
Abstract
Purpose of Review
This review summarizes what has been learned about the interaction between skeletal muscle and bone from mouse models in which BMAL1, a core molecular clock protein has been deleted. Additionally, we highlight several genes which change following loss of BMAL1. The protein products from these genes are secreted from muscle and have a known effect on bone homeostasis.
Recent Findings
Circadian rhythms have been implicated in regulating systems homeostasis through a series of transcriptional-translational feedback loops termed the molecular clock. Recently, skeletal muscle-specific disruption of the molecular clock has been shown to disrupt skeletal muscle metabolism. Additionally, loss of circadian rhythms only in adult muscle has an effect on other tissue systems including bone.
Summary
Our finding that the expression of a subset of skeletal muscle-secreted proteins changes following BMAL1 knockout combined with the current knowledge of muscle-bone crosstalk suggests that skeletal muscle circadian rhythms are important for maintenance of musculoskeletal homeostasis. Future research on this topic may be important for understanding the role of the skeletal muscle molecular clock in a number of diseases such as sarcopenia and osteoporosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Physiology and behavior are temporally coordinated into rhythms coinciding with the 24-h solar cycle [1, 2]. These rhythms, termed circadian (Latin, meaning “about a day”), are present in almost every organism ranging from single cell bacteria to plants and animals [3, 4]. Underlying these rhythms in mammals is a mechanism, termed the molecular clock, which is comprised of a series of interconnected transcriptional-translational feedback loops [5]. This system functions to optimize the timing of cellular events in anticipation of environmental changes such as daylight, food availability, and predator/prey interactions. The molecular clock mechanism is found in virtually all cells within the body including both skeletal muscle and bone [6,7,8,9]. While the intrinsic mechanism is the same across cell types, the molecular clock output is highly tissue-specific [10]. Thus, the ability to synchronize the molecular clock and physiology within a tissue with external day-night/active-inactive cycles provides an evolutionarily conserved method for adapting cells to changing environmental conditions.
The classic relationship between clocks across different tissues historically focused on the top-down ability of the suprachiasmatic nucleus (SCN; termed the master clock) of the hypothalamus to govern the synchronicity of other, peripheral clock systems with its own rhythm [11, 12]. With the rise of models that uncouple the rhythm of the SCN from peripheral clock, it has become apparent that clocks in peripheral tissues are more autonomous than this traditional view [13,14,15,16]. While we know that crosstalk occurs between different tissues, the mechanisms by which clocks in one tissue might influence the physiology of another tissue has not been well studied. In this review, we present the latest findings demonstrating circadian rhythms in skeletal muscle are important for maintenance of its own cellular physiology and its effect on the physiology of other tissues, particularly focusing on the effects of disruption of the skeletal muscle molecular clock on bone health. The existing data, although limited, suggests that skeletal muscle rhythms are important for maintenance of bone [17•]. To date, there is little published work with models of bone cell clock disruption, so understanding bone to muscle crosstalk is unclear [18, 19]. These observations provide a novel role of maintenance of skeletal muscle health in the prevention of bone disease such as osteoporosis.
The Mammalian Molecular Clock
The mechanism underlying circadian rhythms is termed the molecular clock and has been described in detail elsewhere [5, 8]. Operating as the positive arm of the core loop are two members of the PAS-bHLH family of transcription factors, CLOCK (circadian locomotor output control kaput) and Bmal1 (brain muscle arnt-like 1) [20, 21]. These transcription factors heterodimerize and transactivate the negative limb core clock genes (Per1/2/3 and Cry1/2) by binding to E-box elements (5′–CANNTG–3′) in the regulatory regions of these genes. The PER and CRY proteins then accumulate in the cytoplasm, form multimers, and translocate to the nucleus where they inhibit BMAL1:CLOCK transcriptional activity in a process that takes approximately 24 h for a complete feedback cycle.
The orphan nuclear receptors RORα and REV-ERB α/β comprise additional components of the molecular clock. These proteins affect molecular clock function by activating (RORα) or repressing (REV-ERB α/β) Bmal1 transcription [22, 23]. Studies have also implicated kinases (e.g., CK1ε) and E3 ligases (e.g., FBXL3) associated with the proteasome system to tightly regulate the stability and accumulation of PER and CRY proteins [24, 25]. Thus, proper timing of the molecular clock mechanism requires regulation at multiple levels (i.e., transcription, translation, and posttranslational modifications) presenting many targets where environmental cues and physiological function can influence the timing of the molecular clock and cellular circadian rhythms.
In addition to their role in the molecular clock mechanism, components of the molecular clock (Bmal1, Clock, Rev-erb, Rora) have been shown to transcriptionally regulate other genes that are not directly involved in timekeeping. These downstream targets are then designated as clock-controlled genes (CCGs) [1, 26, 27]. While a subset of CCGs is similar in all tissues (e.g., Dbp, Tef), a large percentage of CCGs are tissue-specific [10]. Several tissue-specific transcription factors have been described as targets of the molecular clock [28,29,30,31]. This level of control would set up a transcriptional hierarchy within each tissue by which the core clock controls the expression of tissue-specific transcription factors which in turn acts on tissue-specific transcriptional activity [32]. In this review, we focus on what is known about the tissue-specific functions of the molecular clock in skeletal muscle and how disruption of this mechanism in skeletal muscle affects other tissues.
