status, as well as genetic status at the shared epitope [69]. Thus, defining an individual’s serological subtype may be important from a clinical and research perspective.
Only recently, researchers have begun to expand investigation of gene-environment interactions beyond simply smoking and the shared epitope. For example, one study found an association between textile dust exposure and HLA-DRB1 for RA risk [70]. Combining Epidemiological Investigation of RA and North American RA Consortium cohorts, another study found significant interactions between HLA-DRB1 SE alleles and SNPs associated with ACPA-positive RA [71]. Increasing the complexity even further, another group demonstrated a three way-interaction between alcohol, smoking, and HLA-DRB1 for RA risk [72]. Examining the interaction between other environmental exposures and genetic susceptibility to RA represents an unmet need in the field.
Mechanisms of Disease
While firm scientific evidence shows that genes, environmental exposures, and their interactions can all increase the risk of RA, a central question has become, how? Numerous mechanisms have been recently discovered that might explain some of the observed risk factors. To explain the association between respiratory disease and RA, one mechanism could be direct effects of respiratory pathogens; for example, EBV DNA was identified in synovial tissue of patients with RA [73]. To explain the association between obesity and RA, another study showed aggregates of three or more adipose tissue macrophages, called “crown-like structures,” were much more abundant in RA patients compared to controls. This finding was especially true in early RA and in patients with ACPA positivity [74]. Studies in the past year have also implicated mitochondrial dysfunction [75] and dendritic cells, especially the cDC1 subset, in initiation of inflammatory arthritis [76].
Mechanisms of Disease: Top-Down
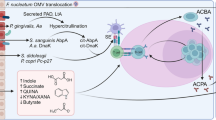
Taking a “top-down” approach to investigate the biological step before RA, called “pre-RA,” has also shed some insights into how RA develops (Fig. 1). Pre-RA is defined as the presence of RA-specific antibodies before clinical disease onset [77]. It confers an increased risk of RA, as high levels of antibodies can increase risk of RA over fourfold [78, 79]. RF was an early RA antibody to be discovered, followed eventually by ACPA, which encompasses all citrullinated peptides (both cyclic and non-cyclic). In fact, single-cell cloning of B cells from RA patients has identified 30 monoclonal ACPAs that are citrulline multi-specific [80]. However, the definition of pre-RA continues to expand beyond RF and ACPA positivity or negativity.

Current research efforts to understand the pathogenesis of RA use either a “top-down” approach, which focuses on pre-RA including anti-posttranslationally modified protein antibodies (AMPAs) and glycosylation, or alternatively a “bottom-up” approach, which focuses on functional genomics
Besides citrullination, other posttranslational modifications including carbamylation (also called homocitrullation) and acetylation may also produce antibodies involved in RA pathogenesis. The antibodies generated by these three processes are ACPAs, anti-carbamylated protein (anti-CarP) antibodies, and anti-lysine acetylated (KAc) antibodies, respectively. In turn, the collection of all these types of antibodies is now called anti-posttranslationally modified protein antibodies (AMPAs). To illustrate their importance, a study of individuals with arthralgia showed that not only RF and ACPA but also CarP increased risk of later diagnosis of inflammatory arthritis [81]. Indeed, a recent basic science study identified numerous carbamylated proteins in RA joints that were recognized by anti-CarP antibodies, supporting their role in RA pathogenesis [82]. In fact, a comprehensive set of antibodies against the entire human proteome, including the citrullinome and homocitrullinome, has been shown to identify close to 92% of all RA cases, compared to only 70% with commercial anti-CCP assays alone [83]. Importantly, monoclonal ACPA from patients was found to also be reactive with anti-CarP and anti-KAc antibodies, demonstrating “multi-reactivity” of these antibodies [84].Thus, the broader term “AMPAs” may be more heavily utilized in research and clinical settings moving forward.
Glycosylation of antibodies may be another form of “pre-RA.” For example, low galactosylation of IgG a median of 4 years before RA onset was associated with increased risk of RA [85]. In addition, glycosylation of IgG ACPA V-domain is also strongly associated with future development of RA, with a hazard ratio over 6 [86]. Like AMPAs, therefore, glycosylation may become a clinical biomarker for RA.
To understand how RA develops, investigators have sought to understand how pre-RA develops. From an epidemiologic standpoint, pre-RA has many of the same risk factors as RA, including female sex, increasing age, and smoking [87, 88]. Genetic predisposition to activation of adaptive immune responses to modified self-antigens may also play a role. For example, HLA-DRB1*14:02 broadens capacity for citrullination, self-peptide presentation, and T cell expansion, increasing risk for ACPA generation and ACPA-positive RA [89].
