
Abstract
Glucocorticoid-induced osteoporosis (GIOP) is one of the most important side effects of glucocorticoid use, as it leads to an increased risk of fractures. Recently, many published studies have focused on the cellular and molecular mechanisms of bone metabolism, the pathophysiology of GIOP, and the intervention options to prevent GIOP. In this review, recent advances in GIOP are summarized, particularly recent progress in our understanding of the mechanisms of GIOP resulting in improved insight that might result in the development of new treatment options in the near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucocorticoids (GCs) are used frequently in a variety of diseases because of their strong anti-inflammatory and immunosuppressive effects. However, corticosteroids have many metabolic side effects, such as insulin resistance, hypertension, glaucoma, and osteoporosis. GC-induced osteoporosis (GIOP) is one of the most devastating side effects because bone loss during long-term GC treatment is generally irreversible and because of its clinical manifestations (eg, vertebral and nonvertebral fractures). In recent years, considerable improvements have been made in our understanding of the pathogenesis of GIOP. Furthermore, new trials report beneficial effects of antiresorptive and anabolic agents. This article reviews the current epidemiology and pathophysiology of GIOP and discusses the treatment possibilities.
Epidemiology
GCs are frequently prescribed in patients with a wide variety of chronic diseases, such as rheumatoid arthritis, polymyalgia rheumatica, inflammatory bowel disease, and chronic obstructive pulmonary disease. It is estimated that 3% of the population 50 years of age and older has used GCs, and this percentage rises to 5.2% among those 80 years of age and older [1]. Treatment with GCs results in bone loss and increased risk of vertebral and nonvertebral fractures [2]. Earlier data demonstrated that 30% of patients with long-term GC use (>6 months) would develop osteoporosis [3]. The bone loss is more pronounced in the trabecular bone, which can be found predominantly in the spine and ribs, and starts within months after initiation of therapy [2, 4]. Earlier studies showed that the bone loss is biphasic, with a rapid initial phase of 3% to 5% in the first year of GC treatment, followed by a slower phase during continued use of 0.5% to 1% annually [5, 6, 7••].
Furthermore, GC use increases the risk of fractures, an effect that appears to be dose dependent [8, 9]. Up to 30% of patients on chronic GC treatment will have an incident fracture [3]. The risk of vertebral fractures is particularly elevated—two to five times, depending on the daily dosage of prednisone; this increase occurs as soon as 3 months after treatment has started [10]. After cessation of GC treatment, the fracture risk gradually returns to baseline and therefore seems to be partially reversible [11]. However, in patients using high doses (>1 g), it took more than 15 months to return to baseline fracture risk. The risk of fractures is increased for women and men and is age dependent. Furthermore, other determinants of bone loss, such as smoking, immobility, and the activity of the underlying disease, also play a role in the increased fracture risk [12].
Although there is an apparent effect of GCs on bone density and fracture risk, the risk of fractures cannot be explained completely by the changes in bone mineral density (BMD). Besides bone loss, an increased fracture risk is supposedly caused by a reduced bone quality [13]. Earlier studies have pointed toward a change (decrease) in the so-called “bone density threshold for vertebral fractures” [14, 15]. After a follow-up of 1 year, patients on GC treatment had significantly more vertebral fractures than controls, while their BMD values were higher.
Mechanisms of Glucocorticoid Action
In recent years, the general mechanism of GC action has become more clear. GCs exert their actions via genomic and nongenomic pathways. Most of their important therapeutic effects are exerted via genomic mechanisms by binding of the GC to cytosolic GC receptors (cGCRs). After binding to the cGCR, the GC/glucocorticoid (GCR) complex translocates to the nucleus, where binding to specific transcription factors leads to induction or inhibition of transcription of certain genes, called transactivation and transrepression, respectively. The GC/GCR complex acts directly with the transcription factors activator protein 1 (AP-1) and nuclear factor-κB, which have an important role in the regulation of many inflammatory genes [16, 17]. Although most of the therapeutic effects of GCs take place by transrepression, transactivation is responsible for a considerable number of metabolic side effects of GC treatment.