The Molecular Clock in Skeletal Muscle
Studies seeking to understand the role of the molecular clock in peripheral tissues have largely involved the use of genetic mouse models targeting loss of Bmal1, as this is the only non-redundant gene within the core feedback loop; thus, one of the first mouse models used to study the effects of circadian rhythm disruption was the germline Bmal1 knockout mouse (Bmal1 KO) [21]. Bmal1 KO mice have a shortened lifespan, exhibit features of advanced aging, and develop significant changes to bone architecture as well a variety of other pathologies [33,34,35,36]. These mice also exhibit significant skeletal muscle weakness and structural pathology in the skeletal muscle with altered myofilament architecture and abnormal mitochondrial volume and function [28]. McDearmon et al. rescued Bmal1 in a skeletal muscle-specific manner on the Bmal1 KO background and found that muscle Bmal1 is sufficient to rescue many of the systemic effects found in Bmal1 KO mice but not rhythmic circadian behavior [37]. These results argue that maintenance of the molecular clock specifically within skeletal muscle is sufficient to prevent a number of different systemic phenotypes.
In order to better understand the output of the skeletal muscle molecular clock, our lab and others have described the skeletal muscle circadian transcriptome [26, 38, 39, 40•]. Miller et al. first identified the skeletal muscle transcriptome using both WT and Clock mutant mice [26]. The Clock mutant mice are characterized by a long period length under constant conditions [41]. Molecular and biochemical studies determined that this mutation in Clock resulted in a dominant negative protein isoform and not a null mutation [42]. Comparing the total gene expression changes of these mice, this group found that both rhythmic and nonrhythmic genes were profoundly affected in the tissues of the Clock mutant mice. This work was followed by several publications using skeletal muscle from C57Bl/6 J and inducible, skeletal muscle-specific Bmal1 knockout mice that identified circadian mRNAs [38, 39, 40•, 43]. These studies have confirmed the tissue specificity of the circadian transcriptome in skeletal muscle through their inclusion of known muscle-specific genes that are largely involved in metabolism, transcriptional regulation, and cellular signaling processes. More than 800 skeletal muscle enriched genes including Myod1, Ucp3, and Myh1 display circadian oscillations in their mRNA levels suggesting a link between the molecular clock and muscle homeostasis. Since MYOD1 is both a myogenic regulatory factor and regulated by the molecular clock, it is an ideal candidate to put forth as a transcription factor to direct the tissue specificity of the molecular clock in skeletal muscle and the need for these rhythms to maintain healthy muscle.
To date, four inducible, skeletal muscle-specific Bmal1 knockout mice have been generated to study the role of the endogenous molecular clock in skeletal muscle [40•, 43,44,45]. While these models use the floxed Bmal1 mouse, they differ through the use of different muscle-specific Cre-recombinase mice. However, it is clear through the study of each of these models that skeletal muscle BMAL1 is necessary for maintenance of skeletal muscle metabolism, particularly glucose handling pathways [43, 46]. Skeletal muscle is a predominant contributor to whole-body glucose handling, and muscle insulin resistance is an early sign of metabolic syndrome. Dyar et al. were the first to report that the muscle clock regulates glucose uptake and metabolism [43]. This was followed with a study from our lab comparing gene expression between C57Bl6/J mice over time of day with an inducible, skeletal muscle-specific Bmal1 knockout mice that found a temporal separation of carbohydrate metabolic pathways from lipid metabolic processes [40•, 46]. Most recently, Peek et al. showed anaerobic glycolysis to be regulated by the interaction of the BMAL1:CLOCK heterodimer with HIFα [44]. While each of these models have shown that the molecular clock regulates metabolic pathways in skeletal muscle, Schroder et al.’s study is the only study to date that reports the effect of skeletal muscle-specific loss of Bmal1 on other tissues [17•].
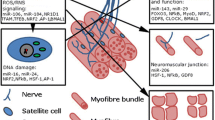
Skeletal Muscle-Bone Coupling
The mechanical relationship between skeletal muscle and bone has been simplified to muscle contractions serving to load and bones acting as attachment sites. This physical coupling of the two tissues is most fully appreciated in development, as they share a common mesenchymal precursor and synchronously develop based on perceived mechanical stimuli [47]. This process continues throughout the adulthood and aging, as bones change their shape and mass due to differing loads from muscle contractions and weight bearing [48]. The relationship between muscle and bone has been outlined in a mechanostat model through which osteocytes monitor deformations, partially resulting from mechanical forces from skeletal muscles, and signal osteoblasts and osteoclasts to change the architecture and mass of the bone accordingly [49]. These adaptations help to maintain homeostasis, as the bone architecture is maintained in an acceptable range. The mechanical coupling would also imply that pathologies in which muscle atrophy occurs would also result in loss of bone mass. This pairing of conditions in muscle and bone is most frequently seen in the aging population in which osteoporosis and sarcopenia are major clinical problems [50].