Finally, innate lymphoid cells are likely crucial for onset of pre-RA, and later RA. A study of individuals with pre-RA showed increased frequency of certain innate lymphoid cells compared to controls [90]. Another study of pre-RA patients showed that having ≥ 5 dominant B cell receptor clones were significantly associated with developing RA and appeared in the synovial tissue, suggesting their role in RA pathogenesis [91]. Furthermore, RA autoreactive B cells were highly multi-reactive, recognizing >3000 peptides modified by either citrullination or carbamylation [92]. This finding suggests that multiple antigen encounters across space and time are involved in the etiology of RA.
Mechanisms of Disease: Bottom-Up
Compared to the “top-down” approach of understanding RA etiology from a pre-RA perspective, another approach is the “bottom-up” approach via functional genomics. Functional genomics is defined as a biological field that attempts to describe gene functions and interactions. The word genomics refers to the genome and RNA sequencing techniques, whereas the word functional refers to the dynamic aspects of gene transcription, translation, and protein interactions rather than static features like DNA. In particular, single-cell RNA sequencing is a powerful technique that sequences genetic information from individual cells to understand their function in the context of their environment. For example, a recent study found that a transcriptome signature especially involving inflammatory pathways, Wnt signaling, and type I interferon increased ability to predict RA [93].
Functional genomics have also shown macrophages may play an important role in RA pathogenesis. Using single-cell RNA sequencing along with fluorescence microscopy, one study performed a spatiotemporal analysis of macrophages in RA tissues, finding that a certain population of tissue-resident macrophages (CX3CR1+) forms an internal barrier at the synovial lining that disappears in RA [94]. A subsequent study using the same technique in humans confirmed that certain macrophage types (MerTKpos) in synovial lining correlated with RA disease activity or remission. That is, they were lost during flare, and regained during remission [95].
Of all cell types however, fibroblasts in particular seem to play a pivotal role in RA etiology based on recent evidence. In a particularly ground-breaking work from the Accelerating Medicines Partnership, Zhang et al. used single-cell RNA sequencing, mass cytometry, and flow cytometry to define the cell populations that drive RA. Out of 18 unique cell populations, they found that sublining fibroblasts and IL1B+ proinflammatory monocytes were particularly expanded in RA synovia. In fact, IL6 largely came from the sublining fibroblasts and IL1B came from the proinflammatory monocytes, implicating these cell types in RA pathogenesis [96•]. A parallel study identified two key fibroblast types: synovial sublining fibroblasts and synovial lining fibroblasts. Proliferation of the former resulted in persistent inflammatory arthritis, whereas proliferation of the latter resembled osteoarthritis [97]. A subsequent study, again using single-cell RNA sequencing, found that the reason the sublining is expanded in RA is NOTCH3 signaling, which drives transcriptional gradients and synovial fibroblast differentiation to mediate inflammation and pathology in RA. They verified these results in a mouse model, where deletion of Notch3 or blocking it with monoclonal antibodies prevented joint damage in inflammatory arthritis [98].
These findings from functional genomics studies not only help explain RA etiology but also have identified discrete targets for its treatment. In particular, fibroblast subtypes may allow for clinical stratification and personalized treatment in RA. These findings may apply to other autoimmune diseases, as inflammatory fibroblasts have been implicated in several other autoimmune diseases including Sjögren’s, idiopathic pulmonary fibrosis, and ulcerative colitis.
Conclusions
In summary, research from the last few years has yielded a revolution in the understanding of RA etiology. In particular, acute and chronic respiratory exposures, obesity, diet and microbiome, genetics, and their interactions markedly increase risk of RA. A combination of these risk factors may be necessary for RA to develop, such as genetic predisposition, compounded by alterations in immunity by obesity, and then ultimately triggered by an insult at a mucosal site such as pulmonary infection or intestinal dysbiosis. Similar risk factors also increase risk of pre-RA, a pre-clinical state characterized by a growing repertoire of anti-posttranslationally modified protein antibodies as well as abnormal glycosylation. Finally, functional genomics approaches have revealed that disruption of synovial macrophages and proliferation of synovial sublining fibroblasts may play a mechanistic role in the pathogenesis of RA. Future epidemiologic, serologic, and transcriptomic studies will likely help to further refine our understanding of RA and its etiology.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Jin Z, Wang D, Zhang H, Liang J, Feng X, Zhao J, et al. Incidence trend of five common musculoskeletal disorders from 1990 to 2017 at the global, regional and national level: results from the global burden of disease study 2017. Ann Rheum Dis. 2020;79(8):1014–22. https://doi.org/10.1136/annrheumdis-2020-217050.
Caplan A. Certain unusual radiological appearances in the chest of coal-miners suffering from rheumatoid arthritis. Thorax. 1953;8(1):29–37. https://doi.org/10.1136/thx.8.1.29.
Thomas DJ, Young A, Gorsuch AN, Bottazzo GF, Cudworth AG. Evidence for an association between rheumatoid arthritis and autoimmune endocrine disease. Ann Rheum Dis. 1983;42(3):297–300. https://doi.org/10.1136/ard.42.3.297.