Apart from genomic actions, nongenomic GC mechanisms can exert rapid clinical effects, which can be divided into three distinct mechanisms [18]. First, GCs exert effects mediated by the cGCR. The binding of GC to cGCR promotes the release of signaling molecules, causing rapid, non-nuclear actions. Second, GCs have effects mediated by the recently discovered membrane-bound GC receptor. Binding of GCs with these membrane-bound GC receptors has been shown to alter transduction pathways within minutes, resulting in apoptosis of the cell. Finally, at very high dosages, unspecific effects are caused by physicochemical interactions between GCs and the cellular membrane. GCs influence ion transport of plasma cells via direct contact with the cell membrane, and direct contact with the mitochondrial membranes causes proton leakage.
Effects of Glucocorticoids on Bone and Fracture Risk
Earlier data on the pathogenesis of GIOP were mainly based on histomorphometric data derived from patients treated with high-dose GCs without protective anti-osteoporotic drugs such as bisphosphonates. In these histomorphometric studies, reduced bone formation was observed, characterized by a low mineral apposition rate, which is related to reduced numbers of osteoblasts, while bone resorption was unchanged or even elevated [19].
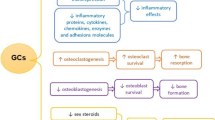
During the past few years, several studies have provided more understanding of the molecular mechanisms involved in GIOP (Fig. 1), which are discussed in more detail below. These include the increased apoptosis of osteoblasts and osteocytes, impaired differentiation of osteoblasts, and increased life span of osteoclasts.

Pathophysiology of glucocorticoid-induced effects on bone cells. BMP—bone morphogenetic protein; Dkk-1—dickkopf-1; GSK3β—glycogen synthase kinase 3β; OPG—osteoprotegerin; PPAR—peroxisome proliferator-activated receptor; RANKL—receptor activator for nuclear factor-κB ligand; Runx2—runt-related protein 2
It has been known for several years that GCs induce apoptosis of osteoblasts and osteocytes. The increased apoptosis of osteoblasts results in a significant reduction in bone formation, and it has been postulated that the loss of osteocytes results in a disrupted osteocyte–canalicular network and failure to respond to bone damage [20]. This may eventually lead to reduced bone strength. However, the mechanism of osteoblast and osteocyte apoptosis was not fully elucidated. Recent data show that GCs induce apoptosis of osteoblasts and osteocytes by activating caspase-3 [21]. Furthermore, apoptosis of osteoblasts was recently found to be related to the activation of glycogen synthase kinase 3β (GSK3β), which plays a role in the Wnt signaling pathway [22]. The Wnt signaling pathway is important in bone metabolism and especially osteoblastogenesis. Normally, binding of Wnt to the low-density lipoprotein receptor–related protein 5 and 6 (LRP5/6) and its co-receptor, frizzled, stabilizes β-catenin, which leads to transcription of target genes and subsequent induction of bone formation. GCs have been shown to suppress this pathway by increasing the production of Wnt pathway inhibitors, such as dickkopf-1 (Dkk-1) [23, 24].
In addition to increased apoptosis of osteoblasts, GCs impair osteoblast function via several pathways. GCs were recently shown to interfere with both the bone morphogenetic protein pathway and the Wnt signaling pathway, thereby inhibiting osteoblast differentiation [25]. As mentioned previously, GCs have been shown to suppress the Wnt signaling pathway by increasing the production of Dkk-1 [23, 24]. Interestingly, silencing Dkk-1 abrogates the GC-induced suppression of osteoblast differentiation [26].
Furthermore, GCs were recently shown to stimulate bone marrow stromal cells—the precursor cells of osteoblasts—to differentiate toward adipocytes instead of osteoblasts. This is mainly mediated through an increased expression of the peroxisome proliferator-activated receptor-γ2 and repression of the osteogenic transcription factor runt-related protein 2 [27]. Recent research suggested that high doses of GCs cause a shift toward adipogenesis via repression of AP-1. Thus, transrepression of AP-1 not only mediates anti-inflammatory actions but also yields reduced bone strength [28].