While the physical coupling of muscle and bone is important for bone health, there is a growing recognition of the secretory capacity of skeletal muscle. Skeletal muscle was shown to release significant amounts of interleukin-6 (IL-6) into the circulation during prolonged exercise [51]. The finding that muscle can secrete factors now termed myokines provided the conceptual framework for how muscles communicate with other organs in vivo. Since this initial study, others have tried to define the skeletal muscle secretome using a number of different models [52, 53, 54, 55, 56, 57•]. While some results differ between these models, a number of the common myokines between these studies are known to affect bone formation and resorption, including IL-6, IL-15, irisin/FNDC5, myostatin, and IGF1. The identification of myokines that affect bone growth provide the best evidence to date that the relationship between skeletal muscle and bone is both mechanical and paracrine in nature.
The Effect of BMAL1 Knockout in Muscle on Bone
Since physical activity follows a circadian pattern, it can be assumed that at least a portion of the load on bone also follows a circadian pattern. Bunger et al. found that the germline deletion of Bmal1 leads to progressive arthropathy but is not essential for bone development [34]. These mice also present with a decreased bone mineral density and decreased muscle force generation [28]. It can be assumed that loss of behavioral rhythmicity and thus rhythmic loading from the muscle on bone plays some role in generating this phenotype. While these findings are important to understand the relationship between these tissues, they do not allow for the study of a particular tissues contribution to the effects of the loss of circadian rhythms on bone. To date, there are limited reports of the impact of loss of Bmal1 targeted to bone specific cell types, but the data suggests that bone resorption may be affected through modulation of RANKL expression in osteoblasts following loss of Bmal1 [18].
Our lab’s finding that skeletal muscle-specific loss of Bmal1 is sufficient to lead to increased bone calcification while maintaining normal cage activity highlights the importance of the skeletal muscle molecular clock in maintaining healthy bone [17•]. iMSBmal1 −/− mice display muscle weakness similar to germline Bmal1 knockout mice. These mice also develop increased calcification of the Achilles’ tendon and reduced cartilage in the foot/ankle and flattened tarsals. This finding was not limited to the hindlimbs, as calcification was also observed in both the ribcage and spine. While the arthropathy is similar to what is found in germline Bmal1 KO mice, the increase in bone calcification was unusual, since most models of muscle weakness display decreased bone calcification. These findings highlight the importance of the skeletal muscle molecular clock for the paracrine relationship between these tissues.
By analyzing published microarray data from Hodge et al., we have identified several myokines that significantly changed expression following skeletal muscle-specific knockout of Bmal1 [40•]. Each of the secreted proteins identified in Table 1 have a known effect on bone, and the mRNA expression levels are significantly changed (p < 0.05) in iMSBmal1 −/− skeletal muscle. Below, we have highlighted the known effects of a few of these myokines and how the changed expression could result in the bone phenotype of adult mice lacking skeletal muscle BMAL1.
Myostatin
Myostatin is a muscle-specific member of the transforming growth factor β superfamily of proteins that is secreted from muscle and is most known for negatively regulating muscle size [74]. Recently, inhibition of the myostatin pathway was shown to increase bone turnover and increased bone mass [69]. Inhibition of the myostatin receptor (ActRIIB) in osteoblasts leads to increased bone formation in mice [75]. Myostatin is also linked to RANKL-induced osteoclast development through promoting the expression of the transcription factor NFATC1 [70•]. Inhibition or loss of myostatin strongly reduces osteoclast formation and bone destruction. Thus, the effect of myostatin on bone is two-fold. It first acts to inhibit osteoblast function through ActRIIB then activates bone resorption through its promotion of osteoclast formation. iMSBmal1 −/− mice display a 30% decrease in myostatin mRNA expression which could explain in part the increased calcification seen in these mice.
IGFBP5
IGFBP5’s role in bone formation is largely known through its ability to bind to the extracellular matrix allowing for IGFs to bind to the surface of bone cells. It is the only IGFBP that has been consistently shown to stimulate osteoblast proliferation [72, 76]. IGFBP5 has also been reported to stimulate osteoclast bone resorption in the presence of osteoblasts [77]. While many of the effects of IGFBP5 on bone formation are positive, overexpression of this protein using an osteocalcin promoter led to decreased bone volume [78]. Thus, it is likely that IGFBP5 works in a dose-dependent manner to regulate bone formation.
There is a 29% decrease in IGFBP5 gene expression following skeletal muscle-specific knockout of Bmal1. The phenotype of these mice could be caused by this change due to a change in the dose of IGFBP5 available on the bone surface. Perhaps at lower doses, IGFBP5 stimulates osteoblast proliferation leading to an increased bone density, while at higher doses, it would lead to an increased stimulation of resorption. Additionally, this protein is positively correlated with growth and bone formation such that it increases during development but decreases in aging and osteoporosis. The apparent decrease in IGFBP5 following skeletal muscle loss of Bmal1 is thus consistent with the idea that the iMSBmal1 −/− mouse is a model of advanced aging.