Frisell T, Holmqvist M, Kallberg H, Klareskog L, Alfredsson L, Askling J. Familial risks and heritability of rheumatoid arthritis: role of rheumatoid factor/anti-citrullinated protein antibody status, number and type of affected relatives, sex, and age. Arthritis Rheum. 2013;65(11):2773–82. https://doi.org/10.1002/art.38097.
Kronzer VL, Crowson CS, Sparks JA, Myasoedova E, Davis J 3rd. Family history of rheumatologic, autoimmune, and non-autoimmune diseases and risk of rheumatoid arthritis. Arthritis Care Res (Hoboken). 2019;73:180–7. https://doi.org/10.1002/acr.24115.
Klockars M, Koskela RS, Järvinen E, Kolari PJ, Rossi A. Silica exposure and rheumatoid arthritis: a follow up study of granite workers 1940-81. Br Med J (Clin Res Ed). 1987;294(6578):997–1000. https://doi.org/10.1136/bmj.294.6578.997.
Mehri F, Jenabi E, Bashirian S, Shahna FG, Khazaei S. The association Between Occupational Exposure to silica and Risk of Developing Rheumatoid Arthritis: A Meta-Analysis. Saf Health Work. 2020;11(2):136–42. https://doi.org/10.1016/j.shaw.2020.02.001.
Ilar A, Klareskog L, Saevarsdottir S, Wiebert P, Askling J, Gustavsson P, et al. Occupational exposure to asbestos and silica and risk of developing rheumatoid arthritis: findings from a Swedish population-based case-control study. RMD Open. 2019;5(2):e000978. https://doi.org/10.1136/rmdopen-2019-000978.
Vessey MP, Villard-Mackintosh L, Yeates D. Oral contraceptives, cigarette smoking and other factors in relation to arthritis. Contraception. 1987;35(5):457–64. https://doi.org/10.1016/0010-7824(87)90082-5.
Hedström AK, Stawiarz L, Klareskog L, Alfredsson L. Smoking and susceptibility to rheumatoid arthritis in a Swedish population-based case-control study. Eur J Epidemiol. 2018;33(4):415–23. https://doi.org/10.1007/s10654-018-0360-5.
Svendsen AJ, Junker P, Houen G, Kyvik KO, Nielsen C, Skytthe A, et al. Incidence of Chronic Persistent Rheumatoid Arthritis and the Impact of Smoking: A Historical Twin Cohort Study. Arthritis Care Res (Hoboken). 2017;69(5):616–24. https://doi.org/10.1002/acr.22987.
Kronzer VL, Crowson CS, Sparks JA, Vassallo R, Davis JM III. Investigating asthma, allergic disease, passive smoke exposure, and risk of rheumatoid arthritis. Arthritis Rheumatol. 2019;71:1217–24. https://doi.org/10.1002/art.40858.
Charoenngam N, Ponvilawan B, Rittiphairoj T, Tornsatitkul S, Wattanachayakul P, Rujirachun P, et al. Patients with asthma have a higher risk of rheumatoid arthritis: A systematic review and meta-analysis. Semin Arthritis Rheum. 2020;50(5):968–76. https://doi.org/10.1016/j.semarthrit.2020.07.015.
Charoenngam N, Ponvilawan B, Rittiphairoj T, Tornsatitkul S, Wattanachayakul P, Rujirachun P, et al. The association between allergic rhinitis and risk of rheumatoid arthritis: A systematic review and meta-analysis. J Evid Based Med. 2020. https://doi.org/10.1111/jebm.12393.
Joo YB, Lim YH, Kim KJ, Park KS, Park YJ. Respiratory viral infections and the risk of rheumatoid arthritis. Arthritis Res Ther. 2019;21(1):199. https://doi.org/10.1186/s13075-019-1977-9.
Chu KA, Chen W, Hsu CY, Hung YM, Wei JC. Increased risk of rheumatoid arthritis among patients with Mycoplasma pneumonia: A nationwide population-based cohort study in Taiwan. PLoS One. 2019;14(1):e0210750. https://doi.org/10.1371/journal.pone.0210750.
Kronzer VL, Westerlind H, Alfredsson L, Crowson CS, Nyberg F, Tornling G, et al. Respiratory diseases as risk factors for seropositive and seronegative rheumatoid arthritis and in relation to smoking. Arthritis Rheumatol. 2020;73:61–8. https://doi.org/10.1002/art.41491.
Zhao N, Smargiassi A, Hatzopoulou M, Colmegna I, Hudson M, Fritzler MJ, et al. Long-term exposure to a mixture of industrial SO(2), NO(2), and PM(2.5) and anti-citrullinated protein antibody positivity. Environ Health. 2020;19(1):86. https://doi.org/10.1186/s12940-020-00637-3.
Wu Q, Xu Z, Dan YL, Cheng J, Zhao CN, Mao YM, et al. Association between traffic-related air pollution and hospital readmissions for rheumatoid arthritis in Hefei, China: A time-series study. Environ Pollut. 2020;268(Pt A):115628. https://doi.org/10.1016/j.envpol.2020.115628.