In contrast to increased apoptosis of osteoblasts and osteocytes, the apoptosis of osteoclasts is reduced during GC treatment. The life span of osteoclasts is extended due to an upregulation of receptor activator for nuclear factor-κB ligand (RANKL) and suppression of osteoprotegerin (OPG) [29]. The GC-mediated suppression of OPG could occur through the Wnt signaling pathway [30]. However, this prolonged life span of osteoclasts may be associated with reduced function. A recent in vitro study showed that direct effects of GCs on osteoclasts result in a suppressed capacity for bone resorption. Such direct effects include, for example, interference with the formation of ruffled border and disruption of the cytoskeleton [31].
Besides direct effects on osteoblasts, osteocytes, and osteoclasts, GCs exert indirect effects on bone. It has been known for a long time that GCs impair bone metabolism via inhibition of calcium resorption in the gastrointestinal tract and inhibition of the renal tubular calcium reabsorption, which may lead to hypocalcemia and a tendency toward hyperparathyroidism. Recently, GCs have been shown to influence the bone mineralization by transrepression of osteocalcin and collagen I, two important proteins of the matrix [32]. Furthermore, GCs may indirectly increase fracture risk via an enhanced risk of falling caused by steroid myopathy [4].
Management of Glucocorticoid-Induced Osteoporosis
General Measures
First, efforts should be made to prescribe GCs in the lowest possible doses and for a short period of time when possible. Patients receiving GC treatment should be advised to stop smoking and limit alcohol intake and should be stimulated to have a sufficient calcium intake and to perform weight-bearing activities daily. Furthermore, an assessment of fall risk is recommended in these patients, who are theoretically at increased risk of falling due to steroid myopathy; a variety of approaches are available [33].
Calcium and vitamin D supplementation have been proven to be essential in the management of GIOP. Patients are advised to have a total calcium intake of at least 1,000 to 1,200 mg/d. An adequate vitamin D level is necessary, and a minimum serum level of greater than 50 nmol/L 25(OH)D3 is advised during the whole year [34]. Vitamin D supplementation in doses of at least 800 IU/d are recommended to achieve these therapeutic levels. However, in some patients, higher doses are needed, probably because some GC users do not go outside often and because it is suggested that GCs interfere with vitamin D absorption. Calcium and vitamin D supplementation are shown to more or less stabilize BMD in patients on chronic GC treatment [35].
Fracture Risk
As discussed previously, the pathophysiology of osteoporotic fragility fractures during GC treatment is multifactorial, as it includes bone- and fall-related factors and is also strongly related to baseline risk. Identifying patients with increased fracture risk solely using bone mass measurements has several disadvantages and shortcomings, such as its age dependency and its inaccuracy in measuring bone quality. Therefore, it has been recommended that fracture risk should be assessed using models that calculate the absolute fracture risk for the individual patient. The disadvantage of using the absolute fracture risk is the lack of consensus regarding when exactly treatment should be initiated. Therefore, any cutoff point will be arbitrary. Several algorithms are presently available to determine how to prevent GIOP and fractures, most based on the daily dosage of prednisone and on the T-score of BMD measurements of the hip and spine. First, the General Practice Research Database, also known as FIGS (Fracture in GIOP Score), has been developed [9]. This model calculates the 5- and 10-year risk of an osteoporotic fracture of the hip, vertebrae, and wrist. The use of this model is somewhat complicated, but this scoring method has the advantage that the underlying disease, the GC dosage, and the fall risk are taken into account. Another frequently used model is the Fracture Risk Assessment (FRAX) tool as proposed by the World Health Organization [36]. This model is unique in that it takes into account the family history and the BMD but excludes the evaluation of the risk factors of falls and the presence or absence of prevalent vertebral deformities, although they are recognized as a risk factor for fractures. Furthermore, GC usage, but not dosage, is recorded in this model. The 10-year fracture risk can be calculated easily on the Internet. Besides being useful in treatment decisions, the assessment of fracture risk may also be useful in improving a patient’s treatment adherence, as it gives the patient a better insight into his or her future fracture risk and into the degree of risk reduction once treatment with anti-osteoporotic drugs is initiated.