TGFB1
Transforming growth factor beta 1 is known to be important for the maintenance and expansion of mesenchymal progenitor cells and osteoblasts through the selective MAPKs and SMAD2/3 pathways [61]. TGF-β1 has been shown to promote matrix production and osteoblast differentiation. In fact, TGF-β1 deficient mice present with reduced bone growth and mineralization [79]. Additionally, this protein reduces osteoblast’s ability to secrete RANKL, a potent osteoclast differentiation factor, limiting osteoclast formation and thus affecting bone mass [80]. TGF-β1 is also a common signaling molecule in a variety of other pathways such as the Wnt signaling pathway in osteoblasts and bone morphogenic protein (BMP) signaling pathway in osteocytes [61]. Thus, the effects of increased amounts of this protein in the circulation should lead to increased bone formation and decreased bone resorption.
A chronic increase in TGFB1 expression by muscle could act in a paracrine fashion to contribute to the bone phenotype seen in iMSBmal1 −/− mice. Since this protein is known to promote bone formation and limit osteoclast formation, it might play a role in the increased calcification of bone in this model. Additionally, the convergence of the TGF-β1 signaling pathway with BMP signaling pathways could explain the reduced cartilage staining and increased calcification of the Achilles’ tendon, as this pathway is known to lead to heterotopic ossification.
Irisin
Irisin is a recently discovered myokine that is cleaved from fibronectin type III domain-containing protein 5 (FNDC5), a membrane-bound protein in skeletal muscle that is induced by exercise [81]. This myokine has been primarily studied through its autocrine functions regulating muscle metabolism and endocrine functions leading to the beiging of white adipose tissue; however, it has recently been shown that it also plays a role in regulating osteoblast function [82, 83]. Colaianni et al. showed that at low doses, this hormone-like myokine enhances osteoblast differentiation and increases cortical bone mineral density; thus, it positively modifies bone geometry [58•].
Fndc5, the precursor gene for irisin, increases nearly 14% in iMSBmal1 −/− mice. While this change is small, we believe that a chronic increase in FNDC5 would lead to biologically significant changes on the bone architecture. Since irisin is known to enhance osteoblast differentiation and modify bone geometry, its increase in iMSBmal1 −/− mice could contribute to both the observed increase in calcification as well as the flattening of the tarsals and misshaping of the tibia and fibula of iMSBmal1 −/− mice.
GDF11
Growth differentiation factor 11 (GDF11) is a member of the TGF-β superfamily of proteins whose deletion results in abnormal skeletal patterning during development [84]. There has been significant controversy over the past 3 years regarding GDF11 and its role in muscle aging and regeneration. Initial reports suggested circulating GDF11 levels decline with age-making strategies that increase GDF11 potentially therapeutic [85,86,87]. This concept, however, has been challenged by the findings of several different labs [88,89,90]. Most recently, it was shown that overexpression of GDF11 in mice induces muscle atrophy, inhibits skeletal muscle regeneration, leads to bone loss, and blocks bone resorption; all outcomes that argue that increasing GDF11 would not be a beneficial strategy for musculoskeletal health [73, 89, 91, 92]. We found that the average expression of Gdf11 decreases approximately 17% in iMSBmal1 −/− mice 5 weeks post knockout of Bmal1. We did not measure circulating GDF11 levels, and while the decline in Gdf11 mRNA expression in the iMSBmal1 −/− muscle might mimic changes with age, it is unclear whether these changes are contributing to the bone phenotype seen in these mice.
Conclusions
The role of the endogenous molecular clock in peripheral tissues, such as skeletal muscle, is a rapidly emerging area of research. Recent studies using different models to induce Bmal1 knockout specifically in adult skeletal muscle have concluded that maintenance of circadian rhythms in skeletal muscle is necessary for metabolic homeostasis in skeletal muscle [40•, 43,44,45]. Since these models have normal behavioral (i.e., activity) rhythms, skeletal muscle-specific Bmal1 knockout mice provide a good model for looking at changes in signaling between muscle and bone without affecting rhythmic loading. Schroder et al. reported changes in the bone calcification and joint cartilage deposition in iMSBmal1 −/− mice that is consistent with the increased bone calcification seen in the germline Bmal1 KO mice [17•, 34]. While muscle weakness is usually accompanied by decreases in bone calcification, this model’s altered bone architecture suggests the changes are more likely due to disrupted cytokine/myokine circulation. Using microarray data from this model, we have identified 14 myokines with a known effect on bone homeostasis whose gene expression in skeletal muscle significantly changes following Bmal1 deletion [40•]. These findings suggest that skeletal muscle BMAL1 is important for maintenance of bone health and highlight the importance of skeletal muscle circadian rhythms in musculoskeletal homeostasis, with implications for aging [93]. Increased knowledge of the relationship between the skeletal muscle molecular clock and muscle-bone crosstalk could lead to a better understanding of aging-related diseases such as sarcopenia and osteoporosis. Additionally, uncovering the pathways underlying this relationship could lead to time of day-based intervention strategies (e.g., exercise and dietary restriction), which alter clock genes in muscle to promote healthy aging.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330(6009):1349–54. doi:10.1126/science.1195027.