Seror R, Henry J, Gusto G, Aubin H, Boutron-Ruault M, Mariette X. Passive smoking in childhood increases risk of developing rheumatoid arthritis. Rheumatology. 2018;58(7):1154–62.
Parks CG, D’Aloisio AA, Sandler DP. Childhood Residential and Agricultural Pesticide Exposures in Relation to Adult-Onset Rheumatoid Arthritis in Women. Am J Epidemiol. 2018;187(2):214–23. https://doi.org/10.1093/aje/kwx224.
Parks CG, Meyer A, Beane Freeman LE, Hofmann JN, Sandler DP. Farming tasks and the development of rheumatoid arthritis in the agricultural health study. Occup Environ Med. 2019;76(4):243–9. https://doi.org/10.1136/oemed-2018-105361.
Ilar A, Alfredsson L, Wiebert P, Klareskog L, Bengtsson C. Occupation and Risk of Developing Rheumatoid Arthritis: Results From a Population-Based Case-Control Study. Arthritis Care Res (Hoboken). 2018;70(4):499–509. https://doi.org/10.1002/acr.23321.
Crowson CS, Matteson EL, Davis JM 3rd, Gabriel SE. Contribution of obesity to the rise in incidence of rheumatoid arthritis. Arthritis Care Res (Hoboken). 2013;65(1):71–7. https://doi.org/10.1002/acr.21660.
Tedeschi SK, Cui J, Arkema EV, Robinson WH, Sokolove J, Lingampalli N, et al. Elevated BMI and antibodies to citrullinated proteins interact to increase rheumatoid arthritis risk and shorten time to diagnosis: A nested case-control study of women in the Nurses’ Health Studies. Semin Arthritis Rheum. 2017;46(6):692–8. https://doi.org/10.1016/j.semarthrit.2016.09.001.
Kokkonen H, Stenlund H, Rantapää-Dahlqvist S. Cardiovascular risk factors predate the onset of symptoms of rheumatoid arthritis: a nested case-control study. Arthritis Res Ther. 2017;19(1):148. https://doi.org/10.1186/s13075-017-1351-8.
Ohno T, Aune D, Heath AK. Adiposity and the risk of rheumatoid arthritis: a systematic review and meta-analysis of cohort studies. Sci Rep. 2020;10(1):16006. https://doi.org/10.1038/s41598-020-71676-6.
Linauskas A, Overvad K, Symmons D, Johansen MB, Stengaard-Pedersen K, de Thurah A. Body Fat Percentage, Waist Circumference, and Obesity As Risk Factors for Rheumatoid Arthritis: A Danish Cohort Study. Arthritis Care Res (Hoboken). 2019;71(6):777–86. https://doi.org/10.1002/acr.23694.
Hedström AK, Klareskog L, Alfredsson L. Interplay between obesity and smoking with regard to RA risk. RMD Open. 2019;5(1):e000856. https://doi.org/10.1136/rmdopen-2018-000856.
• Liu X, Tedeschi SK, Lu B, Zaccardelli A, Speyer CB, Costenbader KH, et al. Long-Term Physical Activity and Subsequent Risk for Rheumatoid Arthritis Among Women: A Prospective Cohort Study. Arthritis Rheumatol. 2019;71(9):1460–71. https://doi.org/10.1002/art.40899. This prospective cohort study of 113,000 women showed that physical activity in the 2-8 years prior to RA diagnosis reduced risk of RA, with a hazard ratio of 0.67 for the highest level of physical activity compared to the lowest. This finding provides another modifiable risk factor for RA besides smoking and weight loss.
Naffaa ME, Rosenberg V, Watad A, Tiosano S, Yavne Y, Chodick G, et al. Adherence to metformin and the onset of rheumatoid arthritis: a population-based cohort study. Scand J Rheumatol. 2020;49(3):173–80. https://doi.org/10.1080/03009742.2019.1695928.
Rosell M, Wesley AM, Rydin K, Klareskog L, Alfredsson L. Dietary fish and fish oil and the risk of rheumatoid arthritis. Epidemiology. 2009;20(6):896–901. https://doi.org/10.1097/EDE.0b013e3181b5f0ce.
Di Giuseppe D, Wallin A, Bottai M, Askling J, Wolk A. Long-term intake of dietary long-chain n-3 polyunsaturated fatty acids and risk of rheumatoid arthritis: a prospective cohort study of women. Ann Rheum Dis. 2014;73(11):1949–53. https://doi.org/10.1136/annrheumdis-2013-203338.
Jin Z, Xiang C, Cai Q, Wei X, He J. Alcohol consumption as a preventive factor for developing rheumatoid arthritis: a dose-response meta-analysis of prospective studies. Ann Rheum Dis. 2014;73(11):1962–7. https://doi.org/10.1136/annrheumdis-2013-203323.