Medication
Bisphosphonates have been shown to be effective in preventing GC-induced bone loss in several randomized controlled trials (Table 1) [5, 6, 37–41]. Alendronate improved lumbar spine BMD in patients on long-term GCs after 1 year, while the BMD decreased in patients receiving placebo [39]. After 2 years, this difference was sustained [5]. Interestingly, the greatest increase in BMD occurred within the first year. Two trials addressed the efficacy of risedronate in preventing GC-induced bone loss [6, 38]. One study included patients starting with GCs and found that risedronate prevented bone loss. The other study included patients on long-term GC treatment and showed that risedronate increased the BMD. The average difference in percentage of BMD change among patients using bisphosphonates was plus 4.6% compared with patients using placebo/calcium [37]. Adding vitamin D supplementation to bisphosphonate treatment further enhanced the beneficial effect to an estimated difference of plus 6%.
Furthermore, for both alendronate and risedronate, a reduction in vertebral fractures has been observed in randomized controlled trials involving patients treated with GCs [5, 41]. Alendronate reduced vertebral fracture risk compared with placebo after 2 years of follow-up: the incidence of morphometric vertebral fractures in patients treated with alendronate was 0.7%, compared with 6.8% in placebo-treated patients (P = 0.026) [5]. No reduction in nonvertebral fractures was observed (incidental fractures, 9.8% vs 5.4% [placebo vs alendronate]). Similarly, a 70% reduction in vertebral fracture risk was found for risedronate as compared with placebo [41]. Again, no significant difference was noted in the incidence of nonvertebral fractures. Recently, zoledronic acid, given yearly by intravenous infusion, was shown to prevent GIOP [7••]. In this randomized controlled trial, zoledronic acid was not compared with placebo, but rather with an active comparator: daily risedronate. Zoledronic acid was found to be more effective than risedronate in both the treatment (zoledronic acid, +4.06% spine BMD vs risedronate, +2.71% spine BMD) and prevention subgroups (zoledronic acid, +2.6% spine BMD vs risedronate, +0.64% spine BMD) after 12 months of treatment. In this trial, no statistically significant difference in fracture rate was observed. Obviously, this is related to the fact that in this trial, a comparison with an anti-osteoporotic drug, not placebo, was performed. The larger increase in BMD in the zoledronate group is difficult to interpret because it is not known whether the larger increase in BMD is also reflected in a larger increase in bone strength and/or a larger reduction in fracture rate. Zoledronic acid should be given by intravenous infusion, which may have some practical considerations and an impact on hospital budget (depending on the health system of each specific country). In addition, the risk of renal damage is higher in patients with compromised kidney function (eg, in older adult patients with moderate kidney function and a superimposed infection), leading to dehydration. The effects of ibandronate were recently studied in men after cardiac transplantation. All men received GCs directly after transplantation, and the mean cumulative dose of cortisone was 17 g after 1 year. Men were randomly assigned to ibandronate or placebo, and after 1 year, spine BMD remained unchanged in the men treated with ibandronate, compared with a decline in spine BMD (-25%) in patients treated with placebo. Furthermore, there was a significant difference in number of morphometric vertebral fractures (ibandronate, 13% vs placebo, 53%) [42]. Thus, ibandronate prevents bone loss and vertebral fractures in cardiac transplantation patients on immunosuppressive therapy, including GCs. No data are available on the effects of strontium ranelate and parathyroid hormone (PTH) 1-84 with respect to the prevention of GIOP.