Schibler U. The 2008 Pittendrigh/Aschoff lecture: peripheral phase coordination in the mammalian circadian timing system. J Biol Rhythm. 2009;24(1):3–15. doi:10.1177/0748730408329383.
Idda ML, Bertolucci C, Vallone D, Gothilf Y, Sanchez-Vazquez FJ, Foulkes NS. Circadian clocks: lessons from fish. Prog Brain Res. 2012;199:41–57. doi:10.1016/B978-0-444-59427-3.00003-4.
Loudon AS. Circadian biology: a 2.5 billion year old clock. Curr Biol. 2012;22(14):R570–1. doi:10.1016/j.cub.2012.06.023.
Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18(3):164–79. doi:10.1038/nrg.2016.150.
Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101(15):5339–46. doi:10.1073/pnas.0308709101.
Schibler U. The daily rhythms of genes, cells and organs. Biological clocks and circadian timing in cells. EMBO Rep. 2005;6:Spec No:S9–13. doi:10.1038/sj.embor.7400424.
Schibler U, Naef F. Cellular oscillators: rhythmic gene expression and metabolism. Curr Opin Cell Biol. 2005;17(2):223–9. doi:10.1016/j.ceb.2005.01.007.
Yamazaki S, Takahashi JS. Real-time luminescence reporting of circadian gene expression in mammals. Methods Enzymol. 2005;393:288–301. doi:10.1016/S0076-6879(05)93012-7.
Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A. 2014;111(45):16219–24. doi:10.1073/pnas.1408886111.
Sollars PJ, Kimble DP, Pickard GE. Restoration of circadian behavior by anterior hypothalamic heterografts. J Neurosci. 1995;15(3 Pt 2):2109–22.
Takahashi JS, DeCoursey PJ, Bauman L, Menaker M. Spectral sensitivity of a novel photoreceptive system mediating entrainment of mammalian circadian rhythms. Nature. 1984;308(5955):186–8.
Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14(23):2950–61.
Zambon AC, McDearmon EL, Salomonis N, Vranizan KM, Johansen KL, Adey D, et al. Time- and exercise-dependent gene regulation in human skeletal muscle. Genome Biol. 2003;4(10):R61. doi:10.1186/gb-2003-4-10-r61.
Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A. 2008;105(39):15172–7. doi:10.1073/pnas.0806717105.
Paschos GK, Ibrahim S, Song WL, Kunieda T, Grant G, Reyes TM, et al. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat Med. 2012;18(12):1768–77. doi:10.1038/nm.2979.
• Schroder EA, Harfmann BD, Zhang X, Srikuea R, England JH, Hodge BA, et al. Intrinsic muscle clock is necessary for musculoskeletal health. J Physiol. 2015;593(24):5387–404. doi:10.1113/JP271436. This is the only study to date to describe the systemic effects, including changes to bone structure, that occur following loss of Bmal1 in adult skeletal muscle.
Takarada T, Xu C, Ochi H, Nakazato R, Yamada D, Nakamura S, Kodama A, Shimba S, Mieda M, Fukasawa K, Ozaki K, Iezaki T, Fujikawa K, Yoneda Y, Numano R, Hida A, Tei H, Takeda S, Eiichi H. Bone resorption is regulated by circadian clock in osteoblasts. J Bone Miner Res. 2017; doi:10.1002/jbmr.3053.
Gorski JP, Huffman NT, Vallejo J, Brotto L, Chittur SV, Breggia A, et al. Deletion of Mbtps1 (Pcsk8, S1p, ski-1) gene in osteocytes stimulates soleus muscle regeneration and increased size and contractile force with age. J Biol Chem. 2016;291(9):4308–22. doi:10.1074/jbc.M115.686626.
Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci U S A. 1998;95(10):5474–9.
Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103(7):1009–17.
Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110(2):251–60.
Sato TK, Panda S, Miraglia LJ, Reyes TM, Rudic RD, McNamara P, et al. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43(4):527–37. doi:10.1016/j.neuron.2004.07.018.
Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152(5):1091–105. doi:10.1016/j.cell.2013.01.055.
Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol. 2007;8(2):139–48. doi:10.1038/nrm2106.
Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A. 2007;104(9):3342–7. doi:10.1073/pnas.0611724104.
Bozek K, Relogio A, Kielbasa SM, Heine M, Dame C, Kramer A, et al. Regulation of clock-controlled genes in mammals. PLoS One. 2009;4(3):e4882. doi:10.1371/journal.pone.0004882.
Andrews JL, Zhang X, McCarthy JJ, McDearmon EL, Hornberger TA, Russell B, et al. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc Natl Acad Sci U S A. 2010;107(44):19090–5. doi:10.1073/pnas.1014523107.
Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, et al. Pancreatic beta cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science. 2015;350(6261):aac4250. doi:10.1126/science.aac4250.
Zhang Y, Fang B, Emmett MJ, Damle M, Sun Z, Feng D, et al. GENE REGULATION. Discrete functions of nuclear receptor Rev-erbalpha couple metabolism to the clock. Science. 2015;348(6242):1488–92. doi:10.1126/science.aab3021.
Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13(7):469–83. doi:10.1038/nrg3242.