Sparks JA, O’Reilly ÉJ, Barbhaiya M, Tedeschi SK, Malspeis S, Lu B, et al. Association of fish intake and smoking with risk of rheumatoid arthritis and age of onset: a prospective cohort study. BMC Musculoskelet Disord. 2019;20(1):2. https://doi.org/10.1186/s12891-018-2381-3.
Baker JF, England BR, Mikuls TR, Hsu JY, George MD, Pedro S, et al. Changes in Alcohol Use and Associations With Disease Activity, Health Status, and Mortality in Rheumatoid Arthritis. Arthritis Care Res (Hoboken). 2020;72(3):301–8. https://doi.org/10.1002/acr.23847.
Lamichhane D, Collins C, Constantinescu F, Walitt B, Pettinger M, Parks C, et al. Coffee and Tea Consumption in Relation to Risk of Rheumatoid Arthritis in the Women’s Health Initiative Observational Cohort. J Clin Rheumatol. 2019;25(3):127–32. https://doi.org/10.1097/rhu.0000000000000788.
Sundström B, Ljung L, Di Giuseppe D. Consumption of Meat and Dairy Products Is Not Associated with the Risk for Rheumatoid Arthritis among Women: A Population-Based Cohort Study. Nutrients. 2019;11(11):2825. https://doi.org/10.3390/nu11112825.
Gan RW, Demoruelle MK, Deane KD, Weisman MH, Buckner JH, Gregersen PK, et al. Omega-3 fatty acids are associated with a lower prevalence of autoantibodies in shared epitope-positive subjects at risk for rheumatoid arthritis. Ann Rheum Dis. 2017;76(1):147–52. https://doi.org/10.1136/annrheumdis-2016-209154.
Hu Y, Sparks JA, Malspeis S, Costenbader KH, Hu FB, Karlson EW, et al. Long-term dietary quality and risk of developing rheumatoid arthritis in women. Ann Rheum Dis. 2017;76(8):1357–64. https://doi.org/10.1136/annrheumdis-2016-210431.
Sparks JA, Barbhaiya M, Tedeschi SK, Leatherwood CL, Tabung FK, Speyer CB, et al. Inflammatory dietary pattern and risk of developing rheumatoid arthritis in women. Clin Rheumatol. 2019;38(1):243–50. https://doi.org/10.1007/s10067-018-4261-5.
Nguyen Y, Salliot C, Gelot A, Gambaretti J, Mariette X, Boutron-Ruault MC, et al. Mediterranean diet and risk of rheumatoid arthritis: findings from the French E3N-EPIC cohort study. Arthritis Rheumatol. 2020;73:69–77. https://doi.org/10.1002/art.41487.
Nelson J, Sjöblom H, Gjertsson I, Ulven SM, Lindqvist HM, Bärebring L. Do Interventions with Diet or Dietary Supplements Reduce the Disease Activity Score in Rheumatoid Arthritis? A Systematic Review of Randomized Controlled Trials. Nutrients. 2020;12(10):2991. https://doi.org/10.3390/nu12102991.
Nezamoleslami S, Ghiasvand R, Feizi A, Salesi M, Pourmasoumi M. The relationship between dietary patterns and rheumatoid arthritis: a case-control study. Nutr Metab (Lond). 2020;17:75. https://doi.org/10.1186/s12986-020-00502-7.
Scher JU, Ubeda C, Equinda M, Khanin R, Buischi Y, Viale A, et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012;64(10):3083–94. https://doi.org/10.1002/art.34539.
Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016;8(1):43. https://doi.org/10.1186/s13073-016-0299-7.
Pianta A, Arvikar S, Strle K, Drouin EE, Wang Q, Costello CE, et al. Evidence of the Immune Relevance of Prevotella copri, a Gut Microbe, in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2017;69(5):964–75. https://doi.org/10.1002/art.40003.
Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. https://doi.org/10.7554/eLife.01202.
Alpizar-Rodriguez D, Lesker TR, Gronow A, Gilbert B, Raemy E, Lamacchia C, et al. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann Rheum Dis. 2019;78(5):590–3. https://doi.org/10.1136/annrheumdis-2018-214514.
Sultan AA, Mallen C, Muller S, Hider S, Scott I, Helliwell T, et al. Antibiotic use and the risk of rheumatoid arthritis: a population-based case-control study. BMC Med. 2019;17(1):154. https://doi.org/10.1186/s12916-019-1394-6.
Armstrong D, Dregan A, Ashworth M, White P, McGee C, de Lusignan S. Influence of prior antibiotic use on risk of rheumatoid arthritis: case control study in general practice. Rheumatology (Oxford). 2020;59(6):1281–7. https://doi.org/10.1093/rheumatology/kez452.
Nguyen Y, Mariette X, Salliot C, Gusto G, Boutron-Ruault MC, Seror R. Chronic diarrhoea and risk of rheumatoid arthritis: findings from the French E3N-EPIC Cohort Study. Rheumatology (Oxford). 2020;59:3767–75. https://doi.org/10.1093/rheumatology/keaa133.