Because the effect of GCs is dominated by their inhibiting effect on bone formation, it is theoretically more or less unexpected that antiresorptive drugs (eg, bisphosphonates) will be useful in GC-treated patients. Recently, the mechanism of bone loss prevention by bisphosphonates has become more clear. Experimental studies demonstrated that bisphosphonates lower the expression of genes that inhibit the mineralization, thereby increasing trabecular bone volume [43]. Theoretically, anabolic agents should be the first choice in GC-treated patients, based on the pathogenesis of GIOP. Clinical studies show that PTH 1-34 (teriparatide) is more effective in preventing GIOP than bisphosphonates. Recently, the anabolic agent teriparatide was compared with the active comparator alendronate in 428 women and men with osteoporosis who received GCs (>5 mg/d) for at least 3 months [44]. After 18 months, the BMD in the lumbar spine increased significantly more in patients receiving teriparatide than in patients receiving alendronate (7.2% vs 3.4%). Remarkably, a difference in the number of patients with new vertebral fractures was observed as well: 0.6% in the teriparatide group versus 6.1% in the alendronate group (P = 0.004). In line with other studies, no difference was found in nonvertebral fracture rate. This finding was confirmed in the follow-up study during another 18 months of treatment [45••]. Interestingly, treatment with teriparatide not only reduced fracture rate but was also shown to reduce back pain and to improve quality of life [46]. No data are available for PTH 1-84 in GC-treated patients. Intervention with PTH 1-34 restored trabecular bone volume, increased bone formation, and increased bone strength in an animal model [43, 47]. PTH treatment counteracts the negative effects of GCs on osteoblast and osteocyte apoptosis. These effects were recently demonstrated to be exerted by increasing the expression of Wnt signaling agonists [43, 47]. Thus far, no clear guidelines exist regarding which GC-treated patients should be offered teriparatide as a first-line drug. Because of the relatively high cost of teriparatide, introduction of teriparatide may have an impact on several budgets. In addition, the inconvenience of injecting teriparatide may limit its use. Nevertheless, teriparatide is attractive not only because of its working mechanism but also because it has been proven to be superior to bisphosphonates in GC-treated patients. Thus, it may be attractive to prescribe teriparatide in patients with a very high risk of fractures (eg, 10-year absolute fracture risk > 20%).
Recent studies on the molecular pathways underlying bone metabolism have identified potential novel therapeutic targets for the management of osteoporosis. For the future, it can be expected that new drugs interfering with the Wnt signaling or RANKL/OPG pathways may prove more effective than current treatment options (eg, bisphosphonates) in reducing GIOP. It is obvious that new anti-osteoporotic drugs for GIOP (eg, denosumab, cathepsin K inhibitors, and monoclonal antibody against sclerostin) should be tested against an active comparator for ethical reasons.
Very recently, the American College of Rheumatology recommendations for the prevention and treatment of GIOP were updated, including renewed recommendations for counseling and monitoring GIOP and updated pharmacotherapeutic interventions [48••]. This update was necessary because of new insights into the value of BMD measurements in identifying patients at risk and because new diagnostic tools and data on therapies were available. These recommendations provide an up-to-date guideline for the management of GIOP for two patient groups: postmenopausal women and men older than 50 years of age, and premenopausal women and men younger than 50 years of age initiating or receiving GC therapy. The subdivision of patients into fracture risk categories (low [<10%], medium [10%–20%], and high [>20%]) results in clear recommendations that are easy to work with in clinical practice. These risk categories were established using the FRAX tool. The FRAX tool is based on a large database, but as mentioned earlier, it unfortunately does not include some important risk factors for fractures, such as daily dose of prednisolone, the presence or absence of prevalent vertebral fractures, and risk of falling. Furthermore, the risk categories were defined by an expert panel rather than based on evidence.
Adherence
Adherence to therapy is also a well-known and important problem in GIOP. A large, retrospective database study recently showed that after 1 year, less than 40% of patients were adherent to treatment [49•]. This low percentage may be explained in several ways. First, anti-osteoporotic drugs are prescribed for prevention of future fractures. In comparable circumstances (eg, patients on blood pressure–lowering drugs), nonadherence is an important issue, whereas in patients with arthritis, adherence is usually better because anti-inflammatory drugs induce relief of pain and inflammation. Second, patients starting on GCs are usually ill and are being informed about their underlying disease, including several possible side effects of GCs, such as hypertension, diabetes, glaucoma, infections, and osteoporosis. Thus, osteoporosis is just one item on the long list of possible side effects, for which it might be necessary to take two or three types of additional drugs. Third, monitoring the effects of anti-osteoporotic drugs is not optimal. The value of BMD testing to monitor therapy and the interval of BMD testing are controversial. Changes in bone markers occur by 2 to 3 months after initiation of treatment with anti-osteoporotic drugs, but measurement of bone marker values is limited in daily clinical practice because of a relatively high analytical variation, circadian rhythm, and costs. It is only possible in some centers that have focused their research on biomarkers and osteoporosis. Finally, (mild) gastrointestinal side effects may occur in 20% to 30% of patients, and 2 to 3 days of fever and flu-like symptoms occur in nearly all patients treated with high-dose bisphosphonates, causing patients to be nonadherent. Furthermore, general factors not directly related to bisphosphonates may come into play. For example, an increased number of prescribed drugs increases an individual’s risk of nonadherence; anti-osteoporotic drugs are usually prescribed in older adults, in whom adherence may be limited due to (early) dementia; and some patients have a general negative opinion against drugs.