Chaix A, Zarrinpar A, Panda S. The circadian coordination of cell biology. J Cell Biol. 2016;215(1):15–25. doi:10.1083/jcb.201603076.
Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006;20(14):1868–73. doi:10.1101/gad.1432206.
Bunger MK, Walisser JA, Sullivan R, Manley PA, Moran SM, Kalscheur VL, et al. Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal-1 locus. Genesis. 2005;41(3):122–32. doi:10.1002/gene.20102.
Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, et al. Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle. 2008;7(9):1197–204. doi:10.4161/cc.7.9.5886.
Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA. Development of dilated cardiomyopathy in Bmal1-deficient mice. Am J Physiol Heart Circ Physiol. 2012;303(4):H475–85. doi:10.1152/ajpheart.00238.2012.
McDearmon EL, Patel KN, Ko CH, Walisser JA, Schook AC, Chong JL, et al. Dissecting the functions of the mammalian clock protein BMAL1 by tissue-specific rescue in mice. Science. 2006;314(5803):1304–8. doi:10.1126/science.1132430.
McCarthy JJ, Andrews JL, McDearmon EL, Campbell KS, Barber BK, Miller BH, et al. Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiol Genomics. 2007;31(1):86–95. doi:10.1152/physiolgenomics.00066.2007.
Pizarro A, Hayer K, Lahens NF, Hogenesch JB. CircaDB: a database of mammalian circadian gene expression profiles. Nucleic Acids Res. 2013;41(Database issue):D1009–13. doi:10.1093/nar/gks1161.
• Hodge BA, Wen Y, Riley LA, Zhang X, England JH, Harfmann BD, et al. The endogenous molecular clock orchestrates the temporal separation of substrate metabolism in skeletal muscle. Skelet Muscle. 2015;5:17. doi:10.1186/s13395-015-0039-5. This paper describes the role of the skeletal muscle molecular clock in temporally regulating genes involved in substrating utilization and storage. The microarrays from this paper were used to determine changes in myokine expression outlined in Table 1.
Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264(5159):719–25.
Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109(3):307–20.
Dyar KA, Ciciliot S, Wright LE, Bienso RS, Tagliazucchi GM, Patel VR, et al. Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol Metab. 2014;3(1):29–41. doi:10.1016/j.molmet.2013.10.005.
Peek CB, Levine DC, Cedernaes J, Taguchi A, Kobayashi Y, Tsai SJ, et al. Circadian clock interaction with HIF1alpha mediates oxygenic metabolism and anaerobic glycolysis in skeletal muscle. Cell Metab. 2017;25(1):86–92. doi:10.1016/j.cmet.2016.09.010.
Nakao R, Shimba S, Oishi K. Muscle Bmal1 is dispensable for the progress of neurogenic muscle atrophy in mice. J Circadian Rhythms. 2016;14(1):1–7. doi:10.5334/jcr.141.
Harfmann BD, Schroder EA, Kachman MT, Hodge BA, Zhang X, Esser KA. Muscle-specific loss of Bmal1 leads to disrupted tissue glucose metabolism and systemic glucose homeostasis. Skelet Muscle. 2016;6:12. doi:10.1186/s13395-016-0082-x.
Olsen BR, Reginato AM, Wang W. Bone development. Annu Rev Cell Dev Biol. 2000;16:191–220. doi:10.1146/annurev.cellbio.16.1.191.
Ferretti JL, Capozza RF, Cointry GR, Garcia SL, Plotkin H, Alvarez Filgueira ML, et al. Gender-related differences in the relationship between densitometric values of whole-body bone mineral content and lean body mass in humans between 2 and 87 years of age. Bone. 1998;22(6):683–90.
Frost HM. Bone’s mechanostat: a 2003 update. Anat Rec A Discov Mol Cell Evol Biol. 2003;275(2):1081–101. doi:10.1002/ar.a.10119.
Go SW, Cha YH, Lee JA, Park HS. Association between sarcopenia, bone density, and health-related quality of life in Korean men. Korean J Fam Med. 2013;34(4):281–8. doi:10.4082/kjfm.2013.34.4.281.
Pedersen BK, Steensberg A, Fischer C, Keller C, Keller P, Plomgaard P, et al. Searching for the exercise factor: is IL-6 a candidate? J Muscle Res Cell Motil. 2003;24(2–3):113–9.
Bortoluzzi S, Scannapieco P, Cestaro A, Danieli GA, Schiaffino S. Computational reconstruction of the human skeletal muscle secretome. Proteins. 2006;62(3):776–92. doi:10.1002/prot.20803.
Catoire M, Mensink M, Kalkhoven E, Schrauwen P, Kersten S. Identification of human exercise-induced myokines using secretome analysis. Physiol Genomics. 2014;46(7):256–67. doi:10.1152/physiolgenomics.00174.2013.
Deshmukh AS, Cox J, Jensen LJ, Meissner F, Mann M. Secretome analysis of lipid-induced insulin resistance in skeletal muscle cells by a combined experimental and bioinformatics workflow. J Proteome Res. 2015;14(11):4885–95. doi:10.1021/acs.jproteome.5b00720.
Pedersen L, Hojman P. Muscle-to-organ cross talk mediated by myokines. Adipocyte. 2012;1(3):164–7. doi:10.4161/adip.20344.