Svendsen AJ, Kyvik KO, Houen G, Junker P, Christensen K, Christiansen L, et al. On the origin of rheumatoid arthritis: the impact of environment and genes--a population based twin study. PLoS One. 2013;8(2):e57304. https://doi.org/10.1371/journal.pone.0057304.
Stahl EA, Wegmann D, Trynka G, Gutierrez-Achury J, Do R, Voight BF, et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet. 2012;44(5):483–9. https://doi.org/10.1038/ng.2232.
Zhou R, Lin X, Li DY, Wang XF, Greenbaum J, Chen YC, et al. Identification of novel genetic loci for osteoporosis and/or rheumatoid arthritis using cFDR approach. PLoS One. 2017;12(8):e0183842. https://doi.org/10.1371/journal.pone.0183842.
Mo XB, Sun YH, Zhang YH, Lei SF. Integrative analysis highlighted susceptibility genes for rheumatoid arthritis. Int Immunopharmacol. 2020;86:106716. https://doi.org/10.1016/j.intimp.2020.106716.
Liu Q, Yang J, He H, Yu Y, Lyu J. Associations between interleukin-10 polymorphisms and susceptibility to rheumatoid arthritis: a meta-analysis and meta-regression. Clin Rheumatol. 2018;37(12):3229–37. https://doi.org/10.1007/s10067-018-4329-2.
Rong H, He X, Wang L, Bai M, Jin T, Wang Y, et al. Association between IL1B polymorphisms and the risk of rheumatoid arthritis. Int Immunopharmacol. 2020;83:106401. https://doi.org/10.1016/j.intimp.2020.106401.
Zhang R, Li H, Bai L, Duan J. Association between T-Cell Immunoglobulin and Mucin Domain 3 (TIM-3) Genetic Polymorphisms and Susceptibility to Autoimmune Diseases. Immunol Invest. 2019;48(6):563–76. https://doi.org/10.1080/08820139.2019.1599009.
Rostami S, Hoff M, Brown MA, Hveem K, Videm V. Comparison of methods to construct a genetic risk score for prediction of rheumatoid arthritis in the population-based Nord-Trondelag Health Study, Norway. Rheumatology (Oxford). 2020;59:1743–51. https://doi.org/10.1093/rheumatology/kez638.
Mo X, Guo Y, Qian Q, Fu M, Zhang H. Phosphorylation-related SNPs influence lipid levels and rheumatoid arthritis risk by altering gene expression and plasma protein levels. Rheumatology (Oxford). 2020;59(4):889–98. https://doi.org/10.1093/rheumatology/kez466.
• Knevel R, le Cessie S, Terao CC, Slowikowski K, Cui J, Huizinga TWJ, et al. Using genetics to prioritize diagnoses for rheumatology outpatients with inflammatory arthritis. Sci Transl Med. 2020;12(545):eaay1548. https://doi.org/10.1126/scitranslmed.aay1548. This study created a genetic risk score for various diseases that cause inflammatory arthritis and then tested it in three cohorts containing a total of 1700 patients. Genetic probabilities discriminated true disease well (area under the curve of 0.7-0.8 in each cohort), providing clinicians with a useful tool to improve disease diagnosis at presentation.
Jiang X, Alfredsson L. Modifiable environmental exposure and risk of rheumatoid arthritis-current evidence from genetic studies. Arthritis Res Ther. 2020;22(1):154. https://doi.org/10.1186/s13075-020-02253-5.
Tang B, Shi H, Alfredsson L, Klareskog L, Padyukov L, Jiang X. Obesity-related traits and the development of rheumatoid arthritis - evidence from genetic data. Arthritis Rheumatol. 2020;73:203–11. https://doi.org/10.1002/art.41517.
Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50(10):3085–92. https://doi.org/10.1002/art.20553.
Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54(1):38–46. https://doi.org/10.1002/art.21575.
Fu J, Nogueira SV, Drongelen VV, Coit P, Ling S, Rosloniec EF, et al. Shared epitope-aryl hydrocarbon receptor crosstalk underlies the mechanism of gene-environment interaction in autoimmune arthritis. Proc Natl Acad Sci U S A. 2018;115(18):4755–60. https://doi.org/10.1073/pnas.1722124115.
Meng W, Zhu Z, Jiang X, Too CL, Uebe S, Jagodic M, et al. DNA methylation mediates genotype and smoking interaction in the development of anti-citrullinated peptide antibody-positive rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):71. https://doi.org/10.1186/s13075-017-1276-2.
Hedstrom AK, Ronnelid J, Klareskog L, Alfredsson L. Complex Relationships of Smoking, HLA-DRB1 Genes, and Serologic Profiles in Patients With Early Rheumatoid Arthritis: Update From a Swedish Population-Based Case-Control Study. Arthritis Rheumatol. 2019;71(9):1504–11. https://doi.org/10.1002/art.40852.