Several measures to improve adherence in GIOP have been suggested, such as the model of “shared decision making” [50] and a specialized “GIOP outpatient clinic” [51]. In the model of shared decision making, the patient and physician discuss the pros and cons of starting anti-osteoporotic drugs. It is important to realize that the arguments used may differ between patients and physicians. Moreover, “patients’ health-believe” is a major factor in therapeutic compliance. Using the absolute fracture risk may be very useful, as it gives patients better insight into their future fracture risk and the degree of risk reduction when treatment with anti-osteoporotic drugs is initiated.
An organized program of care called a glucocorticoid-induced osteoporosis program, in which high fracture risk patients were identified, educated, and intensively monitored, proved to be effective in the treatment of patients on chronic GCs [51]. Patients’ knowledge, lifestyle, and serum vitamin D levels all significantly improved, while adherence to therapy was 91%. The challenge for the future will be the development of implementation strategies to translate these care programs from highly specialized clinics to routine daily clinical care.
Conclusions
GIOP is one of the most important causes of secondary osteoporosis and is associated with an evident clinical impact due to increased fracture risk. Increased apoptosis of osteoblasts and osteocytes and increased life span of osteoclasts leads to decreased BMD and bone strength, and increased insight in recent years into the molecular mechanism has suggested an important role for the RANKL/OPG pathway and Wnt signaling route in the pathophysiology of GIOP. To identify patients with increased fracture risk more accurately, new tools to assess absolute fracture risk are available. GIOP can be partially prevented with the use of oral bisphosphonates (alendronate, risdronate) and zoledronic acid. However, oral bisphosphonates are limited by low adherence rates, and zoledronic acid should be given intravenously. In contrast, PTH 1-34 therapy seems to be superior to oral bisphosphonates but is more expensive. The recent advances in the pathophysiology of GIOP have led to the development of new treatment options that will be available in the (very near) future to reduce the incidence of GIOP and improve the quality of life of these patients.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance, •• Of major importance
Kanis JA, Johansson H, Oden A, et al. A meta-analysis of prior corticosteroid use and fracture risk. J Bone Miner Res. 2004;19:893–9.
van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int. 2002;13:777–87.
Gudbjornsson B, Juliusson UI, Gudjonsson FV. Prevalence of long term steroid treatment and the frequency of decision making to prevent steroid induced osteoporosis in daily clinical practice. Ann Rheum Dis. 2002;61:32–6.
Natsui K, Tanaka K, Suda M, et al. High-dose glucocorticoid treatment induces rapid loss of trabecular bone mineral density and lean body mass. Osteoporos Int. 2006;17:105–8.
Adachi JD, Saag KG, Delmas PD, et al. Two-year effects of alendronate on bone mineral density and vertebral fracture in patients receiving glucocorticoids: a randomized, double-blind, placebo-controlled extension trial. Arthritis Rheum. 2001;44:202–11.
Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 1999;42:2309–18.
•• Reid DM, Devogelaer JP, Saag K, et al.: Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009, 373:1253-63. In this double-blind study, zoledronic acid was compared with risedronate in the prevention of GIOP in men and women receiving treatment with prednisolone. Zoledronic acid increased BMD of the spine more than risedronate and showed a faster inhibitory effect on bone markers. Therefore, this study showed that zoledronic acid is an important additional treatment option for GIOP.