Henningsen J, Rigbolt KT, Blagoev B, Pedersen BK, Kratchmarova I. Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol Cell Proteomics. 2010;9(11):2482–96. doi:10.1074/mcp.M110.002113.
• Perrin L, Loizides-Mangold U, Skarupelova S, Pulimeno P, Chanon S, Robert M, et al. Human skeletal myotubes display a cell-autonomous circadian clock implicated in basal myokine secretion. Mol Metab. 2015;4(11):834–45. doi:10.1016/j.molmet.2015.07.009. This paper was the first to describe the role of the molecular clock in regulating basal myokine secretion.
• Colaianni G, Cuscito C, Mongelli T, Pignataro P, Buccoliero C, Liu P, et al. The myokine irisin increases cortical bone mass. Proc Natl Acad Sci U S A. 2015;112(39):12157–62. doi:10.1073/pnas.1516622112. This paper describes changes in bone mass and strength following weekly injections of irisin, a myokine that is normally secreted following exercise. Findings from this study suggest that the changes to bone mass following exercise are not strictly due to increased loading on the bone, but rather through increased irisin action on osteoblast differentiation.
Hu K, Olsen BR. Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J Clin Invest. 2016;126(2):509–26. doi:10.1172/JCI82585.
Ribeiro N, Sousa SR, Brekken RA, Monteiro F. Role of SPARC in bone remodeling and cancer-related bone metastasis. J Cell Biochem. 2013;115(1):17–26. doi:10.1002/jcb.24649.
Chen G, Deng C, Li YP. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci. 2012;8(2):272–88. doi:10.7150/ijbs.2929.
Lean JM, Murphy C, Fuller K, Chambers TJ. CCL9/MIP-1gamma and its receptor CCR1 are the major chemokine ligand/receptor species expressed by osteoclasts. J Cell Biochem. 2002;87(4):386–93. doi:10.1002/jcb.10319.
Genetos DC, Wong A, Weber TJ, Karin NJ, Yellowley CE. Impaired osteoblast differentiation in annexin A2- and -A5-deficient cells. PLoS One. 2014;9(9):e107482. doi:10.1371/journal.pone.0107482.
Koli K, Wempe F, Sterner-Kock A, Kantola A, Komor M, Hofmann WK, et al. Disruption of LTBP-4 function reduces TGF-beta activation and enhances BMP-4 signaling in the lung. J Cell Biol. 2004;167(1):123–33. doi:10.1083/jcb.200403067.
Amend SR, Uluckan O, Hurchla M, Leib D, Novack DV, Silva M, et al. Thrombospondin-1 regulates bone homeostasis through effects on bone matrix integrity and nitric oxide signaling in osteoclasts. J Bone Miner Res. 2015;30(1):106–15. doi:10.1002/jbmr.2308.
Zhang M, Faugere MC, Malluche H, Rosen CJ, Chernausek SD, Clemens TL. Paracrine overexpression of IGFBP-4 in osteoblasts of transgenic mice decreases bone turnover and causes global growth retardation. J Bone Miner Res. 2003;18(5):836–43. doi:10.1359/jbmr.2003.18.5.836.
Djaafar S, Pierroz DD, Chicheportiche R, Zheng XX, Ferrari SL, Ferrari-Lacraz S. Inhibition of T cell-dependent and RANKL-dependent osteoclastogenic processes associated with high levels of bone mass in interleukin-15 receptor-deficient mice. Arthritis Rheum. 2010;62(11):3300–10. doi:10.1002/art.27645.
Feng S, Madsen SH, Viller NN, Neutzsky-Wulff AV, Geisler C, Karlsson L, et al. Interleukin-15-activated natural killer cells kill autologous osteoclasts via LFA-1, DNAM-1 and TRAIL, and inhibit osteoclast-mediated bone erosion in vitro. Immunology. 2015;145(3):367–79. doi:10.1111/imm.12449.
Bialek P, Parkington J, Li X, Gavin D, Wallace C, Zhang J, et al. A myostatin and activin decoy receptor enhances bone formation in mice. Bone. 2014;60:162–71. doi:10.1016/j.bone.2013.12.002.
• Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med. 2015;21(9):1085–90. doi:10.1038/nm.3917. This paper is the first to describe a direct role of myostatin in osteoclastogenesis. This is the first paper to describe myostatin’s biochemical effect on bone rather than a load-associated effect.
Yang J, Shah R, Robling AG, Templeton E, Yang H, Tracey KJ, et al. HMGB1 is a bone-active cytokine. J Cell Physiol. 2008;214(3):730–9. doi:10.1002/jcp.21268.
Mukherjee A, Rotwein P. Insulin-like growth factor binding protein-5 in osteogenesis: facilitator or inhibitor? Growth Hormon IGF Res. 2007;17(3):179–85. doi:10.1016/j.ghir.2007.01.005.
Liu W, Zhou L, Zhou C, Zhang S, Jing J, Xie L, et al. GDF11 decreases bone mass by stimulating osteoclastogenesis and inhibiting osteoblast differentiation. Nat Commun. 2016;7:12794. doi:10.1038/ncomms12794.
McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387(6628):83–90. doi:10.1038/387083a0.
Park JJ, Berggren JR, Hulver MW, Houmard JA, Hoffman EP. GRB14, GPD1, and GDF8 as potential network collaborators in weight loss-induced improvements in insulin action in human skeletal muscle. Physiol Genomics. 2006;27(2):114–21. doi:10.1152/physiolgenomics.00045.2006.
Mohan S, Nakao Y, Honda Y, Landale E, Leser U, Dony C, et al. Studies on the mechanisms by which insulin-like growth factor (IGF) binding protein-4 (IGFBP-4) and IGFBP-5 modulate IGF actions in bone cells. J Biol Chem. 1995;270(35):20424–31.
Kanatani M, Sugimoto T, Nishiyama K, Chihara K. Stimulatory effect of insulin-like growth factor binding protein-5 on mouse osteoclast formation and osteoclastic bone-resorbing activity. J Bone Miner Res. 2000;15(5):902–10. doi:10.1359/jbmr.2000.15.5.902.
Devlin RD, Du Z, Buccilli V, Jorgetti V, Canalis E. Transgenic mice overexpressing insulin-like growth factor binding protein-5 display transiently decreased osteoblastic function and osteopenia. Endocrinology. 2002;143(10):3955–62. doi:10.1210/en.2002-220129.
Geiser AG, Hummel CW, Draper MW, Henck JW, Cohen IR, Rudmann DG, et al. A new selective estrogen receptor modulator with potent uterine antagonist activity, agonist activity in bone, and minimal ovarian stimulation. Endocrinology. 2005;146(10):4524–35. doi:10.1210/en.2005-0024.
Yasui T, Kadono Y, Nakamura M, Oshima Y, Matsumoto T, Masuda H, et al. Regulation of RANKL-induced osteoclastogenesis by TGF-beta through molecular interaction between Smad3 and Traf6. J Bone Miner Res. 2011;26(7):1447–56. doi:10.1002/jbmr.357.
Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, et al. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 2014;19(2):302–9. doi:10.1016/j.cmet.2013.12.017.
Vaughan RA, Gannon NP, Barberena MA, Garcia-Smith R, Bisoffi M, Mermier CM, et al. Characterization of the metabolic effects of irisin on skeletal muscle in vitro. Diabetes Obes Metab. 2014;16(8):711–8. doi:10.1111/dom.12268.
Colaianni G, Grano M. Role of Irisin on the bone-muscle functional unit. Bonekey Rep. 2015;4:765. doi:10.1038/bonekey.2015.134.
McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Genet. 1999;22(3):260–4. doi:10.1038/10320.
Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344(6184):649–52. doi:10.1126/science.1251152.
Poggioli T, Vujic A, Yang P, Macias-Trevino C, Uygur A, Loffredo FS, et al. Circulating growth differentiation factor 11/8 levels decline with age. Circ Res. 2016;118(1):29–37. doi:10.1161/CIRCRESAHA.115.307521.
Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153(4):828–39. doi:10.1016/j.cell.2013.04.015.
Schafer MJ, Atkinson EJ, Vanderboom PM, Kotajarvi B, White TA, Moore MM, et al. Quantification of GDF11 and myostatin in human aging and cardiovascular disease. Cell Metab. 2016;23(6):1207–15. doi:10.1016/j.cmet.2016.05.023.
Egerman MA, Cadena SM, Gilbert JA, Meyer A, Nelson HN, Swalley SE, et al. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metab. 2015;22(1):164–74. doi:10.1016/j.cmet.2015.05.010.
Rodgers BD, Eldridge JA. Reduced circulating GDF11 is unlikely responsible for age-dependent changes in mouse heart, muscle, and Brain. Endocrinology. 2015;156(11):3885–8. doi:10.1210/en.2015-1628.
Hammers DW, Merscham-Banda M, Hsiao JY, Engst S, Hartman JJ, Sweeney HL. Supraphysiological levels of GDF11 induce striated muscle atrophy. EMBO Mol Med. 2017; doi:10.15252/emmm.201607231.
Lu Q, Tu ML, Li CJ, Zhang L, Jiang TJ, Liu T, et al. GDF11 inhibits bone formation by activating Smad2/3 in bone marrow mesenchymal stem cells. Calcif Tissue Int. 2016;99(5):500–9. doi:10.1007/s00223-016-0173-z.
Duffy JF, Zitting KM, Chinoy ED. Aging and circadian rhythms. Sleep Med Clin. 2015;10(4):423–34. doi:10.1016/j.jsmc.2015.08.002.
Acknowledgements
This work was supported by the University of Florida and the National Institutes of Health grant R01AR066082 to K.A.E.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Karyn Esser and Lance Riley were recipients of grants from National Institutes of Health.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Muscle and Bone
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Riley, L.A., Esser, K.A. The Role of the Molecular Clock in Skeletal Muscle and What It Is Teaching Us About Muscle-Bone Crosstalk. Curr Osteoporos Rep 15, 222–230 (2017). https://doi.org/10.1007/s11914-017-0363-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-017-0363-2