Too CL, Muhamad NA, Ilar A, Padyukov L, Alfredsson L, Klareskog L, et al. Occupational exposure to textile dust increases the risk of rheumatoid arthritis: results from a Malaysian population-based case-control study. Ann Rheum Dis. 2016;75(6):997–1002. https://doi.org/10.1136/annrheumdis-2015-208278.
Diaz-Gallo LM, Ramsköld D, Shchetynsky K, Folkersen L, Chemin K, Brynedal B, et al. Systematic approach demonstrates enrichment of multiple interactions between non-HLA risk variants and HLA-DRB1 risk alleles in rheumatoid arthritis. Ann Rheum Dis. 2018;77(10):1454–62. https://doi.org/10.1136/annrheumdis-2018-213412.
Hedström AK, Hössjer O, Klareskog L, Alfredsson L. Interplay between alcohol, smoking and HLA genes in RA aetiology. RMD Open. 2019;5(1):e000893. https://doi.org/10.1136/rmdopen-2019-000893.
Masuoka S, Kusunoki N, Takamatsu R, Takahashi H, Tsuchiya K, Kawai S, et al. Epstein-Barr virus infection and variants of Epstein-Barr nuclear antigen-1 in synovial tissues of rheumatoid arthritis. PLoS One. 2018;13(12):e0208957. https://doi.org/10.1371/journal.pone.0208957.
Giles JT, Ferrante AW, Broderick R, Zartoshti A, Rose J, Downer K, et al. Adipose Tissue Macrophages in Rheumatoid Arthritis: Prevalence, Disease-Related Indicators, and Associations With Cardiometabolic Risk Factors. Arthritis Care Res (Hoboken). 2018;70(2):175–84. https://doi.org/10.1002/acr.23253.
Khanna S, Padhan P, Jaiswal KS, Jain AP, Ghosh A, Tripathy A, et al. Altered mitochondrial proteome and functional dynamics in patients with rheumatoid arthritis. Mitochondrion. 2020;54:8–14. https://doi.org/10.1016/j.mito.2020.06.005.
Ramos MI, Garcia S, Helder B, Aarrass S, Reedquist KA, Jacobsen SE, et al. cDC1 are required for the initiation of collagen-induced arthritis. J Transl Autoimmun. 2020;3:100066. https://doi.org/10.1016/j.jtauto.2020.100066.
Gerlag DM, Raza K, van Baarsen LG, Brouwer E, Buckley CD, Burmester GR, et al. EULAR recommendations for terminology and research in individuals at risk of rheumatoid arthritis: report from the Study Group for Risk Factors for Rheumatoid Arthritis. Ann Rheum Dis. 2012;71(5):638–41. https://doi.org/10.1136/annrheumdis-2011-200990.
Ford JA, Liu X, Marshall AA, Zaccardelli A, Prado MG, Wiyarand C, et al. Impact of Cyclic Citrullinated Peptide Antibody Level on Progression to Rheumatoid Arthritis in Clinically Tested Cyclic Citrullinated Peptide Antibody-Positive Patients Without Rheumatoid Arthritis. Arthritis Care Res (Hoboken). 2019;71(12):1583–92. https://doi.org/10.1002/acr.23820.
Bemis EA, Demoruelle MK, Seifert JA, Polinski KJ, Weisman MH, Buckner JH, et al. Factors associated with progression to inflammatory arthritis in first-degree relatives of individuals with RA following autoantibody positive screening in a non-clinical setting. Ann Rheum Dis. 2020;80:154–61. https://doi.org/10.1136/annrheumdis-2020-217066.
Titcombe PJ, Wigerblad G, Sippl N, Zhang N, Shmagel AK, Sahlström P, et al. Pathogenic Citrulline-Multispecific B Cell Receptor Clades in Rheumatoid Arthritis. Arthritis Rheumatol. 2018;70(12):1933–45. https://doi.org/10.1002/art.40590.
Ten Brinck RM, van Steenbergen HW, van Delft MAM, Verheul MK, Toes REM, Trouw LA, et al. The risk of individual autoantibodies, autoantibody combinations and levels for arthritis development in clinically suspect arthralgia. Rheumatology (Oxford). 2017;56(12):2145–53. https://doi.org/10.1093/rheumatology/kex340.
Verheul MK, Janssen GMC, de Ru A, Stoeken-Rijsbergen G, Levarht EWN, Kwekkeboom JC, et al. Mass-spectrometric identification of carbamylated proteins present in the joints of rheumatoid arthritis patients and controls. Clin Exp Rheumatol. 2020.
Lo KC, Sullivan E, Bannen RM, Jin H, Rowe M, Li H, et al. Comprehensive Profiling of the Rheumatoid Arthritis Antibody Repertoire. Arthritis Rheumatol. 2020;72(2):242–50. https://doi.org/10.1002/art.41089.
Sahlström P, Hansson M, Steen J, Amara K, Titcombe PJ, Forsström B, et al. Different Hierarchies of Anti-Modified Protein Autoantibody Reactivities in Rheumatoid Arthritis. Arthritis Rheumatol. 2020;72:1643–57. https://doi.org/10.1002/art.41385.