Cooper C, Coupland C, Mitchell M. Rheumatoid arthritis, corticosteroid therapy and hip fracture. Ann Rheum Dis. 1995;54:49–52.
van Staa TP, Geusens P, Pols HA, de Laet C, Leufkens HG, Cooper C. A simple score for estimating the long-term risk of fracture in patients using oral glucocorticoids. QJM. 2005;98:191–8.
van Staa TP. The pathogenesis, epidemiology and management of glucocorticoid-induced osteoporosis. Calcif Tissue Int. 2006;79:129–37.
De Vries F, Bracke M, Leufkens HG, et al. Fracture risk with intermittent high-dose oral glucocorticoid therapy. Arthritis Rheum. 2007;56:208–14.
Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int. 2005;16:581–9.
Lems WF. Bisphosphonates and glucocorticoids: effects on bone quality. Arthritis Rheum. 2007;56:3518–22.
van Staa TP, Leufkens HG, Abenhaim L, et al. Use of oral corticosteroids in the United Kingdom. QJM. 2000;93:105–11.
van Staa TP, Laan RF, Barton IP, et al. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum. 2003;48:3224–9.
McKay LI, Cidlowski JA. Cross-talk between nuclear factor-kappa B and the steroid hormone receptors: mechanisms of mutual antagonism. Mol Endocrinol. 1998;12:45–56.
Vayssiere BM, Dupont S, Choquart A, et al. Synthetic glucocorticoids that dissociate transactivation and AP-1 transrepression exhibit antiinflammatory activity in vivo. Mol Endocrinol. 1997;11:1245–55.
Buttgereit F, Straub RH, Wehling M, Burmester GR. Glucocorticoids in the treatment of rheumatic diseases: an update on the mechanisms of action. Arthritis Rheum. 2004;50:3408–17.
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–82.
O’Brien CA, Jia D, Plotkin LI, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–41.
Liu Y, Porta A, Peng X, et al. Prevention of glucocorticoid-induced apoptosis in osteocytes and osteoblasts by calbindin-D28k. J Bone Miner Res. 2004;19:479–90.
Yun SI, Yoon HY, Jeong SY, Chung YS. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3beta. J Bone Miner Metab. 2009;27:140–8.
Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun. 2005;329:177–81.
Wang FS, Ko JY, Yeh DW, Ke HC, Wu HL. Modulation of Dickkopf-1 attenuates glucocorticoid induction of osteoblast apoptosis, adipocytic differentiation, and bone mass loss. Endocrinology. 2008;149:1793–801.
Pereira RC, Delany AM, Canalis E. Effects of cortisol and bone morphogenetic protein-2 on stromal cell differentiation: correlation with CCAAT-enhancer binding protein expression. Bone. 2002;30:685–91.
Butler JS, Queally JM, Devitt BM, et al. Silencing Dkk1 expression rescues dexamethasone-induced suppression of primary human osteoblast differentiation. BMC Musculoskelet Disord. 2010;11:210.
Shi XM, Blair HC, Yang X, McDonald JM, Cao X. Tandem repeat of C/EBP binding sites mediates PPARgamma2 gene transcription in glucocorticoid-induced adipocyte differentiation. J Cell Biochem. 2000;76:518–27.
Carcamo-Orive I, Gaztelumendi A, Delgado J, et al. Regulation of human bone marrow stromal cell proliferation and differentiation capacity by glucocorticoid receptor and AP-1 crosstalk. J Bone Miner Res. 2010;25:2115–25.
Hofbauer LC, Gori F, Riggs BL, et al. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. 1999;140:4382–9.
Kondo T, Kitazawa R, Yamaguchi A, Kitazawa S. Dexamethasone promotes osteoclastogenesis by inhibiting osteoprotegerin through multiple levels. J Cell Biochem. 2008;103(1):335–45.
Kim HJ, Zhao H, Kitaura H, et al. Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest. 2006;116:2152–60.
Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18:1319–28.
Gillespie LD, Robertson MC, Gillespie WJ, et al.: Interventions for preventing falls in older people living in the community. Cochrane Database Syst Rev 2009, (2):CD007146.
Lips P. Relative value of 25(OH)D and 1, 25(OH)2D measurements. J Bone Miner Res. 2009;22:1668–71.