Gudelj I, Salo PP, Trbojević-Akmačić I, Albers M, Primorac D, Perola M, et al. Low galactosylation of IgG associates with higher risk for future diagnosis of rheumatoid arthritis during 10 years of follow-up. Biochim Biophys Acta Mol Basis Dis. 2018;1864(6 Pt A):2034–9. https://doi.org/10.1016/j.bbadis.2018.03.018.
Hafkenscheid L, de Moel E, Smolik I, Tanner S, Meng X, Jansen BC, et al. N-Linked Glycans in the Variable Domain of IgG Anti-Citrullinated Protein Antibodies Predict the Development of Rheumatoid Arthritis. Arthritis Rheumatol. 2019;71(10):1626–33. https://doi.org/10.1002/art.40920.
Alpizar-Rodriguez D, Brulhart L, Mueller RB, Möller B, Dudler J, Ciurea A, et al. The prevalence of anticitrullinated protein antibodies increases with age in healthy individuals at risk for rheumatoid arthritis. Clin Rheumatol. 2017;36(3):677–82. https://doi.org/10.1007/s10067-017-3547-3.
van Zanten A, Arends S, Roozendaal C, Limburg PC, Maas F, Trouw LA, et al. Presence of anticitrullinated protein antibodies in a large population-based cohort from the Netherlands. Ann Rheum Dis. 2017;76(7):1184–90. https://doi.org/10.1136/annrheumdis-2016-209991.
Scally SW, Law SC, Ting YT, Heemst JV, Sokolove J, Deutsch AJ, et al. Molecular basis for increased susceptibility of Indigenous North Americans to seropositive rheumatoid arthritis. Ann Rheum Dis. 2017;76(11):1915–23. https://doi.org/10.1136/annrheumdis-2017-211300.
Rodríguez-Carrio J, Hähnlein JS, Ramwadhdoebe TH, Semmelink JF, Choi IY, van Lienden KP, et al. Brief Report: Altered Innate Lymphoid Cell Subsets in Human Lymph Node Biopsy Specimens Obtained During the At-Risk and Earliest Phases of Rheumatoid Arthritis. Arthritis Rheumatol. 2017;69(1):70–6. https://doi.org/10.1002/art.39811.
Tak PP, Doorenspleet ME, de Hair MJH, Klarenbeek PL, van Beers-Tas MH, van Kampen AHC, et al. Dominant B cell receptor clones in peripheral blood predict onset of arthritis in individuals at risk for rheumatoid arthritis. Ann Rheum Dis. 2017;76(11):1924–30. https://doi.org/10.1136/annrheumdis-2017-211351.
Steen J, Forsström B, Sahlström P, Odowd V, Israelsson L, Krishnamurthy A, et al. Recognition of Amino Acid Motifs, Rather Than Specific Proteins, by Human Plasma Cell-Derived Monoclonal Antibodies to Posttranslationally Modified Proteins in Rheumatoid Arthritis. Arthritis Rheumatol. 2019;71(2):196–209. https://doi.org/10.1002/art.40699.
Macías-Segura N, Castañeda-Delgado JE, Bastian Y, Santiago-Algarra D, Castillo-Ortiz JD, Alemán-Navarro AL, et al. Transcriptional signature associated with early rheumatoid arthritis and healthy individuals at high risk to develop the disease. PLoS One. 2018;13(3):e0194205. https://doi.org/10.1371/journal.pone.0194205.
Culemann S, Grüneboom A, Nicolás-Ávila J, Weidner D, Lämmle KF, Rothe T, et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature. 2019;572(7771):670–5. https://doi.org/10.1038/s41586-019-1471-1.
Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med. 2020;26(8):1295–306. https://doi.org/10.1038/s41591-020-0939-8.
• Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. 2019;20(7):928–42. https://doi.org/10.1038/s41590-019-0378-1. This study performed single-cell transcriptomics and mass cytometry on 51 samples of synovial tissue from patients with RA or osteoarthritis to identify which cell populations drive joint inflammation. They identified certain types of monocytes and fibroblasts as key mediators of inflammatory arthritis.
Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570(7760):246–51. https://doi.org/10.1038/s41586-019-1263-7.
Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. 2020;582(7811):259–64. https://doi.org/10.1038/s41586-020-2222-z.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
Vanessa Kronzer declares that she has no conflicts of interest.
John Davis reports a research grant from Pfizer and has participated on advisory boards for Abbvie and Sanofi-Genzyme, outside the submitted work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Rheumatoid Arthritis
Rights and permissions
About this article

Cite this article
Kronzer, V.L., Davis, J.M. Etiologies of Rheumatoid Arthritis: Update on Mucosal, Genetic, and Cellular Pathogenesis. Curr Rheumatol Rep 23, 21 (2021). https://doi.org/10.1007/s11926-021-00993-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s11926-021-00993-0