Tang BM, Eslick GD, Nowson C, Smith C, Bensoussan A. Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: a meta-analysis. Lancet. 2007;370:657–66.
Kanis JA, Burlet N, Cooper C, et al. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. 2008;19:399–428.
Amin S, Lavalley MP, Simms RW, Felson DT. The comparative efficacy of drug therapies used for the management of corticosteroid-induced osteoporosis: a meta-regression. J Bone Miner Res. 2002;17:1512–26.
Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res. 2000;15:1006–13.
Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med. 1998;339:292–9.
Stoch SA, Saag KG, Greenwald M, et al. Once-weekly oral alendronate 70 mg in patients with glucocorticoid-induced bone loss: a randomized, placebo-controlled clinical trial. J Rheumatol. 2009;36:1705–14.
Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int. 2000;67:277–85.
Fahrleitner-Pammer A, Piswanger-Soelkner JC, Pieber TR, et al. Ibandronate prevents bone loss and reduces vertebral fracture risk in male cardiac transplant patients: a randomized double-blind, placebo-controlled trial. J Bone Miner Res. 2009;24:1335–44.
Yao W, Cheng Z, Pham A, et al. Glucocorticoid-induced bone loss in mice can be reversed by the actions of parathyroid hormone and risedronate on different pathways for bone formation and mineralization. Arthritis Rheum. 2008;58:3485–97.
Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357:2028–39.
•• Saag KG, Zanchetta JR, Devogelaer JP, et al.: Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: thirty-six-month results of a randomized, double-blind, controlled trial. Arthritis Rheum 2009, 60:3346-55. This article provides the extended results of the randomized controlled trial comparing the anabolic drug teriparatide with the antiresorptive drug alendronate, including data on fracture risk. After 36 months, teriparatide was superior to alendronate in preventing bone loss and in reducing fracture risk in individuals with GIOP.
Langdahl BL, Rajzbaum G, Jakob F, et al. Reduction in fracture rate and back pain and increased quality of life in postmenopausal women treated with teriparatide: 18-month data from the European Forsteo Observational Study (EFOS). Calcif Tissue Int. 2009;85:484–93.
Weinstein RS, Jilka RL, Almeida M, Roberson PK, Manolagas SC. Intermittent parathyroid hormone administration counteracts the adverse effects of glucocorticoids on osteoblast and osteocyte viability, bone formation, and strength in mice. Endocrinology. 2010;151:2641–9.
•• Grossman JM, Gordon R, Ranganath VK, et al.: American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res 2010,62:1515-26. In this article, the new American College of Rheumatology recommendations are presented, offering an up-to-date guideline for GIOP prevention and treatment.
• Netelenbos JC, Geusens PP, Ypma G, Buijs SJ: Adherence and profile of non-persistence in patients treated for osteoporosis-a large-scale, long-term retrospective study in The Netherlands. Osteoporos Int 2010 Sep 14 (Epub ahead of print). Although more knowledge was gained in treatment options for GIOP, this study showed that persistence to osteoporosis medication still is an important factor in daily practice.
de Bekker-Grob EW, Essink-Bot ML, Meerding WJ, Koes BW, Steyerberg EW. Preferences of GPs and patients for preventive osteoporosis drug treatment: a discrete-choice experiment. Pharmacoeconomics. 2009;27:211–9.
Newman ED, Matzko CK, Olenginski TP, et al. Glucocorticoid-Induced Osteoporosis Program (GIOP): a novel, comprehensive, and highly successful care program with improved outcomes at 1 year. Osteoporos Int. 2006;17:1428–34.
Disclosure
Dr. Lems has served as a consultant for MSD, Eli Lilly and Company, Novartis, Servier Laboratories, and Warner Chilcott. Drs. den Uyl and Bultink reported no potential conflicts of interest relevant to this article.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
den Uyl, D., Bultink, I.E.M. & Lems, W.F. Advances in Glucocorticoid-Induced Osteoporosis. Curr Rheumatol Rep 13, 233–240 (2011). https://doi.org/10.1007/s11926-011-0173-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-011-0173-y